

Abstract
Introduction
Dalbavancin is a new lipoglycopeptide that is active against Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus. It has a half-life of 14.4 days, permitting intravenous treatment of acute bacterial skin and skin structure infections without the need for daily dosing.
Objective
The objective of these analyses was to compare the adverse event profile of dalbavancin with that of the comparator agents in the treatment of skin and skin structure infections.
Methods
Data on adverse events and laboratory assessments collected from 3002 patients enrolled in seven late-stage, randomized clinical trials were analyzed for patients receiving dalbavancin or a comparator antibiotic.
Results
Overall adverse event rates were similar or lower for patients receiving dalbavancin (799/1778; 44.9 %) compared with those receiving comparator agents (573/1224; 46.8 %, p = 0.012). The most common treatment-emergent adverse events were nausea, headache, diarrhea, constipation, vomiting, rash, urinary tract infection, pruritus, and insomnia. The duration and timing of the onset of adverse events were similar for patients receiving dalbavancin relative to the comparators.
Conclusion
Dalbavancin exhibits a favorable overall safety profile for treatment of acute bacterial skin and skin structure infections due to Gram-positive bacteria.
Key Points
| Treatment with dalbavancin was well tolerated. |
| The long half-life of dalbavancin did not lead to any safety concerns. |
Introduction
Over the last decade, the USA has witnessed a dramatic increase in the incidence of community-acquired skin infections, an increasing proportion of which are a consequence of methicillin-resistant Staphylococcus aureus (MRSA), reinforcing the need for new and effective antibacterial therapies in this disease [1–4]. Dalbavancin is a lipoglycopeptide with activity against Gram-positive organisms, including MRSA, through interference with bacterial cell wall formation by preventing cross-linking of peptidoglycans. Dalbavancin has a distinctive pharmacokinetic profile, with a terminal half-life of 14.4 days, which allows for infrequent intravenous dosing. It is not metabolized, does not interact with cytochrome P450 enzymes, and is eliminated via both hepatic and renal routes [5]. After entering human studies in 2003, dalbavancin has been studied in 14 phase I, two phase II, and five phase III studies [5–11]. Presented here are safety data from patients enrolled in all seven phase II and phase III studies in the dalbavancin clinical development program that lead to US and European regulatory approval for the treatment of acute bacterial skin and skin structure infections (ABSSSI) [12, 13].
Methods
Studies
The dalbavancin clinical development program was conducted between 2002 and 2013. All phase II and phase III clinical trials for the treatment of uncomplicated and complicated skin infection, catheter-related bloodstream infection and ABSSSI supported by the sponsor and submitted to the regulatory authorities for review and marketing approval are included in the analyses (Table 1). As dalbavancin was not available commercially prior to 2013, these studies represent all clinical trials performed to date. All studies were randomized controlled trials comparing dalbavancin with a comparator agent and were designed as non-inferiority trials. The protocols for all studies were reviewed by the local ethics committees of participating institutions and were consistent with the Declaration of Helsinki. All patients provided written informed consent.
Table 1.
Treatment-related adverse events in randomized and treated patients in phase II/III clinical development program
| Studies | Dalbavancin | Comparator | |||
|---|---|---|---|---|---|
| N | Two dosesa | One doseb | N | Regimen | |
| All phase II/III studies | 1778 | 1428 | 350 | 1224 | |
| Phase II studies | 81 | 54 | 27 | 55 | |
| Catheter-related bloodstream infections | |||||
| VER001-4 [11] | 40 | 34 | 6 | 34 | Vancomycin |
| Skin and skin structure infections | |||||
| VER001-5 [9] | 41 | 20 | 21c | 21 | Standard of care |
| Phase III studies | 1697 | 1374 | 323 | 1169 | |
| Uncomplicated skin and skin structure infection | |||||
| VER001-8 (Data on file, Allergan plc.) [16] | 367 | 94 | 273 | 186 | Cefazolin |
| Complicated skin and skin structure infection | |||||
| VER001-16 (Data on file, Allergan plc.) [16] | 107 | 57 | 50 | 49 | Vancomycin |
| VER001-9 [8] | 571 | 571 | 0 | 283 | Linezolid |
| Acute bacterial skin and skin structure infection | |||||
| DUR001-301 (DISCOVER 1) [10] | 284 | 284 | 0 | 284 | Vancomycin/linezolid |
| DUR001-302 (DISCOVER 2) [10] | 368 | 368 | 0 | 367 | Vancomycin/linezolid |
Comparator agents include cephalosporins, vancomycin, oxacillin, nafcillin and linezolid. Studies VER001-4, 5, and 16 were open-label trials; all others were double blinded. DISCOVER 1 and DISCOVER 2 ClinicalTrials.gov numbers, NCT01339091 and NCT01431339
aIntravenous dalbavancin 1000 mg on day 1 followed by 500 mg on day 8
bIntravenous dalbavancin 1000 mg on day 1
cIntravenous dalbavancin 1100 mg
Dalbavancin was delivered intravenously over 30 min as 1000 mg on day 1 and 500 mg on day 8. In VER001-8 and VER001-16, a total of 273 and 49 patients, respectively, with uncomplicated skin infection were given the option of stopping after the first 1000-mg dose. In VER001-5, a total of 20 patients received a single 1100-mg dose. No adjustment for weight or body mass index (BMI) was required in the clinical program. Comparator agents included vancomycin, linezolid, cefazolin, nafcillin, or oxacillin. Follow-up was performed through day 28 in all studies except the two pivotal studies for ABSSSI (DISCOVER 1 and DISCOVER 2), where the last follow-up visit occurred at day 70.
