

Abstract
Tubulointerstitial nephritis (TIN) is the most common form of renal involvement in IgG4-related disease. It is characterized by a dominant infiltrate of IgG4-positive plasma cells in the interstitium and storiform fibrosis. Demonstration of IgG4-positive plasma cells is essential for diagnosis, but the number of IgG4-positive cells and the ratio of IgG4-positive/IgG-positive plasma cells may vary from case to case and depending on the methods of tissue sampling even in the same case. IgG4-positive plasma cells can be seen in TIN associated with systemic lupus erythematosus, Sjögren syndrome, or anti-neutrophil cytoplasmic antibody–associated vasculitis, which further add diagnostic confusion and difficulties. To have a more clear view of IgG4-TIN and to delineate differential points from other TIN with IgG4-positive plasma cell infiltrates, clinical and histological features of IgG4-TIN and its mimickers were reviewed. In the rear part, cases suggesting overlap of IgG4-TIN and its mimickers and glomerulonephritis associated with IgG4-TIN were briefly described.
Keywords: IgG4-related disease; Lupus nephritis; Sjögren’s syndrome; Anti-neutrophil cytoplasmic antibody-associated vasculitis; Glomerulonephritis, membranous
IgG4-related disease (IgG4-RD) is a systemic fibro-inflammatory disorder involving almost any organ in the body [1-3]. Tubulointerstitial nephritis (TIN) is the most common form of renal involvement, which characterizes a dominant interstitial infiltrate of IgG4-positive plasma cells and storiform fibrosis [1]. Although TIN showing similar histologic features have been reported previously [4], a connection with IgG4-RD demonstrating IgG4-positive cells in the interstitium was first reported in 2004 [5,6]. Since then, case studies and collective reviews on TIN with dominant IgG4-positive cell infiltrate (IgG4-TIN) have been rapidly cumulated during the next 10 years [7,8]. Presently, we have more comprehensive understanding on renal manifestations of this systemic disease, but at the same time, we have come to recognize cases showing variable histology and wide clinical spectrum, some of which do not fit into the narrow spectrum of IgG4-TIN.
IgG4 is unique as it does not activate complements. The role of IgG4 in inflammation and immune deposits has not been clarified yet. Nonetheless, the presence of IgG4-positive plasma cells is a characteristic feature of IgG4-TIN as the name is adopted, and immune deposits may be observed in some cases. The degree of IgG4-positive cell infiltrate and its ratio among the infiltrating cells may vary from case to case and they depend on the sampling methods even in the same case. IgG4-positive plasma cells may be seen in other diseases and may be numerous in some cases of autoimmune diseases [9]. Furthermore, clinical and laboratory features characteristic of IgG4-TIN may be present in TIN of systemic lupus erythematosus (SLE), Sjögren syndrome, or anti-neutrophil cytoplasmic antibody (ANCA)–associated vasculitis. It is important to distinguish TIN cases because therapeutic plans and prognosis may differ depending on the causes.
To have an overview of TIN with IgG4-positive plasma cell infiltrates and to delineate hints for differential diagnosis, clinical and histological features of IgG4-TIN and its mimickers are reviewed. In the rear part, atypical TIN cases showing clinical and laboratory overlaps of IgG4-TIN and its mimickers and glomerulonephritis associated with IgG4-TIN are briefly described.
TUBULOINTERSTITIAL NEPHRITIS IN IMMUNOGLOBULIN G4-RELATED DISEASE
Renal histology is fundamental in the diagnosis of TIN in IgG4-RD. Three features are characteristic: (1) interstitial lymphoplasmacytic infiltrates with dominant IgG4-positive plasma cells; (2) the ratio of IgG4-positive/IgG-positive plasma cells over 40%; and (3) obliterative phlebitis. A cut-off value of >10 IgG4-positive plasma cells/high-power field (HPF) and/or ratio of IgG4-positive/IgG-positive plasma cells >40% was used in the previous Japanese study [10]. Soon after, in the consensus guideline on IgG4-RD in 2012 [11], different cut-off values were applied in the number of IgG4-positive plasma cells according to the type of specimen received. In renal biopsy samples, >10 IgG4-positive plasma cells/HPF are enough, but >30 IgG4-positive plasma cells/HPF are required in nephrectomy specimens. The infiltrate may be patchy in distribution; therefore, the possibility of IgG4-RD should not be excluded based on negative biopsy results, especially in the presence of other supportive clinical and imaging features of IgG4-RD. The IgG4/IgG ratio of plasmacytic infiltration over 40% was maintained in the consensus guideline, which is a reasonable value as it demonstrated a sensitivity of 58.8% and a specificity of 90.2% in a meta-analysis [12]. In the lymphocytic infiltrates, T lymphocytes predominate over B cells. Eosinophils are common and may be numerous in some cases. Inflammatory infiltrates may extend into the renal capsule, which has not been known in TIN of other non-infectious causes [13,14] (Fig. 1A–D). Glomeruli are usually spared, but when glomerulonephritis is associated, membranous nephropathy is the most common [15]. Vascular changes are not common, but renal arteritis was reported in one case associated with TIN [16].
Fig. 1.
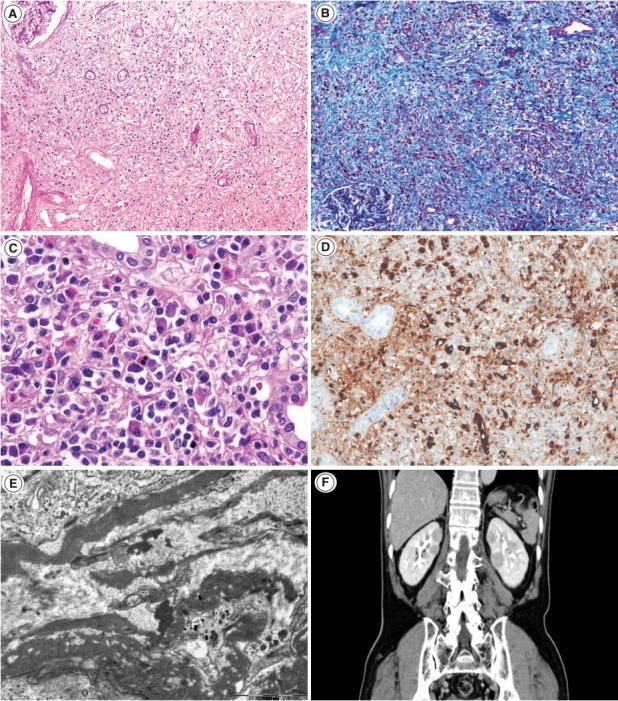
Tubulointerstitial nephritis in IgG4-related disease. (A, B) At lower power, interstitial fibrosis is severe and shows a focal streaming pattern with mixed inflammatory infiltration of lymphocytes and plasma cells (A, periodic-acid Schiff ×100; B, Masson trichrome ×100). (C) In some cases, eosinophil infiltration may be prominent (hematoxylin-eosin. ×400). (D) Many IgG4-positive plasma cells are present in the interstitium (IgG4, ×200). (E) On electron microscopy, fine granular electron-dense deposits are present in the interstitium (×15,000). (F) Contrast-enhanced computed tomography shows patchy multiple round or wedge-shaped parenchymal low-density lesions in both kidneys.
