

Abstract
To date, there are 12 reported cases of hepatoblastoma in trisomy 18 patients, three of whom had a mosaic chromosome pattern. We report on an 18-month-old child who had hemihypertrophy and developmental delay, was found to have hepatoblastoma on surveillance ultrasound scan, and was subsequently diagnosed with mosaic trisomy 18 on array comparative genomic hybridisation from a peripheral blood sample and molecular cytogenetic analysis of the tumour specimen. Although hemihypertrophy has been associated with mosaic trisomies, there are only a couple of published case reports of hemihypertrophy or asymmetry in mosaic trisomy 18 patients and none in the reported cases of hepatoblastoma in a mosaic trisomy 18 setting. We have reviewed the published case reports of hepatoblastoma in trisomy 18 patients and found that they seem to tolerate the intensive treatment very well if there are no significant comorbidities.
Background
Trisomy 18 (Edward syndrome) is the second most common autosomal trisomy in the paediatric age group. The birth prevalence of this disorder is 1 in 3000–8000 live-births. It is a constitutional chromosomal abnormality characterised by multiple congenital anomalies, feeding difficulties and mental retardation. Ninety per cent of affected children die during the first year of life, most often as a result of complex congenital heart diseases and structural brain defects.1 This high level of mortality is uniform throughout the world and is not significantly affected by the differences in health systems in different parts of the world.2 Individuals with mosaic trisomy 18, who make up approximately only 5% of all trisomy 18 cases, carry a trisomy 18 and a euploid cell line. The clinical phenotype is very variable in this group, ranging from the full spectrum of trisomy 18 to a normal phenotype. There is some evidence of non-random association between trisomy 18 and hepatoblastoma, and we report another patient with mosaic trisomy 18 who was successfully treated for hepatoblastoma using a standard approach.
Case presentation
A male child was born at 38 weeks of gestation to a 36-year-old woman by elective caesarean section. Suspicion of Edward syndrome was raised because of antenatal diagnosis of a ventricular septal defect (VSD). The baby's birth weight was 1.8 kg and he stayed in the neonatal unit for 4 weeks needing nasogastric tube feeding. Karyotype showed an apparently normal male (46, XY). There was no deletion of 22q11.2 by florescent in situ hybridisation with the TUPLE1 probe.
Following discharge from the neonatal ward, the baby was followed up by community paediatricians both because of his failure to thrive and concerns about his global developmental delay, which was most striking in speech and language. Bilateral inguinal hernias, which were noted at birth, were repaired. The small patent ductus arteriosus and muscular VSD were considered haemodynamically insignificant. At 6 months of age, the baby was referred to clinical genetics because he was noted to have hemihypertrophy—the left leg being short and thin—and facial asymmetry including prominent right forehead, cheek and jaw. Beckwith-Weidemann syndrome and Silver-Russell syndrome were excluded as possible causes of the hemihypertrophy. A surveillance abdominal ultrasound carried out at the age of 18 months showed a hepatic mass of 4.6×6.5×5.3 cm in segments V and VI. The baby's serum α fetoprotein (AFP) was 2259 ng/mL and biopsy of this mass confirmed an epithelial hepatoblastoma with embryonal and fetal components (figure 1). The tumour was staged as non-metastatic PRETEXT (pre-treatment extent) stage 2 on CT scan (figure 2).
Figure 1.
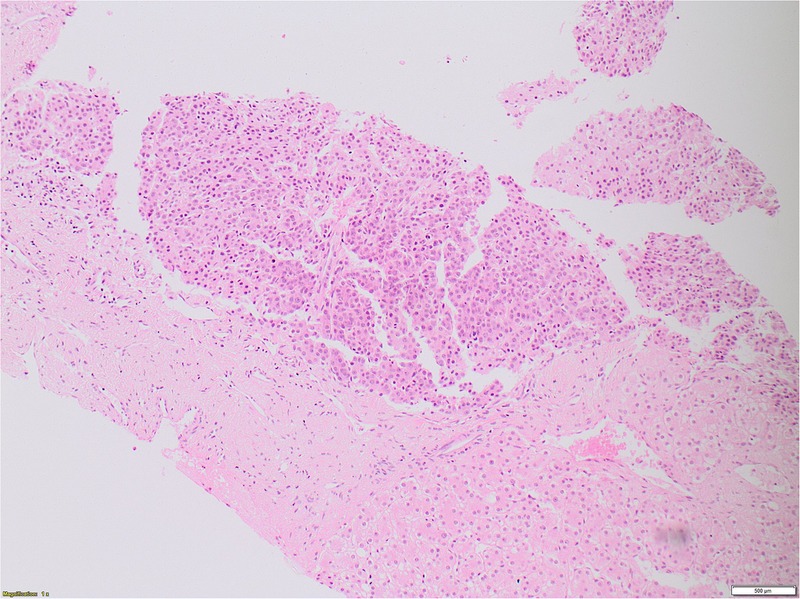
Fetal hepatoblastoma histology.
