

Abstract
Sweet's syndrome (SS) is a neutrophilic dermatosis disorder of unknown aetiology, characterised by acute fever, neutrophilia, painful erythematous papules, nodules and plaques, and an infiltrate consisting predominantly of mature neutrophils in the upper dermis. Classical SS is a rare extra-intestinal manifestation of inflammatory bowel disease (IBD). It is more common in Crohn's disease than in ulcerative colitis (UC). There is a predilection for women, and for patients with colonic disease and active IBD. We report the case of a 39-year-old woman with a flare of moderate severity UC treated with mesalazine who presented with a 5-day history of acute fever, painful papules and plaques on forearms and legs, episcleritis and cervical pain. Skin biopsies showed papillary dermis inflammatory cell infiltration composed mainly of neutrophils, without evidence of leukocytoclastic vasculitis or panniculitis, compatible with SS. The patient had an excellent response to systemic corticosteroids. Symptoms promptly improved and skin lesions resolved after 7 weeks.
Background
Sweet’s syndrome (SS), also referred to as acute febrile neutrophilic dermatosis, is an acute skin disorder of unknown aetiology, described for the first time by Robert Douglas Sweet in 1964. SS is defined by clinical, laboratory and histological criteria. It is characterised by abrupt onset of painful erythematous papules, plaques or nodules and histopathological evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis or panniculitis. Pain associated with cutaneous lesions of SS is often described as tenderness or a burning sensation. Pruritus is a rare feature of this rash. A retrospective study of 77 patients with SS documented pruritus in 18% of patients. The distribution of the cutaneous eruption is often asymmetrical and the most common sites of involvement are the face, neck, trunk and extremities.1 2 Acute fever and leukocytosis with elevated neutrophil count often accompany the cutaneous lesions. Extracutaneous manifestations of SS may occur by neutrophilic infiltration of other organs such as the eye, mouth, muscle, lung, bone, liver, spleen, heart, kidney, central nervous system and gastrointestinal system, resulting in signs and symptoms specific to the site of inflammation.3
After the first description by Sweet, diagnostic criteria for SS were reformulated by Su and Liu in 1986,4 and by von den Driesch in 1994.5 It was divided into three subtypes based on aetiology: classical or idiopathic, malignancy-associated and drug-induced SS. The diagnosis of classic SS requires the presence of both major criteria (abrupt onset of painful erythematous plaques or nodules and histopathological evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis) and two of four minor criteria (fever >38°C; association with an underlying hemoproliferative disorder, solid tumour or inflammatory disease, or preceded by an upper respiratory or gastrointestinal infection; favourable response to treatment with systemic corticosteroids or potassium iodide; three of four abnormal laboratory values at presentation—erythrocyte sedimentation rate >20 mm/1st hour, positive C reactive protein, >8000 leucocytes, >70% neutrophils).1 5 6
The authors report a clinical case of classical SS in a 39-year-old woman with a flare of moderate severity ulcerative colitis (UC) treated with mesalazine. SS is a rare extra-intestinal manifestation of UC that should be borne in mind in the presence of skin lesions, in order to reach prompt diagnosis and establish appropriate treatment.
Case presentation
A 39-year-old Caucasian woman presented with a 5-day history of sudden onset of malaise, fever, painful erythematous papules and pseudovesiculated plaques on forearms and legs, left eye episcleritis and mechanical cervical pain (figure 1A–C). Two months before admission, the patient had developed diarrhoea, sometimes associated with blood (stool frequency 4–5/day), colicky abdominal pain, tenesmus and urgency. She was evaluated in gastroenterology consultation. Colonoscopy performed 1 month before admission to our hospital revealed multiple ulcerative lesions of colonic mucosa involving the rectum, sigmoid, descending and transverse colon. Colonic biopsy showed distortion of crypt architecture, cryptitis, crypt abscesses and a polymorphic inflammatory cell infiltrate in the lamina propria, suggestive of UC. The patient was then diagnosed with an inaugural flare of moderate severity UC. A combination of topic and oral mesalazine was at this point started for remission induction. Symptomatic improvement was seen within a few days, with resolution of abdominal pain, tenesmus, urgency and bleeding. By the time of our hospital admission, the patient presented with four or fewer stools per day, without blood. There were no other symptoms and there was no significant epidemiological context.
Figure 1.

Mucocutaneous manifestations of Sweet's syndrome. (A) Left eye episcleritis. (B) Forearm and (C) leg with erythaematous papules and pseudovesiculated plaques.