Tubulointerstitial immune deposits may be seen in some cases [17]. IgG and C3 are deposited most commonly along the tubular basement membrane [18]. Interstitial immune deposits tend to be restricted in inflamed areas and they are regarded as a late change [18]. By electron microscopy, electron-dense deposits are frequently found in the tubular basement membrane and interstitium [13] (Fig. 1E). In an earlier report of idiopathic hypocomplementemic TIN, which is now regarded as a form of IgG4-TIN, fingerprint organoid deposits were present in the interstitium in two out of nine cases [4]. Glomerular mesangial and Bowman’s capsular deposits were also frequently observed under electron microscopy, even though glomeruli showed no significant changes by light and immunofluorescent microscopy [19].
Most IgG4-TIN patients are males in mid-sixties. Renal involvement was reported in six of 132 (4.5%) [20], 10 of 114 (8.8%),21 54 of 235 (23.0%) [22], and 20 of 57 (35.1%) patients [23] with IgG4-RD. Patients present with acute renal failure, urinary abnormalities, or mass formation with urinary obstruction [7,19]. Urinalysis showed mild proteinuria in 82.6% and hematuria in 34.8% in one collective study [7]. If membranous nephropathy is associated, nephrotic range proteinuria may be present [15]. In the presence of renal failure, serum creatinine levels may be elevated. Serology shows polyclonal hypergammaglobulinemia and elevated IgG4 levels. Serum IgG4 levels (>135 mg/dL) may be elevated in up to 93% of IgG4-TIN [7,8,10], which are higher than the prevalence of about 70% in total IgG4-RD. This high rate may be related to the organ specificity, but it may reflect the increased number of involved organs because other organs are frequently involved at the time of diagnosis [22]. Serum IgG4 levels have been reported to decrease with steroid treatment, and increase with relapse. However, it is neither essential nor specific for the diagnosis. Elevated IgG4 serum levels were reported in 10.8% of SLE and 12.9% of rheumatoid arthritis patients [24]. IgG4 levels may even show a paradoxical response, showing an increase despite effective treatment [25]. Other serologic markers associated with autoimmune diseases or allergies have been frequently reported. Hypocomplementemia, elevated IgE levels, and eosinophilia have been reported in 56.0%–69.6%, 71.4%, and 33.0%–47.8%, respectively [7,8]. Antinuclear antibodies (ANA) and rheumatoid factors, usually in low titers, were reported in 31.0%–69.6% and 38.9%, respectively. Anti-DNA antibody was positive in a few cases.
Although histology may be highly suggestive of IgG4-TIN, confirmatory diagnosis relies on both histological and clinical features. In addition to IgG4-positive plasma cells >10/HPF, the presence of at least one other feature from the imaging studies, serology, or other organ involvement categories was suggested for the diagnosis [8]. Cases showing clinical and laboratory features suggestive of SLE, Sjögren syndrome, or ANCA-associated vasculitis should be excluded [1]. Other organ involvement is frequent at the time of diagnosis of TIN (83.0%–95.7%). Among them, the salivary glands (82.6%), lymph nodes (43.5%), pancreas (39.1%), and lacrimal glands (30.4%) are most frequently involved either synchronously or metachronously (Table 1) [7,8,13,26-28]. In cases of renal involvement without extrarenal manifestation [7], imaging studies may be helpful (Fig. 1F). Four patterns of round or wedge-shaped renal cortical nodules, peripheral cortical lesions, mass-like lesions, and renal pelvic involvement, were reported [23].
Table 1.
Review of IgG4-related tubulointerstitial nephritis
| References | No. of cases | Age (median, yr) | Male:Femlae | Extrarenal lesion | Serum IgG (median, mg/dL) | Serum lgG4 (median, mg/dL) | SCr at biopsy (mean) | Elevated Cr (>1.2 mg/dL) | ANA (+) | RF (+) | Proteinuria (> 1 g/day) | Hematuria | Renal biopsy finding |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Saeki et al. [7] | 23 | 40-83 (64) | 20:3 | Sa (19), LN (10), Pa (9), La (7), Lu (6), Li (1), Pr (1) | 2,721-8,841 (4,387) | 305-4,630 (1,330) | 0.67-6.87 (1.98) | 13/23 | 16/23 | 7/18 | 2/23 | 8/23 | TIN (23/23), MGN (1/23), mild MPGN (3/23), focal segmental EC (1/23) |
| Raissian et al. [8] | 35 | 20-84 (67) | 30:5 | Sa (6), LN (8), Pa (15), La (1), Lu (8), Li (7), RP(3) | - | - | 0.9-9.0 (3.57) | 27/35 | 10/32 | - | 8/27 | 6/27 | TIN (35/35), MGN (2/35), many eosinophils (4/35) |
| Kawano et al. [28] | 20 | 55-83 (70) | 18:2 | Jo (1), La (2), Li (1), LN (5), Lu (6), Ne (1), Pa (7), Pr (2), RP (1), Sa (12) | 1,679-5,380 (3,596) | 408-1,860 (828) | 0.59-7.26 (1.36) | 12/20 | - | - | 2/15 | - | TIN (20/20), MPGN (1), IgAN (1), EC (2), HSPN (2), MGN (3) |
| Yamaguchi et al. [13] | 16 | 45-78 (62) | 12:4 | Pa (8), Sa (7), RP (1), Lu (1), Li (1) | 1,569-6,328 (3,604) | 142-2,120 (958) | 0.84-6.17 (1.6) | 12/16 | - | - | 3/11 | - | TIN (16/16), MGN (2) |
SCr, serum creatinine; Cr, creatinine; ANA, antinuclear antibodies; RF, rheumatoid factor; Sa, salivary gland; LN, lymph node; Pa, pancreas; La, lacrimal gland; Lu, lung; Li, liver; Pr, prostate; TIN, tubulointerstitial nephritis; MGN, membranous nephropathy; MPGN, membranoproliferative glomerulonephritis; EC, endocapillary hypercellularity; RP, retroperitoneum; Jo, joint; Ne, nerve; IgAN, IgA nephropathy; HSPN, Henoch-Schönlein purpura nephritis.