Figure 2.
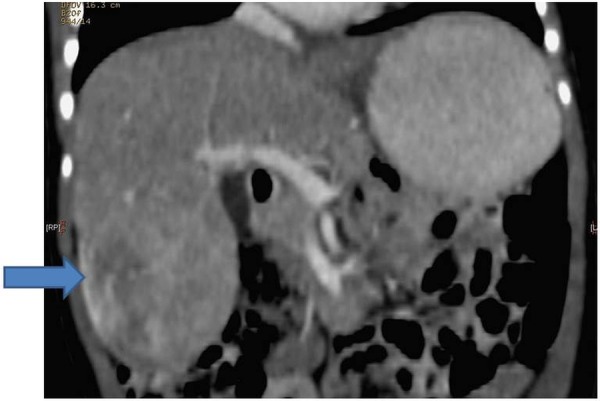
Diagnostic CT of the abdomen.
Treatment
The baby received four courses of neoadjuvant single agent Cisplatin chemotherapy as per the Childhood Liver Tumour Strategy Group (SIOPEL) guidelines. Reassessment MRI scan after four courses of chemotherapy showed a mixed response: there was a significant reduction in the originally identified mass, now measuring 2.7×2.8×2.4 cm, but two new lesions were found in segments IV and VII that were not identified on the original diagnostic CT scan (figures 3 and 4). The baby's AFP had fallen to 50 ng/mL. He proceeded with surgery and the histology demonstrated good response to chemotherapy in the main tumour, which had been removed completely with an intact capsule, but there was evidence of some viable tumour in the segment VII nodule, which approached the resection margins. Because of this histology, it was decided that he should have further adjuvant chemotherapy. He received two courses of PLADO (cisplatin and doxorubicin) chemotherapy as per SIOPEL guidelines. Repeat surveillance MRI at the end of this chemotherapy did not show any further progression and he had a further surgical resection of segment VII. This was a complete resection with no evidence of any viable disease. He had one central line-related bacteraemia episode during the entire treatment and there was no other treatment-related complication. End-of-treatment toxicity work up was unremarkable.
Figure 3.
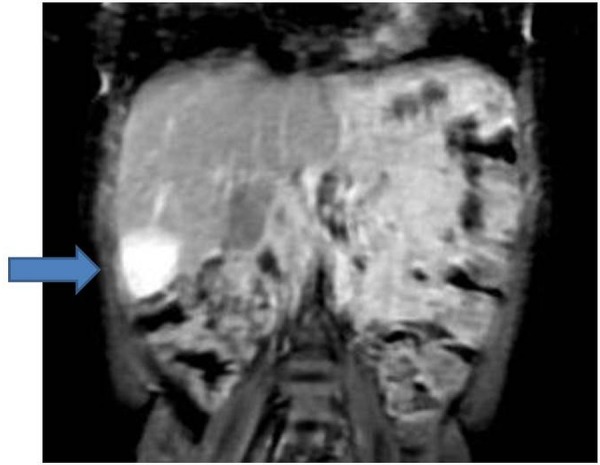
Response assessment MRI of the abdomen.
Figure 4.