Data
All patients in the phase II and phase III studies presented with an infection of the skin or, in one phase II study, a catheter-related bloodstream infection, and are included in the safety analyses. Safety data were collected carefully during each clinical trial. Each protocol outlined the requirements for reporting of adverse events, but, briefly, all observed or volunteered adverse events, regardless of treatment group or suspected causal relationship to study drug, were reported from the time that the patient provided informed consent through the last follow-up visit. Adverse events included clinically significant symptoms and signs, changes in physical examination findings, hypersensitivity, and progression or worsening of underlying disease. Severity was assessed as mild, moderate, or severe based on the investigator’s clinical impression rather than a result of specific criteria for defining the severity of an event. Causality was determined by the investigator for all adverse events as unrelated, unlikely related, possibly related, or probably related to the study drug. Serious adverse events are those that result in death, are life-threatening, require inpatient hospitalization or prolongation of existing hospitalization, result in persistent or significant disability or incapacity, result in congenital anomaly or birth defect, or are assessed as being a medically important event based on medical and scientific judgment. The timing of laboratory assessments, including hematology and biochemistry parameters, was defined in each protocol and performed at baseline and at periodic intervals, including days 3, 8, 14, or 28.
Statistics
In all safety analyses, data are pooled at the patient level across studies, as is typical for the purposes of reporting safety data to regulatory authorities and in retrospective reviews of adverse drug reactions [14, 15]. The safety analyses included patients who were randomized and received a dose of study drug in all the phase II or phase III clinical trials completed through 2013. Patients received the two-dose regimen of dalbavancin or, where permitted in protocols enrolling patients with uncomplicated skin infections, a single 1000-mg dose. For all analyses of treatment-emergent adverse events (TEAEs), if the same adverse event was reported for the same patient more than once, the adverse event was counted only once for that preferred term and at the highest intensity and strongest relationship to study drug. Differences between treatment groups were analyzed using Fisher’s exact test for dichotomous variables (sex, ethnicity, race) and the Wilcoxon rank-sum test for continuous variables (age, BMI, creatinine clearance), prospectively for patient demographics and post hoc, as relevant, for other analyses of interest. Total adverse event comparisons were analyzed using a Cochran–Mantel–Haenszel analysis to adjust for study and a log-normal, Poisson regression model adjusted by study on total number of treatment-related adverse events per patient. No adjustments were made for multiple comparisons, and p values are presented for descriptive purposes only. Safety outcomes are presented as verbatim descriptions of adverse events and were coded using Version 14 or higher of MedDRA®. All statistical analyses were performed using SAS version 9.2.
Results
A total of 1778 patients enrolled in the phase II/III program were randomized and treated with dalbavancin, and 1224 received a comparator agent (Fig. 1, Tables 1 and 2). The median age of all patients receiving dalbavancin was 47.0 years, 17.6 % of patients were aged >65 years, 78 % were White, 64.3 % were from North America, and 71.6 % of patients exceeded a BMI of 25 kg/m2. Demographics were similar in the comparator groups. Small differences between treatment groups were observed in the distribution of race and location and type of infection.
Fig. 1.
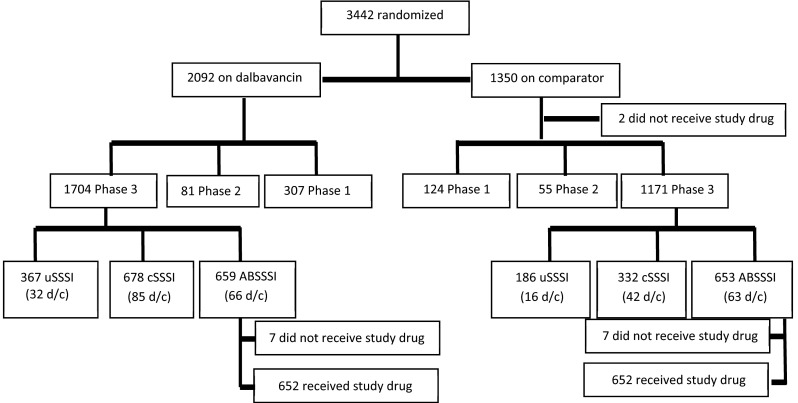
Flow chart of patients included in safety and efficacy analyses. ABSSSI acute bacterial skin and skin structure infection, cSSSI complicated skin and skin structure infection, d/c patients discontinued from the study, uSSSI uncomplicated skin and skin structure infection
Table 2.