IgG4-TIN responds well to steroid therapy with decrease of serum creatinine levels. A recent report of repeated biopsy after steroid treatment showed advanced fibrosis but decreased inflammatory activity with fewer IgG4-positive plasma cells and reduced expression of connective tissue growth factor mRNA [29]. Regarding disease activity, elevated serum IgG4 levels and IgG4-positive plasmablast levels were suggested in one study [30].
TUBULOINTERSTITIAL NEPHRITIS IN LUPUS NEPHRITIS
Lupus nephritis is usually characterized by proliferative glomerulonephritis with massive immune deposits and accompanying mild to moderate interstitial inflammation. Rarely, it may present with predominant TIN without significant glomerular changes [31-37]. Up to now, about 20 cases of predominant lupus TIN have been reported. The patients presented with acute renal failure or renal insufficiency. Interstitial inflammatory infiltrate was composed of mixture of CD4+ and CD8+ T cells, B cells, macrophages, and plasma cells. IgG, C3, and C1q deposits were present in the tubular basement membranes, whereas glomerular deposits were negative or minimal. Electron-dense deposits could be seen in tubular basement membranes and interstitium (Fig. 2).
Fig. 2.
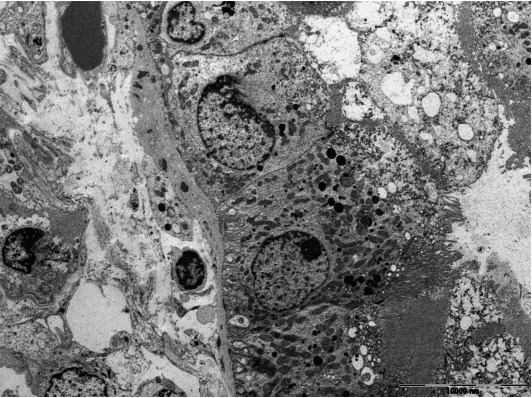
Tubulointerstitial nephritis in lupus nephritis. Granular electron-dense deposits are present in the tubular basement membrane (×3,000).
IgG4-positive plasma cells may infiltrate in the interstitium and IgG4 deposits may be present in the peritubular interstitium and along the tubular basement membrane in lupus TIN [36], similar to IgG4-TIN. In contrast to IgG4-TIN, immune deposits in lupus TIN are rather diffuse. C1q deposits, if prominent, favors lupus TIN. However, if mass-effects on an imaging study or patchy inflammatory infiltrate extending to renal capsule and beyond are present, it is unlikely to be lupus TIN. Massive eosinophil infiltration is also exceptional for lupus.
Distinction between IgG4-RD and SLE depends on the clinical diagnostic criteria, despite some differential histologic features. The American College of Rheumatology (ACR) and Systemic Lupus International Collaborating Clinics (SLICC) criteria may be applied to the diagnosis of SLE. Depending on the criteria applied, a variable proportion of TIN cases with IgG4-positive plasma cells may be categorized into SLE or other autoimmune diseases [10]. However, clinical distinction between IgG4-TIN and lupus TIN is not always simple. Signs of SLE may appear late and may not fulfill the diagnostic criteria at the time of biopsy [38]. Clinical and laboratory features which frequently present in IgG4-RD may also be present in SLE patients [36]. Serum gammaglobulin [35] may be elevated. Serum IgG4 levels were elevated in 10.8% of SLE patients [24]. Retroperitoneal fibrosis may be present [39]. A response to steroid treatment is also good in predominant lupus TIN [35]. Kiyama et al. [40] reported ANA subclasses in SLE and IgG4-RD, demonstrating IgG1, 2, or 3 subclasses in SLE and predominantly IgG2 in IgG4-RD, but very rare or no IgG4 in both conditions.
TUBULOINTERSTITIAL NEPHRITIS IN SJÖGREN SYNDROME
Sjögren syndrome is characterized by keratoconjunctivitis sicca and xerostomia due to immunologic destruction of lacrimal and salivary glands. Renal involvement is infrequent. Distal renal tubular acidosis and acute kidney injury are the main clinical manifestations. Chronic TIN is the most commonly observed form on renal biopsy, but glomerulonephritis may also be observed [41,42]. In TIN of Sjögren syndrome, lymphocytes with a T-cell dominance and macrophages infiltrate in the interstitium along with mild tubulitis and tubular atrophy (Fig. 3A, B). Plasma cells may be numerous but storiform fibrosis is not a feature [43]. Immune deposits may be present in the tubular basement membrane.
Fig. 3.
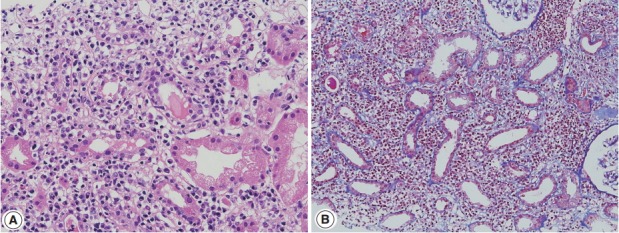
Tubulointerstitial nephritis in Sjögren syndrome. (A) Lymphoplasmacytic infiltrate is present in the interstitium associated with moderate tubulitis (hematoxylin-eosin, ×200). (B) Interstitium is widened by edema and cellular infiltrate without glomerular lesions (Masson trichrome, ×100).
Clinically, symptoms of dry eye and mouth or arthralgia are more frequent in Sjögren syndrome than in IgG4-RD. The presence of anti–SS-A and anti–SS-B antibodies is characteristic and was present in 71% and 54%, respectively, in one large series [41]. Steroid therapy in Sjögren syndrome has limited effects compared with that in IgG4-RD [44]. However, clinical and laboratory features may infrequently overlap with IgG4-TIN. Serum IgG4 levels were reported to be elevated in 7.5% of the patients with primary Sjögren syndrome [24]. One TIN patient with Sjögren syndrome showed elevated serum IgG4 levels and renal interstitial IgG4-positive plasma cell infiltrate [45]. Marked hypocomplementemia was reported in a TIN case of Sjögren syndrome [46]. Some patients had ‘‘pseudolymphoma’’ lesions or autoimmune pancreatitis and sclerosing cholangitis [24]. By contrast, seven of 23 IgG4-TIN cases fulfilled the criteria of Sjögren syndrome [7], and anti–SS-A antibody was present in 4.4% of Mikulicz’s disease [47].