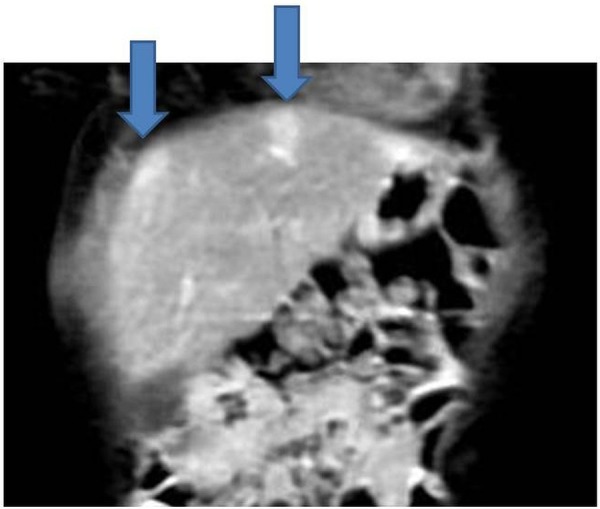
Response assessment MRI of the abdomen.
Outcome and follow-up
On completion of the baby's hepatoblastoma treatment, he had community paediatric, paediatric oncology and clinical genetics follow-up for his ongoing needs related to developmental delay, tumour surveillance and concern about an underlying genetic diagnosis. At 2 years of age, he was reviewed by the genetics team and array CGH was undertaken using an Agilent ISCA (International standards for cytogenomic arrays) 60-K oligoarray. This showed a trisomy 18 cell line present at a level of 50% in blood (figure 5). Reassessment of G-banded chromosomes confirmed the presence of a trisomy 18 cell line at a level of 20% in 30 metaphases scored. Molecular cytogenetic analysis of the tumour then showed trisomy 18 in 22 of 45 cells examined (figure 6). We did not look for evidence of trisomy 18 in any other tissue.
Figure 5.
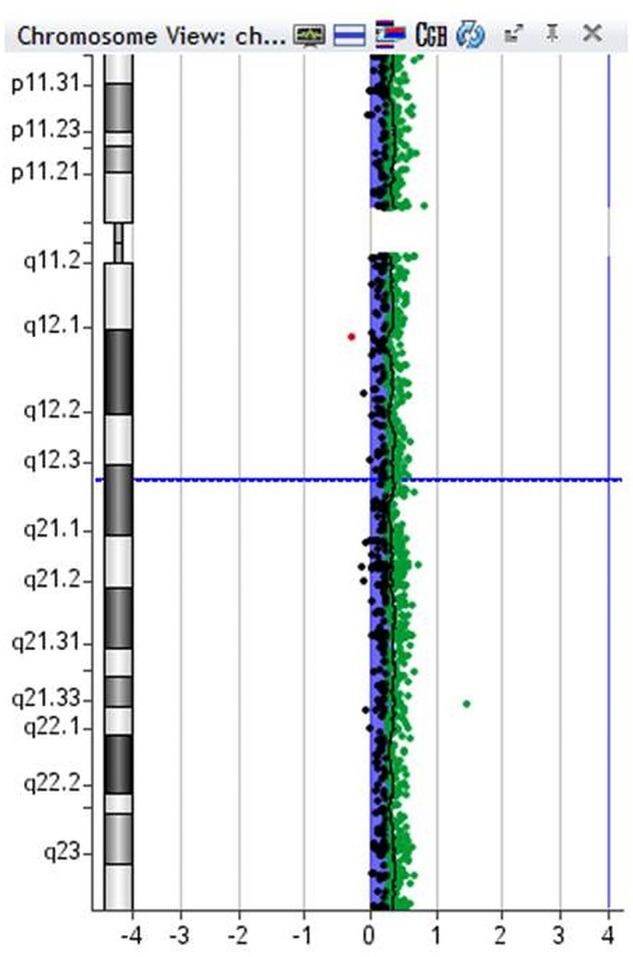
Array comparative genomic hybridisation (CGH) from blood sample.
Figure 6.