Demographics of patients enrolled in phase II/III clinical development program
| Dalbavancin (N = 1778) | Comparator (N = 1224) | |
|---|---|---|
| Age (years) | ||
| N | 1778 | 1224 |
| Mean | 48.3 | 49.2 |
| SD | 16.44 | 16.51 |
| Median | 47.0 | 49.0 |
| Min; max | 16; 93 | 18; 92 |
| Age distribution (years) | ||
| <65 | 1465 (82.4) | 995 (81.3) |
| ≥65 | 313 (17.6) | 229 (18.7) |
| Sex | ||
| Male | 1066 (60.0) | 711 (58.1) |
| Female | 712 (40.0) | 513 (41.9) |
| Race | ||
| Whitea | 1388 (78.1) | 1008 (82.4) |
| Black or African American | 143 (8.0) | 88 (7.2) |
| Asiana | 36 (2.0) | 41 (3.3) |
| American Indian or Alaska Native | 5 (0.3) | 4 (0.3) |
| Native Hawaiian or other Pacific Islander | 1 (0.1) | 1 (0.1) |
| Othera | 205 (11.5) | 82 (6.7) |
| Body mass index (kg/m2) | ||
| N | 1761 | 1218 |
| Mean | 29.9 | 29.4 |
| SD | 8.18 | 7.96 |
| Median | 27.9 | 27.8 |
| Min; max | 14; 98 | 14; 91 |
| Body mass index distribution (kg/m2) | ||
| <18.5 | 23 (1.3) | 18 (1.5) |
| 18.5 to <25 | 465 (26.2) | 359 (29.3) |
| ≥25 | 1273 (71.6) | 841 (68.7) |
| Unknown | 17 (1.0) | 6 (0.5) |
| Indication | ||
| cSSSI/ABSSSIa | 1294 (72.8) | 973 (79.5) |
| uSSSIa | 444 (25.0) | 217 (17.7) |
| Catheter-related bloodstream infections | 40 (2.2) | 34 (2.8) |
| Locationa | ||
| North America | 1143 (64.3) | 689 (56.3) |
| Eastern Europe and South Africa | 395 (22.2) | 389 (31.8) |
| Western Europe | 216 (12.1) | 118 (9.6) |
| Asia–Pacific | 24 (1.3) | 28 (2.3) |
| Temperature ≥38 °C at baselineb, n/N (%) | 549/649 (84.6) | 552/649 (85.0) |
| Median (range)a infection area (cm2) | 324 (26–5100) | 367 (72–3922) |
Data are presented as N (%) unless otherwise indicated
ABSSSI acute bacterial skin and skin structure infection, cSSSI complicated skin and skin structure infection, SD standard deviation, uSSSI uncomplicated skin and skin structure infection
aComparisons with p < 0.05 by Fisher’s exact test
bAmong patients in the ABSSSI studies
Approximately 85 % of patients completed the study drug course of therapy, with the most commonly identified reasons for discontinuation of study medication similarly distributed among worsening clinical status, lost to follow-up, an adverse event, and withdrawal of consent. A total of 89 % of patients completed the study; the most common reasons for not completing the study included withdrawal of consent, death, and lost to follow-up (Table 3).
Table 3.