Medical history was also significant for mild iron deficiency anaemia, essential hypertension and hypothyroidism due to total thyroidectomy for benign multinodular goitre.
The patient was medicated with levothyroxine 100 µg, amlodipine 5 mg and aliskiren/hydrochlorothiazide 300/12.5 mg, all once daily; and mesalazine enemas 4 g two times a day and oral mesalazine (salofalk) 3 g daily in two divided doses.
There was no relevant family or social history.
On admission to our hospital, physical examination revealed tachycardia, a body temperature of 39°C, bilateral disseminated painful erythematous papules and pseudovesiculated plaques on forearms and legs, left eye episcleritis and cervical pain aggravated by movement and local palpation, without neck rigidity.
Investigations
Laboratory evaluation demonstrated elevated inflammatory markers (leucocyte count 12 900/µL with 64.4% neutrophils and 23.3% lymphocytes, C reactive protein 129 mg/L, erythrocyte sedimentation rate 80 mm/1st hour) and microcytic anaemia (haemoglobin 10.9 g/dL, mean cell volume 78.7 fL). Ionogram, liver and renal function tests, arterial blood gasometry and urinalysis were normal.
Blood culture specimens were negative. HIV 1 and 2, Herpes simplex virus 2, varicella-zoster virus and hepatitis C virus were all negative (IgM and IgG). Cytomegalovirus, herpes simplex virus 1, Epstein-Barr virus and parvovirus were IgM negative and IgG positive. The patient was not immune to hepatitis B virus. Weil-Felix test and antistreptolysin-O titre were negative. Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption were negative.
Mantoux test and Interferon-Gamma Release Assay were negative.
Serum immunoglobulin concentrations (IgG, IgA and IgM), antinuclear and antineutrophil cytoplasmic antibodies were negative.
Human chorionic gonadotropin testing was negative.
An infectious cause of diarrhoea was excluded by extensive stool tests including negative results for Clostridium difficile, Escherichia coli, Shigella, Campylobacter, Salmonella, Yersinia and parasites.
Chest X-ray was normal. ECG revealed sinus tachycardia (120 bpm), with no other changes. CT of the abdomen and pelvis revealed oedema and thickening of the descending and sigmoid colon, with multiple infracentimetric mesenteric lymph nodes.
We did not repeat colonoscopy and biopsy as the patient had performed these diagnostic tests 1 month before admission to our hospital, revealing a flare of moderate severity UC and she was currently clinically improved with topical and oral mesalazine.
Ophthalmic examination revealed left eye episcleritis with no other ocular manifestations.
SS was suspected owing to acute fever and cutaneous eruption features, and a skin biopsy specimen was collected from a plaque in the forearm. Histological examination showed papillary dermis inflammatory cell infiltration composed mainly of neutrophils, without evidence of leukocytoclastic vasculitis or panniculitis (figure 2A, B). These findings were compatible with the diagnosis of SS.
Figure 2.
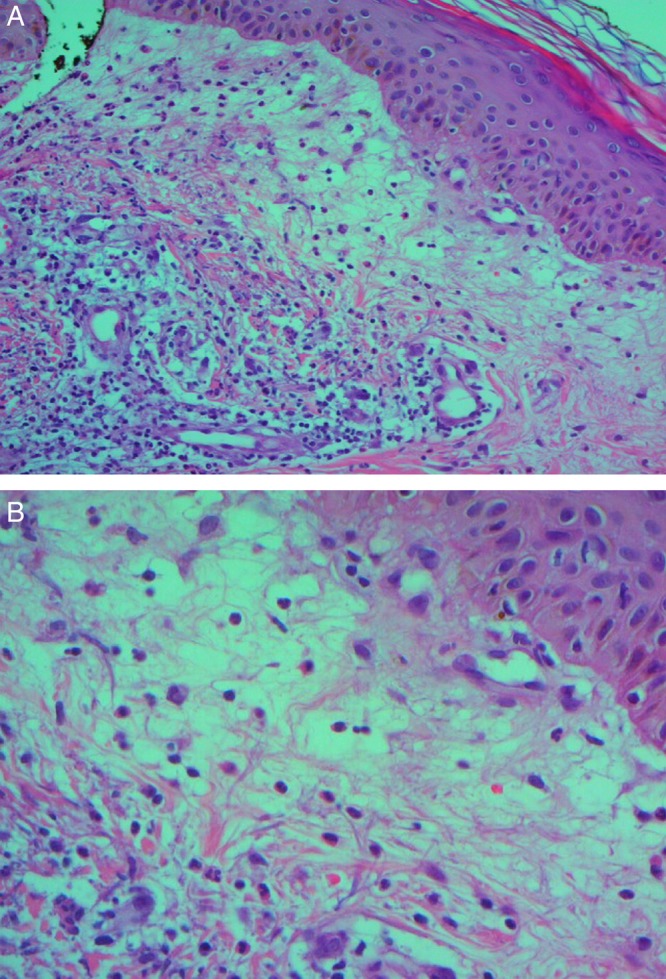
Histological findings of skin biopsy from a plaque in the forearm: neutrophilic infiltration of the superficial dermis tissue. (A) Low-power magnification, H&E stain. (B) High-power magnification, H&E stain.
