

Abstract
Hypophosphatasia is a genetic disorder, characterised by a dysfunctional tissue-non-specific isoenzyme of alkaline phosphatase that impacts bone metabolism and predisposes to osteomalacia or rickets. The clinical presentation is very diverse, depending on the age of onset and the severity of the disease. Several forms of hypophosphatasia are recognised. We present a case of a 50-year-old woman with low impact fractures and loss of teeth at a young age. She also had a low alkaline phosphatase and was diagnosed with adult hypophosphatasia. Although the severe forms of hypophosphatasia are rather rare, the adult form is thought to occur quite frequently. As this condition is not well known by healthcare professionals, the time to diagnosis and initiation of adequate treatment is often postponed. When encountering a patient with low alkaline phosphatase, low bone density or a history of bone fractures, the possibility of hypophosphatasia should be considered.
Background
Hypophosphatasia (HPP) (or phosphoethanolaminuria) is a genetic disorder of the ALPL-gene, coding for tissue-non-specific alkaline phosphatase (TNSALP). HPP affects bone metabolism and predisposes to rickets or osteomalacia. Severe cases of HPP are quite rare, however, the adult form is thought to be far more prevalent. Mornet et al estimated the prevalence of the less severe forms, such as adult HPP, to be 1/6370. In a population with osteomalacia the prevalence will even be higher. This means that HPP is not very rare—the diagnosis is probably missed frequently. Nevertheless, it can easily be diagnosed. By missing this diagnosis patients risk being treated with bisphosphonates, which can aggravate their symptoms; patients will also be denied family screening, which may have profound consequences.
Case presentation
A 50-year-old Caucasian woman presented at our hospital for her recently discovered euthyroid goitre. Her previous medical history revealed stress fractures of the tarsal bones followed by complex regional pain syndrome. She had several surgical interventions for varices on her legs as well as foot surgery after an accident. She had a low impact fracture of the fourth metatarsal of the right foot, which healed very slowly (figure 1). She lost all her teeth in her 20s. Premenopausal osteoporosis was diagnosed by her primary care physician. The patient stopped smoking 10 years earlier and did not take alcohol. Professionally, she was a shop clerk. She used calcium supplements 1 g a day, cholecalciferol 25 000 units per week and alendronate 70 mg once weekly. Blood pressure was 140/70 mm Hg and pulse rate 90/min. She weighed 71.5 kg and her body mass index was 24.8 kg/m². Physical examination was unremarkable, aside from a slightly enlarged thyroid gland.
Figure 1.
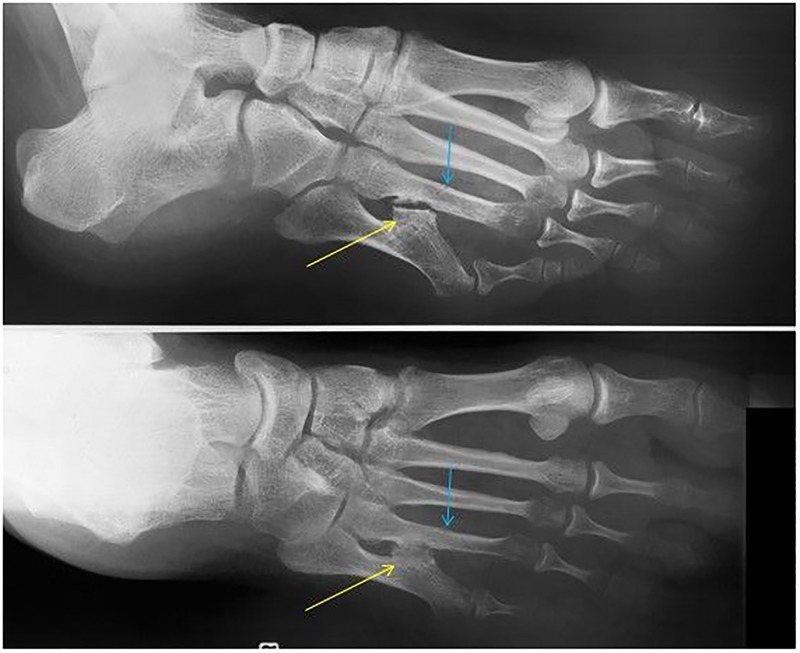
A deformed aspect of the fifth metatarsal with posteromedial bowing and subluxation of the fifth metatarsophalangeal joint. Presence of an exostosis (yellow arrow) at the posteromedial side of the mid-diaphyseal fifth metatarsal, with development of a neo-articulation with the diaphysis of the fourth metatarsal. The blue arrow indicates a sharply delineated transverse mid-diaphyseal fracture line of the fourth metatarsal without displacement. The absence of callus formation and periosteal bone reaction along with the location and shape, fits a recent and complete stress fracture. Note the periarticular demineralisation, mainly in the metatarsal heads 3–5.