Disposition of patients in phase II/III clinical development program
| Dalbavancin | Comparator | |
|---|---|---|
| Total treated | 1778 | 1224 |
| Completed study medication | 1518 (85.0) | 1061 (86.5) |
| Did not complete study medication | 219 (12.3) | 143 (11.7) |
| Treatment failure/worsening clinical status | 25 (1.4) | 16 (1.3) |
| Adverse event | 49 (2.7) | 31 (2.5) |
| Patient withdrew consent or for reason other than adverse event | 21 (1.2) | 19 (1.5) |
| Death | 0 (0.0) | 1 (0.1) |
| Patient lost to follow-up | 27 (1.5) | 13 (1.1) |
| Patient non-compliance | 10 (0.6) | 9 (0.7) |
| Withdrawn at investigator’s discretion | 6 (0.3) | 9 (0.7) |
| Other | 81 (4.5) | 45 (3.7) |
| Completed study | 1595 (89.4) | 1098 (89.6) |
| Did not complete study | 190 (10.6) | 127 (10.4) |
| Adverse event | 0 (0.0) | 1 (0.1) |
| Patient withdrew consent | 42 (2.4) | 22 (1.8) |
| Death | 10 (0.6) | 13 (1.1) |
| Patient lost to follow-up | 107 (6.0) | 67 (5.5) |
| Subject noncompliance | 2 (0.1) | 0 (0.0) |
| Prohibited concomitant medication used | 1 (0.1) | 0 (0.0) |
| Other | 28 (1.6) | 24 (2.0) |
Data are presented as N (%)
Death was infrequent for either dalbavancin or the comparator regimens (0.6 vs. 1.1 %, respectively). Relative to those treated with comparator, patients receiving dalbavancin experienced fewer TEAEs (44.9 vs. 46.8 %, respectively, p = 0.012), fewer treatment-related adverse events (18.4 vs. 20.1 %, respectively, p = 0.014), and fewer treatment-related serious adverse events (0.2 vs. 0.7 %, respectively, p = 0.021). Of the total number of adverse events, including serious adverse events, fewer were judged by the investigator as likely to be related to dalbavancin relative to the comparator (Table 4). The distribution of mild, moderate, and severe events was similar for each regimen.
Table 4.
Adverse events in phase II/III clinical development program
| Dalbavancin (N = 1778) | Comparator (N = 1224) | p value | |
|---|---|---|---|
| Patients with the AE | |||
| TEAEa | 799 (44.9) | 573 (46.8) | 0.012 |
| Treatment-related TEAEa | 328 (18.4) | 246 (20.1) | 0.014 |
| Serious AEs | 109 (6.1) | 80 (6.5) | 0.266 |
| Serious treatment-related AEs | 3 (0.2) | 9 (0.7) | 0.021 |
| Discontinuation due to a TEAEa | 53 (3.0) | 35 (2.9) | 0.857 |
| Discontinuation due to a serious TEAEa | 22 (1.2) | 13 (1.1) | 0.660 |
| Death | 10 (0.6) | 14 (1.1) | 0.087 |
| Number of AEs | |||
| AEs, N (N/patient) | 2386 (1.34) | 1739 (1.42) | |
| Treatment-related AEsb (N, % of total) | 566 (23.7) | 459 (26.4) | 0.0004 |
| Serious AEs (N, % of total) | 135 (5.7) | 100 (5.8) | NS |
| Serious treatment-related AEsc (N, % of total) | 3 (0.1) | 9 (0.5) | 0.036 |
Data are presented as N (%) unless otherwise indicated
AE adverse event, NS not significant, TEAE treatment-emergent adverse event
aCochran–Mantel–Haenszel analysis, adjusted for study
bLog-normal, Poisson regression model adjusted by study on total number of treatment-related adverse events per patient
cFisher’s exact test
Treatment-related serious adverse events occurring in patients treated with dalbavancin included progression of cellulitis, asymptomatic leukopenia in a patient with a baseline white blood cell count of 6.9 × 103 cells/mm3 and a nadir of 3.79 × 103 cells/mm3 that recovered to baseline by day 29, and an anaphylactoid reaction (Table 5). The patient with an anaphylactoid reaction was a 22-year-old White male with a history of reactive airway disease and atopy, who had received general anesthetic agents ~3 h earlier and aztreonam intravenously as a bolus immediately prior to treatment with one dose of intravenous dalbavancin. After 15 min of a 30-min infusion, he developed dyspnea, laryngospasm, and a decrease in blood pressure. The dalbavancin infusion was stopped, and he was treated immediately with epinephrine, midazolam, antihistamines, and a 5-day course of prednisone. Symptoms and signs associated with the event were considered to be completely resolved within 1 h of discontinuation of the infusion and did not recur.
Table 5.
Treatment-related serious adverse events in phase II/III clinical development program
| Treatment-related serious adverse event | Dalbavancin (N = 1778) | Comparator (N = 1224) |
|---|---|---|
| Total | 3 (0.2) | 9 (0.7) |
| Leukopenia | 1 (0.1) | 0 |
| Anaphylactoid reaction | 1 (0.1) | 0 |
| Cellulitis | 1 (0.1) | 1 (0.1) |
| Renal failure acute | 0 | 2 (0.2) |
| Gastrointestinal disorder | 0 | 1 (0.1) |
| Face edema | 0 | 1 (0.1) |
| Pancytopenia | 0 | 1 (0.1) |
| Thrombocytopenia | 0 | 1 (0.1) |
| Nephropathy toxic | 0 | 1 (0.1) |
| Pancreatitis acute | 0 | 1 (0.1) |
The most common adverse events with onset after starting study drug treatment were nausea, headache, diarrhea, constipation, vomiting, rash, urinary tract infection, pruritus, and insomnia (Table 6). Nausea, diarrhea, and pruritus were the most common treatment-related adverse events, with an incidence of approximately ≥2 %. The duration of adverse events was similar for dalbavancin and the comparator regimens, with a median of 3.0 and 4.0 days and a mean of 7.7 and 8.0 days, respectively (Fig. 2). Additionally, the mean, median, and range of the time to the onset of adverse events were similar on each regimen (Fig. 3). An increase of adverse events was observed at day 8, seen with both the comparator and dalbavancin; this is most likely because the protocols required a visit on day 8 and not necessarily on days 4–7, allowing for a greater opportunity to report an event on that day.