TUBULOINTERSTITIAL NEPHRITIS IN ANTI-NEUTROPHIL CYTOPLASMIC ANTIBODY-ASSOCIATED VASCULITIS
Tubulointerstitial inflammation is frequently associated with ANCA-associated vasculitis. Interstitial inflammatory infiltrate is composed of lymphocytes, plasma cells, and some neutrophils. In typical cases, glomerular crescents or necrotizing lesions are commonly found with or without vasculitis and the distinction from IgG4-RD is not difficult. IgG4-positive plasma cells may be present, but the number of IgG4-positive plasma cells and/or IgG4-positive/IgG-positive plasma cell ratio is usually not high [9]. In addition, elevated C-reactive protein (CRP) or erythrocyte sedimentation rate (ESR) is in favor of ANCA-associated vasculitis.
Rarely, IgG4-dominant TIN may present with concomitant cytoplasmic ANCA and antibody to proteinase 3 [48,49]. Among the three types of ANCA-associated vasculitis, eosinophilic granulomatosis with polyangiitis (EGPA; Churg-Strauss syndrome) shows a close similarity with IgG4-RD, in terms of upper airway involvement and eosinophilia. It may be related to up-regulation of Th2 cytokines associated with increased IgG4 response [2]. Yamamoto et al. [50] reported elevated serum IgG4 levels and increased IgG4-positive/IgG-positive plasma cell ratio and also IgG4 renal infiltrate in EGPA patients. Vaglio et al. [51] showed that IgG4 levels correlated with the disease activity in EGPA patients. Chang et al. [52] showed increased IgG4-positive cells in 18.6% of 43 cases of granulomatosis with polyangiitis (Wegener’s granulomatosis) including four kidney samples, but the cases were limited to sinonasal or orbital/periorbital biopsies. A case of ANCA-negative EGPA showed salivary gland swelling, high serum IgG4 levels, membranous nephropathy with eosinophil-rich TIN, and leukocytoclastic vasculitis [53].
TUBULOINTERSTITIAL NEPHRITIS IN OTHER CONDITIONS SIMULATING IMMUNOGLOBULIN G4-TUBULOINTERSTITIAL NEPHRITIS
Tubulointerstitial nephritis and uveitis with dominant IgG4-positive plasma cells was suggested as a form of IgG4-TIN [55], but it was not supported by others [56]. Sakairi et al. [57] reported a case of ANCA-negative renal small-vessel vasculitis with IgG4-TIN. Recently, a case of multiple organ involvement remarkably similar to that of IgG4-RD was reported. The patient showed multiple hypodense renal lesions in radiographic examination and lymphoplasmacytic infiltrates with storiform fibrosis, but did not have accompanying elevated serum IgG4 and IgG4-positive plasma cell infiltration [58]. IgG4-TIN was reported in a patient with chronic lymphocytic leukemia [59], and in a renal allograft patient [60].
CLINICAL AND HISTOLOGICAL OVERLAP BETWEEN IMMUNOGLOBULIN G4-TUBULOINTERSTITIAL NEPHRITIS AND TUBULOINTERSTITIAL NEPHRITIS OF AUTOIMMUNE DISEASES
Cases showing clinical, laboratory, and histological overlap of IgG4-TIN and TIN of autoimmune diseases have been introduced in previous sections [24,45,47,53], and summarized in Table 2. These overlapping features may partly have roots in autoimmune mechanisms. IgG4-RD demonstrates immunologic derangement in cytokine profiles and activation of regulatory T cells [2]. Practically, the overlap causes dilemma and difficulties in diagnosis. Even though each disease accompanies unique histological features, confirmatory diagnosis is made by the diagnostic criteria of each disease. The ACR and SLICC criteria for SLE have been well established and used, but they have been modified by consensus among the experts, statistical results, and pathogenetic mechanisms [61]. Sjögren syndrome also has complex diagnostic criteria incorporating clinical features and histological findings. As mentioned previously, the diagnostic criteria of IgG4-RD are also composed of a combination of histological, clinical, imaging, and laboratory features. Furthermore, these complex diagnostic criteria may evolve as our understanding of the pathogenetic mechanisms and clinical course of the disease extends, as in SLE. In fact, some cases that had originally been diagnosed as rheumatoid arthritis, Sjögren syndrome or antiphospholipid syndrome were re-categorized into IgG4-RD [62]. Even if we apply the above diagnostic criteria, symptoms are protean and laboratory abnormalities and organ involvement may vary in each individual patient. Clinical and laboratory findings supportive of autoimmune diseases or IgG4-RD may not be apparent at the time of biopsy [45]. Furthermore, in some cases it is not possible to distinguish IgG4-TIN from TIN of autoimmune diseases due to overlapping clinical and laboratory features. Cases in this gray zone should be collected and reserved in a separate category until we have more clear understanding on the pathogenetic mechanisms of IgG4-RD and so we can unveil the possible link between them.
Table 2.
Cases showing overlap features between IgG4-TIN and TIN of autoimmune diseases
| Age | Sex | Serum IgG (mg/dL) | Serum IgG4 (mg/dL) | Antibody | SCr (mg/dL) | Proteinuria | Systemic complications | IgG4/HPF | IgG4/IgG (%) | IF | EM | Diagnosis of kidney biopsy | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| lgG4-RD and Sjogren's syndrome | 49 | F | 6,000 | 2,790 | Anti-SS-A (1:16) | - | - | Chronic hepatitis (3), portal hypertension (2), retroperitoneal ficrosis (1), renal involve (1) | 21.7 | 42.1 | - | - | - | Yamamoto et al. [47] |
| 43 | F | 1,898 | 188 | Anti-SS-A (1:16) | - | - | - | - | - | - | - | |||
| 48 | F | 3,009 | 768 | Anti-SS-A (1:4) | . | . | 11.3 | 23.5 | . | . | . | |||
| 56 | F | 1,890 | 694 | Anti-SS-A (1:16) | - | - | 9.7 | 19.4 | - | - | - | |||
| 59 | M | 1,880 | 339 | Anti-SS-A (1:64) | - | - | - | - | - | - | - | |||
| 73 | M | 1,912 | 374 | Anti-SS-A (1:16) | - | - | 6.7 | 21.3 | - | - | - | |||
| 61 | M | 2,558 | 774 | Anti-SS-A (1:16) | - | - | 14.7 | 33.4 | - | - | - | |||
| lgG4-RD and Sjogren's syndrome | 62 | F | 8,478 | 647 | ANA (1:10,240, homogeneous), anti-SS-A (+), anti-SS-B (+) | 0.92 | 0.82 g/gCr | General malaise, dry mouth, Raynaud’s phenomenon, anemia, lower extremity weakness, hypergammaglobulinemia | 15 | - | No immunoglobulin or complement deposition | - | Chronic plasma cell-rich TIN | Kawano et al. [45] |
| lgG4-RD and Churg-Strauss Syndrome | 68 | F | 1,997 | 275 | ANCA (–), anti–SS-A (–), RF (+) | 0.9 | 1.2 g/day | Asthma, multifocal pulmonary infiltrates, marked eosinophilia, a rash on feet, right median nerve paralysis, salivary gland swelling | - | 10 | IgG, C3 (granular, capillary), lgG1, lgG4 (+) | Electron-dense to electron-lucent subepithelial deposits in glomerular capillary walls | MGN (stage lll-IV) with eosinophil-rich TIN | Ayuzawa et al. [53] |
| IgG4-RD and Lupus nephritis | 71 | F | lgG1:1,230, lgG2:735, lgG3:418 | 37.1 | ANA (1.320 homogeneous) | 9.65 | 2.6 g/day | Abdominal pain, vomiting, diarrhea, epigastric tenderness, bilateral lower extremity pitting edema, marked leukocytosis, hypoalbuminemia, no skin changes | 13 | - | IgG, K, L (2+, granular, mesangial), IgM, IgA, C3 (1+, granular, mesangial) | Small paramesangial and scattered small electron dense to electron lucent subepithelial and intramembranous deposits | IgG4-related TIN with MGN, and/or lupus membranous nephritis with TIN | Zaarour et al. [54] |
| ANCA (–), anti–SS-A (–), anti–SS-B (–), anti-dsDNS (–), anti-Sm (–), anti-GBM (–) |
IgG4-TIN, tubulointerstitial nephritis with dominant IgG4-positive cell infiltrate; SCr, serum creatinine; HPF, high-power field; IF, immunofluorescence; EM, electron microscopy; IgG4-RD, IgG4-related disease; ANA, antinuclear antibodies; TIN, tubulointerstitial nephritis; ANCA, anti-neutrophil cytoplasmic antibody; RF, rheumatoid factor; MGN, membranous nephropathy; GBM, glomerular basement membrane; K. kappa light chain; L, lambda light chain.