Investigations
Standard blood work up was performed for a general check-up. The laboratory findings showed a normal full blood count, normal biochemistry but an unmeasurable low ALP of less than 0.34 μkat/L (0.65–2.30) and low 25-OH-vitamin D of 24.7 nmol/L (78–156). Calcium, phosphate and albumin were within normal ranges. Subsequent analysis showed elevated vitamin B6 (pyridoxine) of 378 nmol/L (26–102). Previous radiographic findings were compatible with a fracture of the fourth metatarsal bone of the right foot and signs of osteoporosis. Skeletal scintigraphy revealed increased uptake near the medial cuneiform bone of the right foot. Since the biology results and the previous medical history were suggestive of adult HPP, a genetic analysis was conducted to investigate the ALPL-gene, coding for TNSALP. This showed a heterozygote mutation c. 484A>G (P. Gly162Ser), confirming the diagnosis of hypophosphatasia. Genetic analysis is, however, not strictly necessary to make the diagnosis of HPP.
Differential diagnosis
The low ALP that was picked up in a standard blood test, together with the patient’s medical record of a low impact fracture, teeth loss at a young age and premenopausal osteoporosis, prompted us to consider the diagnosis of HPP. Subsequently, a vitamin B6 analysis was carried out and came back higher than normal, confirming our suspicion. We decided to conduct a genetic analysis to find the responsible mutation; this test was positive and confirmed the diagnosis of HPP. Genetic analysis is not strictly necessary to make the diagnosis. It is enough to have a compelling medical history of fractures or teeth loss, low ALP that cannot be explained otherwise and X-ray or bone densitometry showing low-density bone mass.
The differential diagnosis of a low ALP depends, of course, on the age of the patient and includes: pernicious anaemia, multiple myeloma, starvation, coeliac disease, milk alkali syndrome, hypothyroidism, Cushing's disease, ia, Wilson's disease, vitamin D intoxication, vitamin C deficiency, magnesium or zinc deficiency, radioactive heavy metals, massive transfusion, osteogenesis imperfecta type 2, improperly collected blood sample, clofibrate therapy, inappropriate reference range and cardiac bypass.
When these differentials are ruled out and low ALP is associated with bone fractures, non-healing fractures, teeth loss at a young age and low bone density, the diagnosis of HPP is more likely. Low ALP in itself is not sufficient to make a definite diagnosis of HPP. It should also be noted that bisphosphonates also can lower bone-specific ALP levels, but total ALP rarely falls below the lower limit of the normal reference range.1
Treatment
Bisphosphonates were discontinued as they could worsen the clinical course of the disease. Oral vitamin D supplements were continued as the patient's vitamin D level was very low. Vitamin D and calcium supplements are only warranted in the case of a proven deficiency. In patients with muscle aches, classical analgaesics can be prescribed. In 2012, a trial was conducted with ENB-0040 (asfotase alfa), a recombinant human TNSALP that specifically targets the bone. This enzyme replacement therapy showed promising results in children, but is not yet available for clinical use. The only other treatment option would be the use of teriparatide, a human recombinant parathyroid hormone. The patient was stable and did not develop new fractures, so there was no need for further treatment beyond vitamin D supplements.