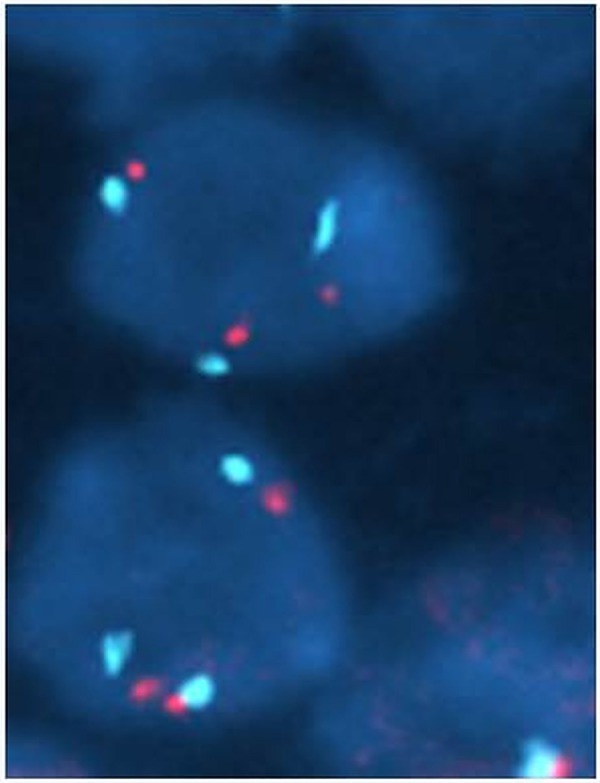
Fluoresence in situ hybridisation (FISH) studies from tumour cells.
The child's current follow-up is by paediatric oncology with support from the community paediatric team. He remains well clinically with normal AFP and surveillance MRI scans not showing any evidence of disease recurrence (figure 7). He therefore remains in complete remission over 3½ years from end of treatment and we are optimistic that he has been cured of his hepatoblastoma.
Figure 7.
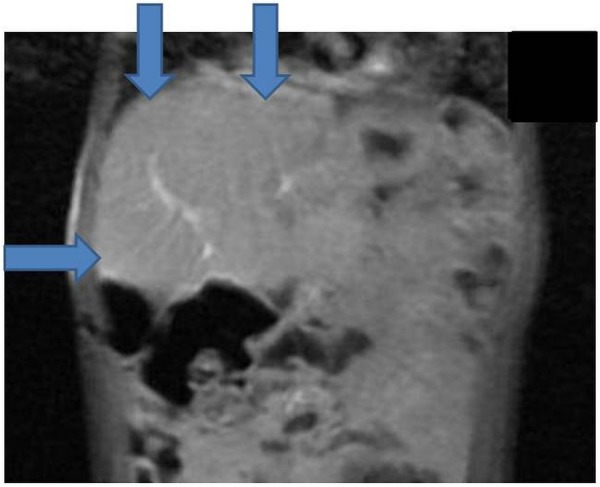
End of treatment MRI of the abdomen.
Discussion
Hepatoblastoma is the most common malignant neoplasm of the liver in young children. It mostly presents in infancy with abdominal distension and a hepatic mass. The serum AFP is elevated and can be diagnostic when associated with typical clinical and radiological appearances. A complete surgical resection, determined by PRETEXT staging, is essential to optimise the chances of long-term cure and, in Europe, this is almost always preceded by neoadjuvant chemotherapy.3 4
Following an extended literature search we found 13 reported cases, including our patient's, of hepatoblastoma in trisomy 18 patients, and their important characteristics have been summarised in table 1.1 3 5–13 Of these 13 patients, 6 died within 1–5 months of presentation, either because of tumour progression or a cardiac complication related to trisomy 18. Seven were still alive at the time of individual publications and remained disease-free with a follow-up period ranging from 16 months to more than 7 years. Of these seven patients, four had a trisomy 18 mosaic pattern,1 3 6 as in the current case.
Table 1.