GLOMERULONEPHRITIS ASSOCIATED WITH IMMUNOGLOBULIN G4-TUBULOINTERSTITIAL NEPHRITIS
Glomerulonephritis may develop in the setting of IgG4-RD. Membranous nephropathy is the most common, and about 30 cases have been reported in the English literature so far [26,63-69]. Membranous nephropathy presented concurrently with TIN in most case reports, but in one collective series, four of nine cases presented with only glomerulonephritis [15]. Nephrotic syndrome is a frequent manifestation at the time of biopsy [15]. Glomerular histology is similar to that of idiopathic membranous nephropathy, except for a negative staining against phospholipase A2 receptor (PLA2R) antibodies. By light microscopy, glomerular cellularity is normal and the basement membrane is mildly thickened with occasional spikes (Fig. 4A). Immunofluorescence shows granular staining of IgG along the peripheral capillary walls (Fig. 4B). IgG4 is usually dominant among the IgG subclasses and C3 deposits are frequent. On electron microscopy, electron dense-deposits are present mainly in the subepithelial and occasionally in the intramembranous areas, ranging from stage I to III.
Fig. 4.
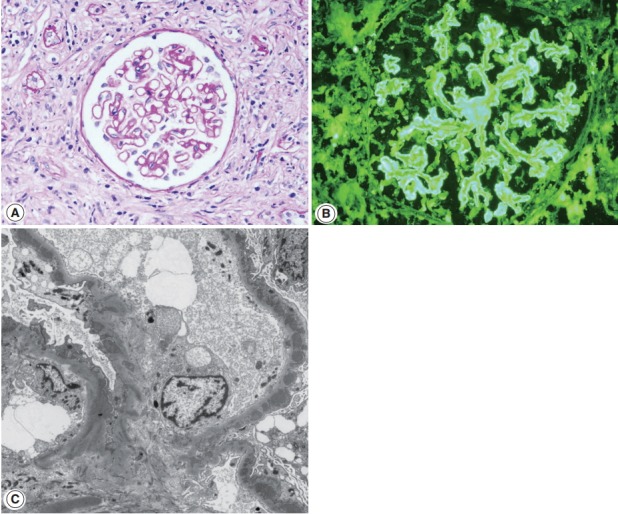
Membranous nephropathy associated with IgG4-TIN. (A) Glomerular basement membrane is thickened with occasional spikes (periodic acid-Schiff, ×200). (B) Granular staining of IgG is present along the peripheral capillary walls (IgG, ×200). (C) Subepithelial electron-dense deposits are seen (×3,000).
Membranous nephropathy of IgG4-RD is sometimes difficult to differentiate from lupus membranous nephritis. Although glomerular IgA, IgM, or C1q deposits are rare in IgG4-RD, strong C1q deposits have been reported in a few cases of IgG4-RD. Mesangial or subendothelial deposits were present in three and four of nine cases of membranous nephropathy associated with IgG4-RD, respectively [15]. Tubuloreticular inclusions, which are regarded as a characteristic feature of lupus nephritis [70], were observed in one case of IgG4-RD [15]. Some authors proposed differential IgG subclass staining, which demonstrated strong IgG2 staining in lupus but not in IgG4-RD. However, it has not been verified in other studies [15]. IgG3 staining intensity similar to or exceeding that of IgG4 [71] or IgG2 [72] was reported in membranous lupus nephritis, but its comparison with that in IgG4-RD has not been done. Similar to TIN, distinction between membranous nephropathy associated with IgG4-RD and lupus membranous nephritis could not be made with certainty in a few cases. In a recent report of a case of membranous nephropathy, clinical features of both SLE and IgG4-RD were present [69]. Features suggestive of SLE were vitiligo, elevated ESR and CRP, hypocomplementemia, positive ANA and weakly positive anti-dsDNA antibody and atypical pANCA, while increased serum IgG4 levels, sialadenitis, lymphadenopathy, large kidneys, and marked hepatomegaly favored IgG4-RD. We experienced a case of membranous nephropathy in a patient satisfying ARA and SLICC criteria for SLE, but the patient also had retroperitoneal fibrosis.
Except for membranous nephropathy, Henoch-Schönlein purpura nephritis [73-75], membranoproliferative [76], and endocapillary proliferative glomerulonephritis [77] have been reported anecdotally. Cases with Henoch-Schönlein purpura nephritis had mesangial IgA deposition in addition to TIN. It is unclear whether these glomerulonephritides have a direct relation with IgG4-TIN or develop co-incidentally. In a case of endocapillary proliferative glomerulonephritis with crescent formation, the patient had hydroureteronephrosis due to retroperitoneal fibrosis and elevated circulating immune complexes [77], which might raise a suspicion of urinary tract infection causing glomerulonephritis.