Outcome and follow-up
The patient is seen regularly at our outpatient centre and is doing well. As of now, her symptoms are under control and there is no need for further, more specific therapy. A family screening is being conducted to rule out a TNSALP mutation in her family. Bone densitometry will be carried out at regular intervals to monitor the patient's bone density evolution. Also, 1,25-OH2-vitamin D and calcium levels are monitored.
Discussion
HPP (or phosphoethanolaminuria) was first described in 1948 by Rathbun, a Canadian paediatrician. He presented the case of a 9-week-old boy with rickets and epilepsy.2 Since then, six distinct clinical forms of HPP have been recognised, according to the age of onset and the severity of the disease: benign perinatal, lethal perinatal, infantile, childhood, adult and odontohypophosphatasia.3 A seventh form is known as pseudohypophosphatasia. This variant has comparable clinical and biochemical traits as those of infantile hypophosphatasia, although it differs in TNSALP levels and activity; these are normal or even elevated in vitro, but there is impaired enzymatic activity in vivo. Our patient was diagnosed with adult HPP. This case report illustrates the proteiform clinical presentation of HPP and highlights the therapeutical implications of this disease. HPP is a hereditary condition that is caused by either one or two mutations in the ALPL-gene, encoding TNSALP. A deficiency of TNSALP leads to inadequate bone mineralisation. There are three different types of ALP: intestinal ALP, fetal ALP and tissue-non-specific ALP. Defective bone mineralisation is the hallmark of HPP.
The prevalence of severe cases is estimated to be 1/300 000; less severe forms are thought to be more prevalent. In a study of a European population, the prevalence of less severe forms is estimated to be 1/6370.4
The phenotypical variability in HPP can be explained by the extent of residual enzymatic activity of the TNSALP associated with the identified mutations. Patients with a mild form of HPP have a mutation with some residual enzymatic activity, whereas patients with a severe clinical form have a mutation that leads to a complete loss of enzymatic activity. Some patients carrying an ALPL-gene mutation, however, remain asymptomatic. At present, there are 285 known mutations described in the ‘ALPL-gene mutations database’, which can be looked up at the following url: http://www.sesep.uvsq.fr/03_hypo_mutations.php. Severe forms are always inherited in an autosomal recessive way; the less severe forms can be inherited either in an autosomal dominant or autosomal recessive manner.
TNSALP is an ectoenzyme responsible for cleaving the extracellular substrates, inorganic pyrophosphate (PPi) and pyridoxal-5′-phosphate (PLP). Whether phosphoethanolamine (PEA) is a substrate in vivo remains unclear.5 PPi is produced by glycoprotein-I, a cell membrane bound enzyme. PPi is then hydrolysed by TNSALP into inorganic phosphate (Pi). Pi is needed to crystallise hydroxyapatite; in contrast, PPi suppresses the development of hydroxyapatite crystals and promotes chondrocalcinosis.6 The phosphorylation of pyridoxine (vitamin B6) results in PLP, the biological active metabolite of vitamin B6. When PLP is hydrolysed by TNSALP, the unphosphorylated pyridoxal (PL) is able to traverse the blood–brain barrier, where it can be phosphorylated intracellularly. This is the reason why pyridoxine-responsive seizures are an indication of the severity of the disease and the possible lethal prognosis.7 The clinical expression varies from a total lack of bone mineralisation, which leads to stillbirth, to stress fractures and premature deciduous or permanent teeth loss in adults, as was the case in our patient.8 A description of the clinical presentation of each form of HPP is given in table 1.
Table 1.