Differential diagnosis
The clinical differential diagnosis of SS is dependent on skin lesion morphology—these lesions can mimic the morphology of several other mucocutaneous and systemic conditions. Hence, on admission, the differential diagnosis included infectious and inflammatory disorders (bacterial sepsis, herpes simplex virus infection, varicella-zoster virus infection, panniculitis, syphilis, tuberculosis, SS, erythema nodosum and pyoderma gangrenosum), leukocytoclastic vasculitis, autoimmune diseases (Behçet's disease, dermatomyositis and lupus erythematosus), drug eruptions and, less likely, neoplastic conditions (leukaemia cutis, lymphoma and metastatic tumour). The supplementary study excluded most of these pathologies and made the diagnosis of SS.
Treatment
The patient presented with acute malaise, fever, cutaneous eruption and elevated inflammatory markers, as well as having watery diarrhoea, so empirical therapy with oral ciprofloxacin 500 mg every 12 h and oral metronidazole 500 mg every 8 h was initiated assuming the possibility of an infectious colitis. Ciprofloxacin covered Salmonella species (sp), Shigella sp, Campylobacter jejuni and Escherichia coli. Metronidazole covered Clostridium difficile, Giardia and Entamoeba histolytica. Ophthalmic diclofenac was initiated in the context of left eye episcleritis.
Despite this therapy, there was sustained fever and rising inflammatory markers (leucocyte count 19 800/µL with 81% neutrophils, C reactive protein 322 mg/L). As clinical and histological findings of skin biopsy were suggestive of SS, treatment with systemic corticosteroids was initiated and antibiotics were discontinued. Intravenous methylprednisolone sodium succinate at a dose of 0.8 mg/kg/day was begun with rapid improvement of skin lesions (figure 3), episcleritis and cervical pain, complete resolution of fever and fall of the inflammatory markers. The patient was discharged on the fifth day of corticosteroid therapy, with a prednisolone tapering schedule and oral mesalazine.
Figure 3.
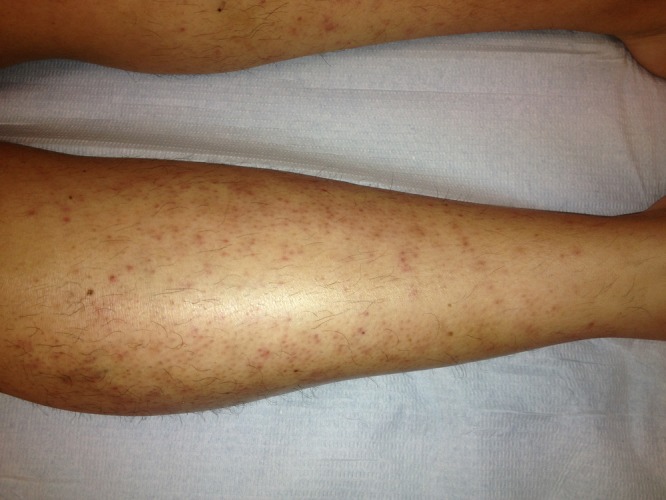
Leg skin lesions after 4 days of corticosteroid therapy.
Outcome and follow-up
Treatment with oral mesalazine 3 g/day and prednisolone 55 mg/day was carried out for 3 weeks and subsequently progressive corticosteroid weaning over 1 month was performed, maintaining oral mesalazine. There was complete resolution of skin lesions and episcleritis, with no further manifestations of SS. Diarrhoea and anaemia also resolved and colonoscopy performed 6 months after the patient’s discharge showed quiescent UC.