Footnotes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Saeki T, Kawano M. IgG4-related kidney disease. Kidney Int. 2014;85:251–7. doi: 10.1038/ki.2013.393. [DOI] [PubMed] [Google Scholar]
- 2.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 3.Divatia M, Kim SA, Ro JY. IgG4-related sclerosing disease, an emerging entity: a review of a multi-system disease. Yonsei Med J. 2012;53:15–34. doi: 10.3349/ymj.2012.53.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kambham N, Markowitz GS, Tanji N, Mansukhani MM, Orazi A, D’Agati VD. Idiopathic hypocomplementemic interstitial nephritis with extensive tubulointerstitial deposits. Am J Kidney Dis. 2001;37:388–99. doi: 10.1053/ajkd.2001.21320. [DOI] [PubMed] [Google Scholar]
- 5.Uchiyama-Tanaka Y, Mori Y, Kimura T, et al. Acute tubulointerstitial nephritis associated with autoimmune-related pancreatitis. Am J Kidney Dis. 2004;43:e18–25. doi: 10.1053/j.ajkd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19:474–6. doi: 10.1093/ndt/gfg477. [DOI] [PubMed] [Google Scholar]
- 7.Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016–23. doi: 10.1038/ki.2010.271. [DOI] [PubMed] [Google Scholar]
- 8.Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22:1343–52. doi: 10.1681/ASN.2011010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houghton DC, Troxell ML. An abundance of IgG4+ plasma cells is not specific for IgG4-related tubulointerstitial nephritis. Mod Pathol. 2011;24:1480–7. doi: 10.1038/modpathol.2011.101. [DOI] [PubMed] [Google Scholar]
- 10.Kawano M, Saeki T, Nakashima H, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615–26. doi: 10.1007/s10157-011-0521-2. [DOI] [PubMed] [Google Scholar]
- 11.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–92. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 12.Deng C, Li W, Chen S, et al. Histopathological diagnostic value of the IgG4+/IgG+ ratio of plasmacytic infiltration for IgG4-related diseases: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2015;94:e579. doi: 10.1097/MD.0000000000000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi Y, Kanetsuna Y, Honda K, et al. Characteristic tubulointerstitial nephritis in IgG4-related disease. Hum Pathol. 2012;43:536–49. doi: 10.1016/j.humpath.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Yoshita K, Kawano M, Mizushima I, et al. Light-microscopic characteristics of IgG4-related tubulointerstitial nephritis: distinction from non-IgG4-related tubulointerstitial nephritis. Nephrol Dial Transplant. 2012;27:2755–61. doi: 10.1093/ndt/gfr761. [DOI] [PubMed] [Google Scholar]
- 15.Alexander MP, Larsen CP, Gibson IW, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int. 2013;83:455–62. doi: 10.1038/ki.2012.382. [DOI] [PubMed] [Google Scholar]
- 16.Sharma SG, Vlase HL, D’Agati VD. IgG4-related tubulointerstitial nephritis with plasma cell-rich renal arteritis. Am J Kidney Dis. 2013;61:638–43. doi: 10.1053/j.ajkd.2012.07.031. [DOI] [PubMed] [Google Scholar]
- 17.Nagamachi S, Ohsawa I, Sato N, et al. Immune complex-mediated complement activation in a patient with IgG4-related tubulointerstitial nephritis. Case Rep Nephrol Urol. 2011;1:7–14. doi: 10.1159/000330664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornell LD, Chicano SL, Deshpande V, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol. 2007;31:1586–97. doi: 10.1097/PAS.0b013e318059b87c. [DOI] [PubMed] [Google Scholar]
- 19.Nishi S, Imai N, Yoshida K, Ito Y, Saeki T. Clinicopathological findings of immunoglobulin G4-related kidney disease. Clin Exp Nephrol. 2011;15:810–9. doi: 10.1007/s10157-011-0526-x. [DOI] [PubMed] [Google Scholar]
- 20.Koizumi S, Kamisawa T, Kuruma S, et al. Organ correlation in IgG4-related diseases. J Korean Med Sci. 2015;30:743–8. doi: 10.3346/jkms.2015.30.6.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812–9. doi: 10.1097/PAS.0b013e3181f7266b. [DOI] [PubMed] [Google Scholar]
- 22.Inoue D, Yoshida K, Yoneda N, et al. IgG4-related disease: dataset of 235 consecutive patients. Medicine (Baltimore) 2015;94:e680. doi: 10.1097/MD.0000000000000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlachou PA, Khalili K, Jang HJ, Fischer S, Hirschfield GM, Kim TK. IgG4-related sclerosing disease: autoimmune pancreatitis and extrapancreatic manifestations. Radiographics. 2011;31:1379–402. doi: 10.1148/rg.315105735. [DOI] [PubMed] [Google Scholar]
- 24.Mavragani CP, Fragoulis GE, Rontogianni D, Kanariou M, Moutsopoulos HM. Elevated IgG4 serum levels among primary Sjogren’s syndrome patients: do they unmask underlying IgG4-related disease? Arthritis Care Res (Hoboken) 2014;66:773–7. doi: 10.1002/acr.22216. [DOI] [PubMed] [Google Scholar]
- 25.Goh TL, Cicovic A, Sapsford T, Semple D. A case of immunoglobulin G4 (IgG4) tubulointerstitial nephritis with delayed elevation of serum IgG4 levels. Intern Med J. 2015;45:788–90. doi: 10.1111/imj.12803. [DOI] [PubMed] [Google Scholar]
- 26.Watson SJ, Jenkins DA, Bellamy CO. Nephropathy in IgG4-related systemic disease. Am J Surg Pathol. 2006;30:1472–7. doi: 10.1097/01.pas.0000213308.43929.97. [DOI] [PubMed] [Google Scholar]
- 27.Yoneda K, Murata K, Katayama K, et al. Tubulointerstitial nephritis associated with IgG4-related autoimmune disease. Am J Kidney Dis. 2007;50:455–62. doi: 10.1053/j.ajkd.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 28.Kawano M, Mizushima I, Yamaguchi Y, et al. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:609795. doi: 10.1155/2012/609795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arai H, Hayashi H, Takahashi K, et al. Tubulointerstitial fibrosis in patients with IgG4-related kidney disease: pathological findings on repeat renal biopsy. Rheumatol Int. 2015;35:1093–101. doi: 10.1007/s00296-014-3153-5. [DOI] [PubMed] [Google Scholar]