Table 6.
Treatment-emergent adverse events at >2 % in either treatment regimen in phase II/III clinical development program
| Dalbavancin (N = 1778) | Comparator (N = 1224) | |
|---|---|---|
| Treatment-emergent adverse events | 799 (44.9) | 573 (46.8) |
| Nausea | 98 (5.5) | 78 (6.4) |
| Headache | 83 (4.7) | 59 (4.8) |
| Diarrhea | 79 (4.4) | 72 (5.9) |
| Constipation | 52 (2.9) | 30 (2.5) |
| Vomiting | 50 (2.8) | 37 (3.0) |
| Rash | 38 (2.1) | 22 (1.8) |
| Urinary tract infection | 36 (2.0) | 16 (1.3) |
| Pruritus | 32 (1.8) | 35 (2.9) |
| Insomnia | 27 (1.5) | 30 (2.5) |
| Treatment-related and treatment-emergent adverse events | ||
| Nausea | 49 (2.8) | 40 (3.3) |
| Diarrhea | 45 (2.5) | 45 (3.7) |
| Pruritus* | 11 (0.6) | 23 (1.9) |
* p = 0.001
Fig. 2.

Duration of adverse events
Fig. 3.

Day of onset of adverse events (AE). TEAE treatment-emergent AE
Adverse event rates with dalbavancin or comparator were similar when assessed by age, sex, or race, as were those related to impaired renal or hepatic function (Table 7). Similar numbers of patients experienced an infusion-related adverse event; however, given that dalbavancin is dosed only on day 1 and day 8, restricting the reported adverse events to only those 2 days of active dalbavancin infusion (i.e., not including placebo infusions given on other days during the blinded clinical trials) identified far fewer infusion-associated events in patients treated with dalbavancin (12 events in 1778 patients vs. 53 events in 1224 comparator patients). Red man syndrome was not identified in any patient receiving dalbavancin in the phase II/III clinical program, although two such cases were reported in phase I studies.
Table 7.
Patients with adverse events of special interest in phase II/III clinical development program
| Dalbavancin | Comparator | |
|---|---|---|
| Infusion-associated AEs | 40/1778 (2.2) | 38/1224 (3.1) |
| Number of infusion-associated events | 48 | 55 |
| Number of events on day of active infusion | 12 | 53 |
| Renal-associated AEs | 33/1778 (1.9) | 24/1224 (2.0) |
| Treatment-related renal-associated AEs | 3 (0.2) | 5 (0.4) |
| Serious renal-associated AEs | 3 (0.2) | 6 (0.5) |
| Treatment-related serious renal-associated AEs | 0 | 3 (0.2) |
| Hepatobiliary AEs | 19/1778 (1.1) | 9/1224 (0.7) |
| Treatment-related treatment-emergent hepatobiliary AEs | 6 (0.3) | 1 (0.1) |
| Serious treatment-emergent hepatobiliary AEs | ||
| All treatment-emergent hepatobiliary AEs | 3 (0.2) | 2 (0.2) |
| Treatment-related hepatobiliary AEs | 0 | 0 |
| TEAE by age, sex, or race | ||
| <65 years of age | 641/1465 (43.8) | 465/995 (46.7) |
| ≥65 years of age | 158/313 (50.5) | 108/229 (47.2) |
| Male | 449/1066 (42.1) | 308/711 (43.3) |
| Female | 350/712 (49.2) | 265/513 (51.7) |
| White | 579/1388 (41.7) | 448/1008 (44.4) |
| Black | 90/143 (62.9) | 58/88 (65.9) |
| Other | 130/247 (52.6) | 67/127 (52.8) |
Data are presented as n (%) or n/N (%) unless otherwise indicated
AE adverse event, TEAE treatment-emergent adverse event
Laboratory assessments performed during the course of the clinical trials demonstrated generally similar laboratory changes post-baseline in patients treated with either dalbavancin or comparator (Table 8). No overall difference was observed in the post-baseline measurements of alanine transaminase (ALT) among patients enrolled in the phase II or III studies, including subjects with an elevated ALT measurement at baseline. Slightly more patients in the dalbavancin treatment group with a normal ALT value at baseline had a post-baseline ALT elevation greater than three times the upper limit of normal (0.8 vs. 0.2 % receiving dalbavancin and comparator, respectively), but fewer patients had a similar degree of elevation if the ALT was abnormal at baseline (12.7 vs. 14.2 % receiving dalbavancin and comparator, respectively). In eight of these 12 patients with a normal ALT at baseline, one or more significant underlying hepatic disorders were identified, such as hepatitis C, hepatitis B, acute cholecystitis, or active alcohol abuse as measured by carbohydrate-deficient transferrin. No case met the criteria for Hy’s law. With regard to renal function, an increase in creatinine was less frequently observed in patients receiving dalbavancin, and was most evident in subjects with a normal creatinine at baseline.