Case reports of hepatoblastoma in trisomy 18
| Authors | Age (m) | Sex | Birth weight (g) | Karyotype | Histology | Chemotherapy | Surgery | Clinical outcome |
|---|---|---|---|---|---|---|---|---|
| Dasouki and Barr8 | 33 | F | 1860 | 47,XX,+18 | NA | None | None | Died within 3 weeks |
| Mamlok et al9 | 3 | F | 1800 | 47,XX,+18 | Embryonal type | None | None | Died of heart failure |
| Tanaka et al6 | 24 | F | 1750 | 47,XX,+18 mosaic | Fetal type | None | Right lobectomy | Alive 7 years post surgery |
| Bove et al10 | 21 | F | 3300 | 47,XX,+18 | Mixed type | None | Right lobectomy | Died at 5 months from presentation |
| Teraguchi et al11 | 7 | F | 2722 | 47,XX,+18 | Fetal type | None | Partial lobectomy | Alive 2 years post surgery |
| Maruyama et al12 | 3 | F | 2464 | 47,XX,+18 | Fetal type | None | None | Died of heart failure |
| Kitanovski et al13 | 6 | F | 1630 | 47,XX,+18 | Fetal type | None | None | Died within 1 month of diagnosis |
| Fernandez et al1 | 9 | M | N/A | 47, XY,+18 mosaic | Fetal type | Cisplatin, 5FU, vincristine, adriamycin | Liver transplant | Alive 2 years after transplant |
| Uekusa et al5 | 18 | M | 2538 | 47, XY,+18 | Fetal type | Adriamycin, cisplatin | Right lobectomy | Alive 18 months after treatment |
| Pereira et al3 | 120 | F | 680 | 47,XX,+18 mosaic | Fetal type | Cisplatin, 5FU, vincristine—only one course | Left lobectomy | Alive 2 years after surgery |
| Tan et al7 | 12 | F | 1320 | 47,XX,+18 | Fetal type | None | Right hemihepatectomy | Alive 16 months after surgery |
| Tan et al7 | 7 | F | 1870 | 47,XX,+18 | Mixed type | None | Right hemihepatectomy | Died of heart failure |
| Present case | 18 | M | 1800 | 47,XY,+18 mosaic | Epithelial type | Cisplatin, adriamycin | Partial lobectomy—twice | Alive 44 months from end of treatment |
5FU, 5 fluorouracil; G, grams; M, months; NA, not available.
Low birth weight and left to right cardiac shunts were a common finding. Hemihypertrophy, however, was not documented in any of the cases and, to our knowledge, our index patient is the first to have hepatoblastoma in a setting of trisomy 18 mosaicism and hemihypertrophy. Hemihypertrophy and asymmetry have been reported as an association of trisomy 18.14 Hemihypertrophy can occur in children with BWS and, depending on the aetiology, those children may be at risk of Wilms’ tumour and hepatoblastoma. The risk of Wilms’ tumour in children with isolated hemihypertrophy is <5% and only those with paternal uniparental disomy 11p15 or isolated H19 hypermethylation are advised to have surveillance.15 This child underwent screening by abdominal US as he was deemed to be at risk of Wilms’ tumour by virtue of his non-isolated hemihypertrophy of unknown cause. Although chromosomal mosaicism is a known cause of asymmetry, it is not standard practice to offer tumour surveillance to this group of patients.3 It is interesting to speculate that this patient would not have been undergoing surveillance had his diagnosis of mosaic trisomy 18 been known.
There is sufficient evidence in the literature to suggest that fetal histology carries a better prognosis in hepatoblastoma as compared to other histiotypes, and this was well reflected in this small cohort of hepatoblastoma patients in trisomy 18 settings.16 17 Surgery was successfully attempted in all seven of the living patients. Two had surgery alone as curative treatment; four received standard chemotherapy in addition to surgery, including one with a successful orthotopic liver transplant. One patient had an attenuated cycle of chemotherapy, which was interrupted because of complications with infections, and proceeded with curative surgery on recovery.
Learning points.
It appears that this particular group of hepatoblastoma patients have a good outcome, provided there are no associated life limiting cardiac or other structural defects.
In any child with asymmetry, mosaicism should always be considered. It might be necessary to study the karyotype in a greater number of cells or to study different cell lines, for example, fibroblasts. Array technology is able to detect mosaicism and is replacing karyotyping as a first-line investigation.
Isolated asymmetric growth is not a standard indication for tumour screening in children but this child was offered screening because of a constellation of unexplained features and might have led to early diagnosis.
Acknowledgments
The authors acknowledge Dr Kaye Platt for providing supplementary radiology images for this case report, and Dr Darren Fowler for providing histology images.