- 30.Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67:2466–75. doi: 10.1002/art.39205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cunningham E, Provost T, Brentjens J, Reichlin M, Venuto RC. Acute renal failure secondary to interstitial lupus nephritis. Arch Intern Med. 1978;138:1560–1. [PubMed] [Google Scholar]
- 32.Case records of the Massachusetts General Hospital Weekly clinicopathological exercise. Case 53-1976. N Engl J Med. 1976;295:1526–32. doi: 10.1056/NEJM197612302952710. [DOI] [PubMed] [Google Scholar]
- 33.Gur H, Kopolovic Y, Gross DJ. Chronic predominant interstitial nephritis in a patient with systemic lupus erythematosus: a follow up of three years and review of the literature. Ann Rheum Dis. 1987;46:617–23. doi: 10.1136/ard.46.8.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tron F, Ganeval D, Droz D. Immunologically-mediated acute renal failure of nonglomerular origin in the course of systemic lupus erythematosus [SLE]: report of two cases. Am J Med. 1979;67:529–32. doi: 10.1016/0002-9343(79)90806-4. [DOI] [PubMed] [Google Scholar]
- 35.Mori Y, Kishimoto N, Yamahara H, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus nephritis. Clin Exp Nephrol. 2005;9:79–84. doi: 10.1007/s10157-004-0338-3. [DOI] [PubMed] [Google Scholar]
- 36.Omokawa A, Wakui H, Okuyama S, et al. Predominant tubulointerstitial nephritis in a patient with systemic lupus erythematosus: phenotype of infiltrating cells. Clin Nephrol. 2008;69:436–44. doi: 10.5414/cnp69436. [DOI] [PubMed] [Google Scholar]
- 37.Singh AK, Ucci A, Madias NE. Predominant tubulointerstitial lupus nephritis. Am J Kidney Dis. 1996;27:273–8. doi: 10.1016/s0272-6386(96)90553-3. [DOI] [PubMed] [Google Scholar]
- 38.Alpers CE, Hopper J, Jr, Bernstein MJ, Biava CG. Late development of systemic lupus erythematosus in patients with glomerular “fingerprint” deposits. Ann Intern Med. 1984;100:66–8. doi: 10.7326/0003-4819-100-1-66. [DOI] [PubMed] [Google Scholar]
- 39.Okada H, Takahira S, Sugahara S, Nakamoto H, Suzuki H. Retroperitoneal fibrosis and systemic lupus erythematosus. Nephrol Dial Transplant. 1999;14:1300–2. doi: 10.1093/ndt/14.5.1300. [DOI] [PubMed] [Google Scholar]
- 40.Kiyama K, Yoshifuji H, Kandou T, et al. Screening for IgG4-type anti-nuclear antibodies in IgG4-related disease. BMC Musculoskelet Disord. 2015;16:129. doi: 10.1186/s12891-015-0584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramos-Casals M, Brito-Zerón P, Seror R, et al. Characterization of systemic disease in primary Sjogren’s syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology (Oxford) 2015;54:2230–8. doi: 10.1093/rheumatology/kev200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kidder D, Rutherford E, Kipgen D, Fleming S, Geddes C, Stewart GA. Kidney biopsy findings in primary Sjögren syndrome. Nephrol Dial Transplant. 2015;30:1363–9. doi: 10.1093/ndt/gfv042. [DOI] [PubMed] [Google Scholar]
- 43.Pijpe J, Vissink A, Van der Wal JE, Kallenberg CG. Interstitial nephritis with infiltration of IgG-kappa positive plasma cells in a patient with Sjögren’s syndrome. Rheumatology (Oxford) 2004;43:108–10. doi: 10.1093/rheumatology/keg429. [DOI] [PubMed] [Google Scholar]
- 44.Umehara H, Okazaki K, Masaki Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22:1–14. doi: 10.1007/s10165-011-0508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawano M, Suzuki Y, Yamada K, et al. Primary Sjögren’s syndrome with chronic tubulointerstitial nephritis and lymphadenopathy mimicking IgG4-related disease. Mod Rheumatol. 2015;25:637–41. doi: 10.3109/14397595.2013.844303. [DOI] [PubMed] [Google Scholar]
- 46.Yukawa N, Tsuboi N, Yukawa S, et al. Marked hypocomplementemia and tubulointerstitial nephritis in a male patient with Sjögren’s syndrome. Mod Rheumatol. 2004;14:164–8. doi: 10.1007/s10165-004-0285-6. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto M, Takahashi H, Shinomura Y. Are Sjögren’s syndrome and IgG4-related disease able to coexist? Mod Rheumatol. 2015;25:970–1. doi: 10.3109/14397595.2014.948950. [DOI] [PubMed] [Google Scholar]
- 48.Kronbichler A, Gut N, Zwerina J, Neuwirt H, Rudnicki M, Mayer G. Extending the spectrum of a chameleon: IgG4-related disease appearing as interstitial nephritis and mimicking anti-neutrophil cytoplasmic antibody-associated vasculitis. Rheumatology (Oxford) 2015;54:1936–8. doi: 10.1093/rheumatology/kev248. [DOI] [PubMed] [Google Scholar]
- 49.Perez Alamino R, Martínez C, Espinoza LR. IgG4-associated vasculitis. Curr Rheumatol Rep. 2013;15:348. doi: 10.1007/s11926-013-0348-9. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto M, Takahashi H, Suzuki C, et al. Analysis of serum IgG subclasses in Churg-Strauss syndrome: the meaning of elevated serum levels of IgG4. Intern Med. 2010;49:1365–70. doi: 10.2169/internalmedicine.49.3532. [DOI] [PubMed] [Google Scholar]
- 51.Vaglio A, Strehl JD, Manger B, et al. IgG4 immune response in Churg-Strauss syndrome. Ann Rheum Dis. 2012;71:390–3. doi: 10.1136/ard.2011.155382. [DOI] [PubMed] [Google Scholar]
- 52.Chang SY, Keogh KA, Lewis JE, et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener’s): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol. 2013;44:2432–7. doi: 10.1016/j.humpath.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 53.Ayuzawa N, Ubara Y, Keiichi S, et al. Churg-Strauss syndrome with a clinical condition similar to IgG4-related kidney disease: a case report. Intern Med. 2012;51:1233–8. doi: 10.2169/internalmedicine.51.6074. [DOI] [PubMed] [Google Scholar]