Table 8.
Selected laboratory measurements (phase II/III clinical development program)
| Organ system | Parameter | Criteria, post-baseline | Dalbavancin | Comparator |
|---|---|---|---|---|
| Renal | Creatinine | ≥1.5 × ULN and ≥twofold ↑ | 3 (0.2) | 6 (0.6) |
| >ULN but normal at baseline* | 51 (3.6) | 63 (6.6) | ||
| >ULN but high at baseline | 69 (46.3) | 63 (52.5) | ||
| Hematologic | Hematocrit | ≤0.8 × LLN and ≥0.25-fold ↓ | 5 (0.3) | 4 (0.4) |
| Platelets | ≤0.6 × LLN and ≥0.4-fold ↓ | 2 (0.1) | 4 (0.4) | |
| WBC | ≤0.5 × LLN and ≥0.75-fold ↓ | 1 (0.1) | 1 (0.1) | |
| Hepatic | ||||
| All patients | ALT | Total | 1707 | 1186 |
| >ULN** | 417 (24.4) | 307 (25.9) | ||
| >3 × ULN | 44 (2.6) | 31 (2.6) | ||
| >5 × ULN | 9 (0.5) | 9 (0.8) | ||
| >10 × ULN | 4 (0.2) | 2 (0.2) | ||
| Normal baseline | ALT | Total | 1437 | 975 |
| >ULN** | 218 (15.2) | 139 (14.3) | ||
| >3 × ULN | 12 (0.8) | 2 (0.2) | ||
| >5 × ULN | 5 (0.3) | 1 (0.1) | ||
| >10 × ULN | 3 (0.2) | 0 | ||
| Elevated baseline | ALT | Total | 237 | 197 |
| >ULN** | 173 (73.0) | 155 (78.7) | ||
| >3 × ULN | 30 (12.7) | 28 (14.2) | ||
| >5 × ULN | 4 (1.7) | 8 (4.1) | ||
| >10 × ULN | 1(0.4) | 2 (1.0) | ||
ALT alanine aminotransferase, LLN lower limit of normal, ULN upper limit of normal, WBC white blood cell
* p = 0.001, ** p > 0.05
Of special interest is a comparison of the rate of adverse events in patients receiving the glycopeptide vancomycin and the lipoglycopeptide dalbavancin. The DISCOVER studies compared dalbavancin with vancomycin, with an option to switch to oral linezolid to complete 10–14 days of total therapy. In these two studies combined, 54 patients who received vancomycin for at least 10 days were compared with 637 who received two doses of intravenous dalbavancin and, to control for any demographic variables associated with continuation of intravenous therapy only, 61 who received dalbavancin and at least 10 days of intravenous placebo (Table 9). Fewer TEAEs (p = 0.08) and serious TEAEs (p = 0.03) were reported in patients treated with dalbavancin. In an assessment of potential study drug impact on renal function in this population, nephrotoxicity rates were lower in patients receiving dalbavancin than in those receiving vancomycin for at least 10 days [dalbavancin 3.3 vs. vancomycin 9.3 % (p = 0.06)], supported by rates of nephrotoxicity in a smaller subset of patients, controlling for factors related to continuation of intravenous therapy [dalbavancin 1.7 % vs. vancomycin 9.3 % (p = 0.21)].
Table 9.