Footnotes
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Fernandez KS, Baum R, Fung B et al. Chemoresistant hepatoblastoma in a patient with mosaic trisomy 18 treated with orthotopic liver transplantation. Pediatr Blood Cancer 2011;56:498–500. 10.1002/pbc.22768 [DOI] [PubMed] [Google Scholar]
- 2.Lin HY, Lin SP, Chen YJ et al. Clinical characteristics and survival of trisomy 18 in a medical centre in Taipei, 1988–2004. Am J Med Genet A 2006;140A:945–51. 10.1002/ajmg.a.31173 [DOI] [PubMed] [Google Scholar]
- 3.Pereira EM, Marion R, Ramesh KH et al. Hepatoblastoma in a mosaic Trisomy 18 patient. J Pediatr Hematol Oncol 2012;34:e145–8. 10.1097/MPH.0b013e3182459ee8 [DOI] [PubMed] [Google Scholar]
- 4.Roebuck DJ, Aronson D, Clapuyt P et al. 2005 PREEXT: a revised staging system for primary malignant liver tumors of childhood developed by the SIOPEL group. Pediatr Radiol 2007;37:123–32. 10.1007/s00247-006-0361-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uekusa S, Sugito K, Kawashima H et al. Successful treatment for hepatoblastoma in a 1-year-old boy with Trisomy 18. Pediatr Int 2012;54:428–30. 10.1111/j.1442-200X.2011.03528.x [DOI] [PubMed] [Google Scholar]
- 6.Tanaka K, Uemoto S, Asonuma K et al. Hepatoblastoma in a 2-Year-Old Girl with Trisomy 18. Eur J Pediatr Surg 1992;2:298–300. 10.1055/s-2008-1063464 [DOI] [PubMed] [Google Scholar]
- 7.Tan ZH, Lai A, Chen CK et al. Association of trisomy 18 with hepatoblastoma and its implications. Eur J Pediatr 2014;173:1595–8. 10.1007/s00431-013-2147-8 [DOI] [PubMed] [Google Scholar]
- 8.Dasouki M, Barr M Jr. Trisomy 18 and hepatic neoplasia. Am J Med Genet 1987;27:203–5. 10.1002/ajmg.1320270122 [DOI] [PubMed] [Google Scholar]
- 9.Mamlok V, Nichols M, Lockhart L et al. Trisomy 18 and hepatoblastoma. Am J Med Genet 1989;33:125–6. 10.1002/ajmg.1320330119 [DOI] [PubMed] [Google Scholar]
- 10.Bove KE, Soukup S, Ballard ET et al. Hepatoblastoma in a child with trisomy 18: cytogenetics, liver anomalies, and literature review. Pediatr Pathol Lab Med 1996;16:253–62. [PubMed] [Google Scholar]
- 11.Teraguchi M, Nogi S, Ikemoto Y et al. Multiple hepatoblastomas associated with trisomy 18 in a 3-year-old girl. Pediatr Hematol Oncol 1997;14:463–7. 10.3109/08880019709028777 [DOI] [PubMed] [Google Scholar]
- 12.Maruyama K, Ikeda H, Koizumi T. Hepatoblastoma associated with trisomy 18 syndrome: a case report and a review of the literature. Pediatr Int 2001;43:302–5. 10.1046/j.1442-200x.2001.01380.x [DOI] [PubMed] [Google Scholar]
- 13.Kitanovski L, Ovcak Z, Jazbec J. Multifocal hepatoblastoma in a 6-month-old girl with trisomy 18: a case report. J Med Case Rep 2009;3:8319 10.4076/1752-1947-3-8319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitas AL, Paiva M, Cordeiro AI et al. Mosaic trisomy 18 in a five-month-old infant. Case Rep Pediatr 2013;2013:929861 10.1155/2013/929861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott RH, Walker L, Olsen ØE et al. Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 2006;91:995–9. 10.1136/adc.2006.101295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herzog CE, Andrassy RJ, Eftekhari F. Childhood cancers: hepatoblastoma. Oncologist 2000;5:445–53. 10.1634/theoncologist.5-6-445 [DOI] [PubMed] [Google Scholar]
- 17.Haas JE, Feusner JH, Finegold MJ. Small cell undifferentiated histology in hepatoblastoma may be unfavorable. Cancer 2001;92:3130–4. [DOI] [PubMed] [Google Scholar]