The clinical presentation of the different forms of HPP
| Perinatal lethal HPP | Impaired bone mineralisation in utero, osteochondral spurs protruding from arms or legs, rachitis deformation of the thorax and hypoplastic lung. These patients survive maximally a few days |
| Perinatal benign HPP | Bone mineralisation defects in utero with improvement of bone defects during pregnancy, bone shortening and bowing of the limbs |
| Infantile HPP | Symptoms develop during the first 6 months of life. Rachitis deformation of the thorax and metaphyses, premature craniosynostosis with intracranial hypertension, hypercalcaemia with irritability, anorexia, vomiting, polydipsia, dehydration, hypotonia and constipation, short stature in adulthood, premature loss of deciduous teeth |
| Childhood HPP | Most heterogeneous form of HPP. Enlarged joints, delay in walking, waddling gait, short stature, failure to thrive, intracranial hypertension, fractures, bone pain, premature teeth loss, spontaneous remission possible |
| Adult HPP | Presentation during adulthood, stress fractures of the metatarsal bones, pseudofractures of the femur, chondrocalcinosis and predisposition to osteoarthropathy, premature loss of deciduous and permanent teeth |
| Odonto-HPP | Affects patients of all ages. No skeletal abnormalities. Reduced alveolar bone, premature exfoliation of primary teeth, severe caries. Loose teeth on examination |
HPP, hypophosphatasia.
Musculoskeletal pain is also a well-known feature accompanying hypophosphatasia. The diagnosis of HPP is based on clinical, laboratory and radiographic findings. Total serum ALP is the screening test of choice. Low total serum ALP in conjunction with clinical features (pathological fractures, teeth loss at a young age, skeletal pain) and a low bone mineralisation level is highly suggestive for the diagnosis of adult hypophosphatasia. Other causes of low ALP should be ruled out. Calcium and phosphorus levels are normal or elevated in hypophosphatasia. Bone biopsy can be negative in adult HPP;9 although particular histological changes in the bone of patients with HPP have been reported, comprising an excess of unmineralised bone matrix that can appear in a patchy distribution.
Mutations in the ALPL-gene are not demonstrated in all patients with HPP, and DNA-analysis is therefore not obligatory for making the diagnosis. Increased levels of serum PLP, serum PPi and urinary PEA can further support the diagnosis, but are in themselves not pathognomonic for this disorder.
Treatment is supportive and comprises of, in the first place: non-steroidal anti-inflammatory drugs for musculoskeletal pain, orthopaedic interventions in case of fractures, and dental follow-up to maintain speech and chew function for adequate nutrition. Supplemental calcium and vitamin D are not helpful, with the exception of a demonstrated deficit. Bisphosphonates should be avoided in HPP as they can aggravate the disease manifestations and cause fractures. Bisphosphonates are chemically stable analogues of PPi, they decrease bone metabolism and can bind zinc and magnesium ions, thus impeding ALP function.10 The administration of intact parathyroid hormone, teriparatide, has been shown to be beneficial in adult hypophosphatasia.11 In 2012, a study of a trial was published in which ENB-0040 (asfotase alfa), developed by Enobia Pharma (Canada), was used in 11 children.12 Asfotase alfa is a recombinant human TNSALP that specifically targets the bone. This study with enzyme replacement therapy showed promising results. Recognising hypophosphatasia is important, as it influences therapeutical decisions and makes family screening possible.
In conclusion, we present a case of adult HPP, a hereditary cause of defective bone mineralisation. HPP is not well known by physicians and the less severe forms are underestimated in medical practice. The diagnosis should be considered in the presence of low total ALP, radiographic findings (stress fractures, pseudofractures, bone demineralisation) and a history of low impact bone fractures or teeth loss at a young age.
Patient's perspective.