Discussion
A classical subtype constitutes the majority of cases of SS and most frequently occurs between the ages of 30–60 years. It is defined as SS that meets the established diagnostic criteria and is not associated with malignancy or drug exposure. It may occur in the setting of a variety of medical conditions, namely infections (particularly upper respiratory tract and gastrointestinal infections, usually developing 1–3 weeks after infection), autoimmune diseases (Behçet’s syndrome, rheumatoid arthritis, sarcoidosis, autoimmune thyroid disease, systemic lupus erythematosus, dermatomyositis), inflammatory bowel disease (IBD) (UC and Crohn’s disease) and pregnancy. The pathogenesis of SS may be multifactorial and remains to be definitively determined. Hypersensitivity reactions to bacterial, viral, neoplastic and other antigens, inflammatory cytokine overproduction and inappropriate regulation (including interleukin 1, 3, 6 and 8, granulocyte macrophage-colony stimulating factor and interferon-γ) and genetic susceptibility (abnormalities in chromosome 3q) are possible contributing factors.1 6 7
The association between IBD and SS was first described in 1988. SS is a rare extra-intestinal manifestation of IBD, more common in Crohn’s disease (70%) than in UC (30%). There is a significant predominance in women (87%), patients with colonic disease (100%) and those with other extra-intestinal features (77%). SS is associated with active IBD in 67–80%, but may precede the onset of intestinal symptoms in 21%, and has been also described after proctocolectomy for UC. Higher prevalence among females may be related to hormonal and genetic mechanisms, but this association is not totally understood.8–10
Our patient had been diagnosed with a flare of UC 2 months before admission to our hospital, with symptomatic improvement seen within a few days of therapy with topical and oral mesalazine, maintaining four or fewer stools per day, without blood. Three weeks after starting mesalazine, she developed typical SS skin lesions, manifestations suggestive of neutrophilic infiltration of other organs (eye and muscle) and a systemic inflammatory response syndrome (malaise, fever, tachycardia, leukocytosis), meeting all diagnostic criteria for classical SS.
Ocular inflammation is a common extracutaneous manifestation of classical SS occurring in 17–72% of patients. Possible ocular manifestations include episcleritis, scleritis, iritis, glaucoma, dacryoadenitis, choroiditis and peripheral ulcerative keratitis. The musculoskeletal system is another frequent site of involvement, manifesting as myalgias, arthralgias or arthritis. Intestinal neutrophilic inflammation may result in diarrhoea. Uncommon extracutaneous manifestations of SS include neutrophilic alveolitis, pleural effusions, airway obstruction, myocarditis, aortic stenosis, coronary artery occlusion, aortitis, hepatomegaly, hepatitis, splenomegaly, mesangial glomerulonephritis, haematuria, proteinuria, encephalitis, aseptic meningitis and sterile osteomyelitis.1 3 11–13
SS associated with IBD is a particularly challenging diagnosis since nearly one-third of patients with IBD develop skin lesions (specific manifestations of IBD, reactive cutaneous disorders, secondary to malnutrition and malabsorption, secondary to drug therapy or miscellaneous). Reactive cutaneous disorders include SS, erythema nodosum (EN), pyoderma gangrenosum, cutaneous polyarteritis nodosa and pyodermatitis. EN is the most frequent skin manifestation associated with IBD, occurring in 11% of cases, more commonly in colonic Crohn’s disease than in UC. It typically presents suddenly as multiple, deep-red, tender, painful nodules distributed over the shins, but can also appear on the thighs, trunk and upper extremities. Histologically, EN is a panniculitis involving inflammation of septa in the subcutaneous fat tissue, usually without associated vasculitis, although small vessel inflammatory changes occasionally occur.14 15