Summary of total adverse events for patients receiving >10 days of active or placebo intravenous dosing in DISCOVER
| Number of patients who experienced at least one | Dalbavancin (N = 61) | Vancomycin (N = 54) | p value |
|---|---|---|---|
| TEAE | 18 (29.5) | 25 (46.3) | 0.08 |
| TEAE leading to premature discontinuation of study drug | 1 (1.6) | 0 | 1.0 |
| Drug-related TEAE | 3 (4.9) | 3 (5.6) | 1.00 |
| Serious TEAE | 1 (1.6) | 7 (13.0) | 0.03 |
| Serious TEAE leading to premature study drug discontinuation | 0 | 0 | 1.0 |
| Drug-related serious TEAE | 0 | 0 | 1.0 |
| Serious TEAE leading to death | 0 | 1 (1.9) | 0.23 |
| Nephrotoxicity on therapya | |||
| Safety population | 21/637 (3.3) | 31/638 (4.9) | 0.16 |
| All dalbavancin patients versus IV vancomycin only | 21/637 (3.3) | 5/54 (9.3) | 0.06 |
| Patients who received only IV therapy and no oral therapyb | 1/58 (1.7) | 5/54 (9.3) | 0.21 |
Data are presented as n (%) unless otherwise indicated
IV intravenous, TEAE treatment-emergent adverse event
aNephrotoxicity defined as a 50 % increase from baseline serum creatinine or an absolute increase in serum creatinine of 0.5 mg/dl; p value by Fischer’s exact test
bDalbavancin-active (and IV vancomycin-placebo) versus IV vancomycin-active (and dalbavancin-placebo) without receiving oral linezolid/placebo
Discussion
The safety of dalbavancin has been assessed in 14 clinical studies, including seven phase II or phase III studies of skin infection. The adverse event profile of dalbavancin has been rigorously studied in these clinical trials, leading to the regulatory approval for the indication of ABSSSI in the USA and the EU. With a database of 1778 patients receiving dalbavancin, it is possible to exclude with 95 % confidence the occurrence of any as yet unobserved adverse event at an incidence of >0.2 %, providing reassurance that the data collected thus far will be useful in informing the benefit/risk assessment of dalbavancin. Further refinements of the safety profile will follow with the addition of new clinical trial data and a rigorous post-marketing surveillance program.
The rate of adverse events for patients treated with dalbavancin was similar or slightly lower relative to a pool of comparator agents that included cephalosporins, vancomycin, oxacillin, nafcillin, and linezolid, whether from any cause or related to drug, based on the investigator’s assessment of causality. The types of treatment-related adverse events observed are those commonly seen in patients enrolled in clinical trials, including nausea, diarrhea, and pruritus. While a reduction in adverse event rates of 2–3 % may be of limited clinical significance to the treatment decision for an individual patient, it may be more meaningful on a population basis and, at a minimum, provide some reassurance that dalbavancin is safe and well tolerated relative to alternative treatment options.
One concern that could be raised regarding any drug with a long half-life is that adverse events, when they occur, could last longer, be more severe and possibly occur later, than would occur with a drug with a short half-life. However, patients treated with dalbavancin had a mean duration of adverse events that was similar to that of the much shorter-acting comparators, and these events did not occur any later post-baseline than did events occurring with the comparator, even though follow-up visits occurred up to 70 days after the last scheduled visit.
No difference in the adverse event profile was seen when patients were assessed by age, sex, or race. No difference was also seen in the rate of either renal- or hepatic-associated adverse events. Fewer patients treated with dalbavancin had an elevation in creatinine assessed either against the pooled comparators or against vancomycin, specifically. Similar numbers of patients had abnormalities in hematologic indices.
Increases in serum levels of liver enzymes [ALT, aspartate transaminase (AST)], associated with microscopic findings (histiocytic vacuolation and focal hepatocyte necrosis) in the liver, were noted in toxicology studies in rats and dogs when dalbavancin was administered daily for 28–90 days. In addition, renal toxicity characterized by increases in blood urea nitrogen (BUN) and creatinine and microscopic kidney findings was observed in rats and dogs at doses 5–7 times higher than the expected human dose on an exposure basis. In the clinical program, the majority of the patients with elevated transaminases were found to have other underlying conditions associated with a higher likelihood of hepatic transaminase elevations, even with a normal ALT at baseline, and, overall, they occurred at similar rates with dalbavancin and the comparator.
Our analyses have some limitations. Presentations of pooled safety data prior to marketing authorization are only the first step in the collection of important safety information. Safety data collected from the post-marketing experience will ultimately contribute significantly to our understanding of safety and tolerability, both because the drug is now used in more typical and less controlled circumstances than seen in a clinical trial and because the number of patients exposed to the drug will increase dramatically. It should be noted that one phase II study and one phase III study have not been previously published but are included in the safety assessments for completeness; both studies were performed in patients with skin infections, and the safety outcomes within each trial were similar to the overall experience (Data on file, Allergan plc.). The presentation of these safety data could have been alternatively displayed as relative risk with controls for heterogeneity, given that the patients were enrolled in many studies, not one large trial. However, ultimately, given that the patients were selected for entry into the studies with many criteria common to each trial, data were collected via a standardized data-collection instrument and statistical analyses performed through one set of programs, the overall interpretation of outcomes would not be materially different if analyzed with a different approach. Lastly, it should also be pointed out that approximately 20 % of patients in this pooled analysis had an uncomplicated skin infection and received a single dose of dalbavancin 1000 mg. Though they did not receive a total dose of dalbavancin 1500 mg, they had a significant duration of exposure to drug over the observation period given the compound’s long half-life, and including the outcomes from these patients provides greater precision for our estimates of any late adverse sequelae.