I lost my teeth very early, in my mid-20s. I also had multiple stress fractures of my feet. These problems exist for several years now. I never thought of having a medical condition provoking these problems. When I got the diagnosis of hypophosphatasia, I was very surprised. I had never heard of hypophosphatasia. Of course, I did a lot of research on the internet to learn more about my condition. I am very glad to know that I have hypophosphatasia, I finally know why I lost my teeth so early and why I had stress fractures. Knowing I have this disease does not affect the way I live my life, nevertheless, I find it important to realise I have a genetic disease. This gives me the opportunity to let my children get screened for hypophosphatasia too.
Learning points.
An adult patient with a low alkaline phosphatase level and osteomalacia or teeth loss at a young age should raise suspicion of the possibility of adult hypophosphatasia.
Increased levels of serum PLP (pyridoxal-5’-phosphate), serum PPi (inorganic pyrophosphate) and urinary PEA (phosphoethanolamine) can support the diagnosis of hypophosphatasia but are not pathognomonic for this disorder.
The lower reference limit of alkaline phosphatase is often ignored by physicians although it could be of clinical importance.
Bisphosphonates could possibly worsen the clinical course of a patient with hypophosphatasia, their use is thus contraindicated in this context.
Enzyme replacement therapy with recombinant human tissue-non-specific alkaline phosphatase is a promising new therapeutic approach that could be the mainstay of treatment in the near future.
Acknowledgments
Dr Yannick De Brucker, radiologist, was kind enough to provide us with the X-ray of the foot and also wrote the legend for it. Dr Denayer Ellen helped us with the genetic analysis.
Footnotes
Contributors: JB wrote most of the manuscript. BB diagnosed the patient and helped in writing the manuscript. MM performed the genetic analysis and helped in writing the genetics part of the manuscript. BV helped in writing the manuscript and also revised it.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer review.
References
- 1.McKiernan FE, Berg RL, Fuehrer J. Clinical and radiographic findings in adults with persistent hypophosphatasemia. J Bone Miner Res 2014; 29:1651–60. 10.1002/jbmr.2178 [DOI] [PubMed] [Google Scholar]
- 2.Rathbun J. Hypophosphatasia; a new developmental anomaly. Am J Dis Child 1948;75:822–31. 10.1001/archpedi.1948.02030020840003 [DOI] [PubMed] [Google Scholar]
- 3.Mornet E. Hypophosphatasia. Best Pract Res Clin Rheumatol 2008;22:113–27. 10.1016/j.berh.2007.11.003 [DOI] [PubMed] [Google Scholar]
- 4.Mornet E, Yvard A, Taillandier A et al. . A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet 2011;75:439–45. 10.1111/j.1469-1809.2011.00642.x [DOI] [PubMed] [Google Scholar]
- 5.Millan JL. Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2006. [Google Scholar]
- 6.Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys 1984;231:1–8. 10.1016/0003-9861(84)90356-4 [DOI] [PubMed] [Google Scholar]
- 7.Whyte PM. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci 2010;1192:190–200. 10.1111/j.1749-6632.2010.05387.x [DOI] [PubMed] [Google Scholar]
- 8.Taillandier A, Domingues C, De Cazanove C et al. . Molecular diagnosis of hypophosphatasia and differential diagnosis by targeted next generation sequencing. Mol Genet Metab 2015;116:215–20. 10.1016/j.ymgme.2015.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thacker RV, Whyte MP, Eisman JA et al. . Genetics of bone biology and skeletal disease. Waltham: Academic Press, 2013. [Google Scholar]
- 10.Sutton RA, Mumm S, Coburn SP et al. . “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res 2012;27:987–94. 10.1002/jbmr.1565 [DOI] [PubMed] [Google Scholar]
- 11.Whyte MP, Mumm S, Deal C. Adult hypophosphatasia treated with teriparatide. J Clin Endocrinol Metab 2007;92:1203–8. 10.1210/jc.2006-1902 [DOI] [PubMed] [Google Scholar]
- 12.Whyte MP, Greenberg CR, Salman NJ et al. . Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 2012;366:904–13. 10.1056/NEJMoa1106173 [DOI] [PubMed] [Google Scholar]