Untreated SS lesions can persist for weeks to months and may eventually resolve in some patients with classical SS. Recurrence of the dermatosis can occur in approximately one-third of cases. The mainstay of treatment for SS is systemic corticosteroid therapy. Dermatosis-related symptoms promptly improve and skin manifestations may resolve subsequently after a 6-week course of steroid treatment. In adults, systemic corticosteroid therapy often begins with 0.5–1 mg/kg/day of prednisolone as a single oral morning dose until clinical response. Then the dose can be tapered to 10 mg/day within 4–6 weeks, but some patients may require 2–3 months of treatment. For resistant disease, intravenous methylprednisolone sodium succinate at a dose of up to 1000 mg/day may be initially necessary for 3–5 days, with subsequent tapering oral dose of corticosteroid or other immunosuppressant agent. Topical or intralesional corticosteroids are used to treat localised and small numbers of SS lesions, without systemic manifestations of the disease. For adults who cannot be treated with systemic glucocorticoids, other first-line systemic treatments are colchicine (0.5 mg orally three times a day for 10–21 days) and potassium iodide (300 mg orally three times a day). Second-line therapy with indomethacin (150 mg orally for 7 days followed by 100 mg orally for 14 days) or with immunosuppressants, such as cyclosporine (2–10 mg/kg/day, tapering by 2 mg/kg/day every 2 days and discontinuing on day 21) in monotherapy or in combination with steroids, dapsone (1.5–2 mg/Kg/day), azathioprine (1.5–2 mg/kg/day), cyclophosphamide (1–1.5 mg/kg/day) and tumour necrosis factor (TNF)-α antagonists, have been reported to be successful in refractory SS cases.1 9 16 17 There have been case reports of patients with Crohn’s disease and refractory SS responding to infliximab, a monoclonal anti-TNF-α antibody.18–21 TNF-α antagonists such as etanercept, infliximab and adalimumab should be considered in resistant or highly relapsing SS cases.22 Curiously, some of the drugs used for the treatment of SS have also been observed, though rarely, to trigger the condition, namely etanercept, infliximab, adalimumab and lenalidomide.23 Anakinra, an anti-interleukin-1 receptor antagonist, is a promising therapeutic to refractory SS.23 24
Learning points.
Sweet’s syndrome (SS) is defined by clinical, laboratory and histological criteria. It is characterised by abrupt onset of painful erythematous papules, plaques or nodules and histopathological evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis or panniculitis. Extracutaneous manifestations of SS may occur by neutrophilic infiltration of other organs, resulting in signs and symptoms specific to the site of inflammation.
The clinical differential diagnosis of SS is dependent on skin lesion morphology—these lesions can mimic the morphology of several other mucocutaneous and systemic conditions.
SS associated with inflammatory bowel disease (IBD) is a particularly challenging diagnosis since nearly one-third of the patients with IBD develop skin lesions.
SS is a rare extra-intestinal manifestation of IBD, more common in Crohn’s disease than in ulcerative colitis. It is associated with active IBD in 67–80%, but may precede the onset of intestinal symptoms in 21% of individuals.
The mainstay of treatment for SS is systemic corticosteroid therapy. Dermatosis-related symptoms promptly improve and skin manifestations may resolve subsequently after a 6-week course of treatment.
Acknowledgments
The authors wish to express their gratitude to Dr Herberto Bettencourt of the Anatomical Pathology Department, Dr Joana Rocha of the Dermatology Department and Dr Carlos Menezes of the Ophthalmology Department of Hospital Pedro Hispano, for their clinical contribution.
Footnotes
Contributors: RLC, MS and CC were responsible for the patient's management during hospitalisation and follow-up after discharge, collected all significant clinical information and drafted this manuscript. AF reviewed, redrafted and made significant contribution to the final version. All the authors have given final approval of this version.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Cohen PR. Sweet's syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis 2007;2:34 10.1186/1750-1172-2-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rochet NM, Chavan RN, Cappel MA et al. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol 2013;69:557–64. 10.1016/j.jaad.2013.06.023 [DOI] [PubMed] [Google Scholar]
- 3.Moschella SL, Davis MD. Neutrophilic dermatoses. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 1. Spain: Mosby Elsevier, 2008:379–93. [Google Scholar]