- 54.Zaarour M, Weerasinghe C, Eter A, El-Sayegh S, El-Charabaty E. An overlapping case of lupus nephritis and IgG4-related kidney disease. J Clin Med Res. 2015;7:575–81. doi: 10.14740/jocmr2189w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugimoto T, Tanaka Y, Morita Y, Kume S, Uzu T, Kashiwagi A. Is tubulointerstitial nephritis and uveitis syndrome associated with IgG4-related systemic disease? Nephrology (Carlton) 2008;13:89. doi: 10.1111/j.1440-1797.2007.00870.x. [DOI] [PubMed] [Google Scholar]
- 56.Houghton D, Troxell M, Fox E, Rosenbaum J. TINU (tubulointerstitial nephritis and uveitis) syndrome is not usually associated with IgG4 sclerosing disease. Am J Kidney Dis. 2012;59:583–4. doi: 10.1053/j.ajkd.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 57.Sakairi T, Okabe S, Hiromura K, et al. A case of ANCA-negative renal small-vessel vasculitis with tubulointerstitial infiltration of IgG4-positive plasma cells. Mod Rheumatol. 2014 May 20; doi: 10.3109/14397595.2014.915510. [Epub]. http://dx.doi.org/10.3109/14397595.2014.915510. [DOI] [PubMed] [Google Scholar]
- 58.Hara S, Kawano M, Mizushima I, et al. A condition closely mimicking IgG4-related disease despite the absence of serum IgG4 elevation and IgG4-positive plasma cell infiltration. Mod Rheumatol. 2014 Jun 2; doi: 10.3109/14397595.2014.916836. [Epub]. http://dx.doi.org/10.3109/14397595.2014.916836. [DOI] [PubMed] [Google Scholar]
- 59.Malone AF, Sparks MA, Howell DN, Middleton JP, Smith SR, Lehrich RW. IgG4-related tubulointerstitial nephritis associated with chronic lymphocytic leukemia. J Nephrol. 2013;26:1195–8. doi: 10.5301/jn.5000298. [DOI] [PubMed] [Google Scholar]
- 60.Nishikawa K, Takeda A, Masui S, et al. A case of IgG4-positive plasma cell-rich tubulointerstitial nephritis in a kidney allograft mimicking IgG4-related kidney disease. Nephrology (Carlton) 2014;19 Suppl 3:52–6. doi: 10.1111/nep.12250. [DOI] [PubMed] [Google Scholar]
- 61.Rekvig OP, Van der Vlag J. The pathogenesis and diagnosis of systemic lupus erythematosus: still not resolved. Semin Immunopathol. 2014;36:301–11. doi: 10.1007/s00281-014-0428-6. [DOI] [PubMed] [Google Scholar]
- 62.Soliotis F, Mavragani CP, Plastiras SC, Rontogianni D, Skopouli FN, Moutsopoulos HM. IgG4-related disease: a rheumatologist’s perspective. Clin Exp Rheumatol. 2014;32:724–7. [PubMed] [Google Scholar]
- 63.Cravedi P, Abbate M, Gagliardini E, et al. Membranous nephropathy associated with IgG4-related disease. Am J Kidney Dis. 2011;58:272–5. doi: 10.1053/j.ajkd.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 64.Fervenza FC, Downer G, Beck LH, Jr, Sethi S. IgG4-related tubulointerstitial nephritis with membranous nephropathy. Am J Kidney Dis. 2011;58:320–4. doi: 10.1053/j.ajkd.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 65.Jindal N, Yadav D, Passero C, et al. Membranous nephropathy: a rare renal manifestation of IgG4-related systemic disease. Clin Nephrol. 2012;77:321–8. doi: 10.5414/cn107037. [DOI] [PubMed] [Google Scholar]
- 66.Li XL, Yan TK, Li HF, et al. IgG4-related membranous nephropathy with high blood and low urine IgG4/IgG ratio: a case report and review of the literature. Clin Rheumatol. 2014;33:145–8. doi: 10.1007/s10067-013-2406-0. [DOI] [PubMed] [Google Scholar]
- 67.Palmisano A, Corradi D, Carnevali ML, et al. Chronic periaortitis associated with membranous nephropathy: clues to common pathogenetic mechanisms. Clin Nephrol. 2010;74:485–90. doi: 10.5414/cnp74485. [DOI] [PubMed] [Google Scholar]
- 68.Saeki T, Imai N, Ito T, Yamazaki H, Nishi S. Membranous nephropathy associated with IgG4-related systemic disease and without autoimmune pancreatitis. Clin Nephrol. 2009;71:173–8. doi: 10.5414/cnp71173. [DOI] [PubMed] [Google Scholar]
- 69.Stylianou K, Maragkaki E, Tzanakakis M, Stratakis S, Gakiopoulou H, Daphnis E. Acute interstitial nephritis and membranous nephropathy in the context of IgG4-related disease. Case Rep Nephrol Dial. 2015;5:44–8. doi: 10.1159/000369924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jennette JC, Iskandar SS, Dalldorf FG. Pathologic differentiation between lupus and nonlupus membranous glomerulopathy. Kidney Int. 1983;24:377–85. doi: 10.1038/ki.1983.170. [DOI] [PubMed] [Google Scholar]
- 71.Haas M. IgG subclass deposits in glomeruli of lupus and nonlupus membranous nephropathies. Am J Kidney Dis. 1994;23:358–64. doi: 10.1016/s0272-6386(12)80997-8. [DOI] [PubMed] [Google Scholar]
- 72.Omokawa A, Komatsuda A, Nara M, et al. Distribution of glomerular IgG subclass deposits in patients with membranous nephropathy and anti-U1 ribonucleoprotein antibody. Nephrol Dial Transplant. 2012;27:1937–41. doi: 10.1093/ndt/gfr571. [DOI] [PubMed] [Google Scholar]
- 73.Tamai R, Hasegawa Y, Hisano S, Miyake K, Nakashima H, Saito T. A case of IgG4-related tubulointerstitial nephritis concurrent with Henoch-Schonlein purpura nephritis. Allergy Asthma Clin Immunol. 2011;7:5. doi: 10.1186/1710-1492-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ito K, Yamada K, Mizushima I, et al. Henoch-Schönlein purpura nephritis in a patient with IgG4-related disease: a possible association. Clin Nephrol. 2013;79:246–52. doi: 10.5414/CN107114. [DOI] [PubMed] [Google Scholar]
- 75.Yang H, Choi SK, Kim B, et al. IgG4-related tubulointerstitial nephritis accompanied by Henoch-Schonlein purpura. Korean J Med. 2014;87:96–100. [Google Scholar]
- 76.Morimoto J, Hasegawa Y, Fukushima H, et al. Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis. Intern Med. 2009;48:157–62. doi: 10.2169/internalmedicine.48.1339. [DOI] [PubMed] [Google Scholar]
- 77.Katano K, Hayatsu Y, Matsuda T, et al. Endocapillary proliferative glomerulonephritis with crescent formation and concurrent tubulointerstitial nephritis complicating retroperitoneal fibrosis with a high serum level of IgG4. Clin Nephrol. 2007;68:308–14. doi: 10.5414/cnp68308. [DOI] [PubMed] [Google Scholar]