Conclusion
Relative to other options for treatment of Gram-positive infections of the skin, dalbavancin was safe and well tolerated in an extensive clinical trial database of patients with skin and skin structure infection. Collection and analysis of post-marketing surveillance data will help further refine this favorable overall safety profile.
Acknowledgments
The authors would like to acknowledge the contributions of Michael Zelasky and Karthik Gupta for help in data management and Shaina Barnes for administrative support.
This paper was presented at IDWeek 2013, 2–6 October 2013, San Francisco, CA, USA and IDWeek 2014, 8–12 October 2014, Philadelphia, PA, USA.
Compliance with Ethical Standards
Funding
This work was supported by Durata Therapeutics, Inc.
Conflict of interest
George H. Talbot reports receiving fees through Talbot Advisors LLC for serving on advisory boards and consulting for Durata Therapeutics and owning stock or stock options for Durata Therapeutics. Helen W. Boucher serves as an advisor to Merck, Nabriva, and Achtellion. Mark Wilcox reports receiving consulting fees from Astra-Zeneca, Bayer, Cerexa, Durata, The Medicines Company, Motif Biosciences, Nabriva, Paratek and Pfizer; lecture fees from Astra-Zeneca and Pfizer; and grant support from Pfizer. Sailaja Puttagunta and Michael W. Dunne report being employees of and owning stock in Durata Therapeutics. There are no other potential conflicts of interest relevant to this article.
Ethics approval
All clinical trials were reviewed and approved by the local ethics committee. All patients provided signed informed consent.
References
- 1.Pallin DJ, Egan DJ, Pelletier AJ, et al. Increased US emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann Emerg Med. 2008;51(3):292–298. doi: 10.1016/j.annemergmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 2.David MZ, Daum RS, Bayer AS, et al. Staphylococcus aureus bacteremia at five U.S. Academic Medical Centers, 2008–2011: significant geographic variation in community-onset infections. Clin Infect Dis. 2014;59(6):798–807. doi: 10.1093/cid/ciu410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011 doi: 10.1093/cid/ciq146. [DOI] [PubMed] [Google Scholar]
- 4.Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med. 2007;357(4):380–390. doi: 10.1056/NEJMcp070747. [DOI] [PubMed] [Google Scholar]
- 5.Dorr MB, Jabes D, Cavaleri M, et al. Human pharmacokinetics and rationale for once-weekly dosing of dalbavancin, a semi-synthetic glycopeptide. J Antimicrob Chemother 2005;55(Suppl 2): ii25–30. [DOI] [PubMed]
- 6.Dunne MW, Zhou M, Borje Darpo B. A thorough QT study with dalbavancin: a novel lipoglycopeptide antibiotic for the treatment of acute bacterial skin and skin-structure infections. Int J Antimicrob Agents. 2015;45(4):393–8. [DOI] [PubMed]
- 7.Dunne MW, Puttagunta S, Sprenger CR, et al. Extended-duration dosing and distribution of dalbavancin into bone and articular tissue. Antimicrob Agents Chemother. 2015;59(4):1849–1855. doi: 10.1128/AAC.04550-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jauregui LE, Babazadeh S, Seltzer E, et al. Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41(10):1407–1415. doi: 10.1086/497271. [DOI] [PubMed] [Google Scholar]
- 9.Seltzer E, Dorr MB, Goldstein BP, et al. Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections. Clin Infect Dis. 2003;37(10):1298–1303. doi: 10.1086/379015. [DOI] [PubMed] [Google Scholar]
- 10.Boucher HW, Wilcox M, Talbot GH, et al. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169–2178. doi: 10.1056/NEJMoa1310480. [DOI] [PubMed] [Google Scholar]
- 11.Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by gram-positive pathogens. Clin Infect Dis. 2005;40:374–380. doi: 10.1086/427283. [DOI] [PubMed] [Google Scholar]
- 12.Dalvance® prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021883s000lbl.pdf. Accessed 22 Sept 2015.
- 13.Dalbavancin summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002840/WC500183869.pdf. Accessed 22 Sept 2015.
- 14.Guidance for industry and investigators safety reporting requirements for INDs and BA/BE Studies. US Department of Health and Human Services, Food and Drug Administration. December 2012. http://www.fda.gov/downloads/Drugs/.../Guidances/UCM227351.pdf
- 15.Bouvy JC, De Bruin ML, Koopmanschap MA. Epidemiology of adverse drug reactions in Europe: a review of recent observational studies. Drug Saf. 2015;38(5):437–453. doi: 10.1007/s40264-015-0281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalbavancin for injection for treatment of acute bacterial skin and skin structure infections. NDA 021-883 briefing document. Presented to the FDA Anti-Infective Drugs Advisory Committee 31 March 2014. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM390793.pdf. Accessed 22 Sept 2015.