- 4.Su WP, Liu HN. Diagnostic criteria for Sweet's syndrome. Cutis 1986;37:167–74. [PubMed] [Google Scholar]
- 5.von den Driesch P. Sweet's syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol 1994;31:535–56. 10.1016/S0190-9622(94)70215-2 [DOI] [PubMed] [Google Scholar]
- 6.Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug induced Sweet's syndrome. J Am Acad Dermatol 1996;34:918–23. 10.1016/S0190-9622(96)90080-8 [DOI] [PubMed] [Google Scholar]
- 7.Shin OR, Lee Y, Bak S et al. Gastroenterology: sweet's syndrome in a patient with acutely exacerbated ulcerative colitis. J Gastroenterol Hepatol 2015;30:965 10.1111/jgh.12911 [DOI] [PubMed] [Google Scholar]
- 8.Travis S, Innes N, Davies MG et al. Sweet's syndrome: an unusual cutaneous feature of Crohn's disease or ulcerative colitis. Eur J Gastroenterol Hepatol 1997;9:715–20. 10.1097/00042737-199707000-00013 [DOI] [PubMed] [Google Scholar]
- 9.Ali M, Duerksen DR. Ulcerative colitis and Sweet's syndrome: a case report and review of the literature. Can J Gastroenterol 2008;22:296–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diaz-Peromingo JA, Garcia-Suarez F, Sanchez-Leira J et al. Sweet's syndrome in a patient with acute ulcerative colitis: presentation of a case and review of the literature. Yale J Biol Med 2001;74:165–8. [PMC free article] [PubMed] [Google Scholar]
- 11.Gottlieb CC, Mishra A, Belliveau D et al. Ocular involvement in acute febrile neutrophilic dermatosis (Sweet syndrome): new cases and review of the literature. Surv Ophthalmol 2008;53:219–26. 10.1016/j.survophthal.2008.02.006 [DOI] [PubMed] [Google Scholar]
- 12.Cohen PR, Hongsmann H, Kurzrock R. Acute febrile neutrophilic dermatosis (Sweet syndrome). In: Goldsmith LA, Katz SI, Gilchrest BA et al., eds. Fitzpatrick's dermatology in general medicine. 8th edn Vol 1 McGraw Hill, 2012:362. [Google Scholar]
- 13.Vanourny J, Swick BL. Sweet syndrome with systemic inflammatory response syndrome. Arch Dermatol 2012;148:969–70. 10.1001/archdermatol.2012.766 [DOI] [PubMed] [Google Scholar]
- 14.Pellicer Z, Santiago JM, Rodriguez A et al. Management of cutaneous disorders related to inflammatory bowel disease. Ann Gastroenterol 2012;25:21–6. [PMC free article] [PubMed] [Google Scholar]
- 15.Vavricka SR, Schoepfer A, Scharl M et al. Extraintestinal manifestations of inflammatory bowel disease. Inflamm Bowel Dis 2015;21:1982–92. 10.1097/MIB.0000000000000392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marzano AV, Menicanti C, Crosti C et al. Neutrophilic dermatoses and inflammatory bowel diseases. G Ital Dermatol Venereol 2013;148: 185–96. [PubMed] [Google Scholar]
- 17.Terai T, Sugimoto M, Osawa S et al. Successful treatment of ulcerative colitis complicated by Sweet's syndrome by corticosteroid therapy and leukocytapheresis. Clin J Gastroenterol 2011;4:151–6. 10.1007/s12328-011-0215-z [DOI] [PubMed] [Google Scholar]
- 18.Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol 2009;10:301–12. 10.2165/11310730-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 19.Schadt CR, Callen JP. Management of neutrophilic dermatoses. Dermatol Ther 2012;25:158–72. 10.1111/j.1529-8019.2012.01488.x [DOI] [PubMed] [Google Scholar]
- 20.Foster EN, Nguyen KK, Sheikh RA et al. Crohn's disease associated with Sweet's syndrome and Sjogren's syndrome treated with infliximab. Clin Dev Immunol 2005;12:145–9. 10.1080/17402520500134254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahier JF, Lion L, Dewit O et al. Regression of Sweet's syndrome associated with Crohn's disease after anti-tumour necrosis factor therapy. Acta Gastroenterol Belg 2005;68:376–9. [PubMed] [Google Scholar]
- 22.Angelo VM, Rim SI, Simone S et al. Autoinflammatory skin disorders in inflammatory bowel diseases, pyoderma gangrenosum and Sweet's syndrome: a comprehensive review and disease classification criteria. Clin Rev Allergy Immunol 2013;45:202–10. 10.1007/s12016-012-8351-x [DOI] [PubMed] [Google Scholar]
- 23.Cohen PR, Anzalone CL. Acute febrile neutrophilic dermatosis (Sweet's syndrome). Curr Opin Hematol 2013;20:26–35. 10.1097/MOH.0b013e32835ad132 [DOI] [PubMed] [Google Scholar]
- 24.Maalouf D, Battistella M, Bouaziz JD. Neutrophilic dermatosis: disease mechanism and treatment. Curr Opin Hematol 2015;22:23–9. 10.1097/MOH.0000000000000100 [DOI] [PubMed] [Google Scholar]