

Abstract
Sarcomas are a group of cancers that arise from transformed cells of mesenchymal origin. They can be classified into over 50 subtypes, accounting for approximately 1% of adult and 15% of pediatric cancers. Wide surgical resection, radiotherapy, and chemotherapy are the most common treatments for the majority of sarcomas. Among these therapies, chemotherapy can palliate symptoms and prolong life for some sarcoma patients. However, sarcoma cells can have intrinsic or acquired resistance after treatment with chemotherapeutics drugs, leading to the development of multidrug resistance (MDR). MDR attenuates the efficacy of anticancer drugs and results in treatment failure for sarcomas. Therefore, overcoming MDR is an unmet need for sarcoma therapy. Certain protein kinases demonstrate aberrant expression and/or activity in sarcoma cells, which have been found to be involved in the regulation of sarcoma cell progression, such as cell cycle, apoptosis, and survival. Inhibiting these protein kinases may not only decrease the proliferation and growth of sarcoma cells, but also reverse their resistance to chemotherapeutic drugs to subsequently reduce the doses of anticancer drugs and decrease drug side-effects. The discovery of novel strategies targeting protein kinases opens a door to a new area of sarcoma research and provides insight into the mechanisms of MDR in chemotherapy. This review will focus on the recent studies in targeting protein kinase to reverse chemotherapeutic drug resistance in sarcoma.
Keywords: multidrug resistance (MDR), protein kinase, sarcoma
Introduction
Sarcomas are a heterogenous group of malignant tumors that arise from transformed cells of mesenchymal origin. Sarcomas are divided into two main groups: soft tissue sarcomas and bone sarcomas. Soft tissue sarcomas (STS) originate in connective tissues such as fat, muscle, nerve, tendon, the lining of joints, blood vessels, or lymph vessels. There are more than 50 different histological subtypes of STS, such as undifferentiated pleomorphic sarcoma (UPS), rhabdomyosarcoma, liposarcoma, angiosarcoma, synovial sarcoma, leiomyosarcoma, and others. Bone sarcomas develop from bone, and include osteosarcoma, Ewing sarcoma, and chondrosarcoma. There are 15,000 new sarcoma cases per year in the United States, consisting of 12,000 cases of STS and 3,000 cases of bone sarcomas1. The 5-year overall survival rate is approximately 50–80% for sarcomas2.
Surgical resection, radiotherapy, and systemic chemotherapy comprise the standard treatments for sarcoma. The application of multi-agent chemotherapy and appropriate surgical resection has significantly improved the survival rate and quality of life after treatment for patients with certain types of sarcoma, including osteosarcoma, rhabdomyosarcoma, and Ewing sarcoma. For example, major progress has been made in the treatment of patients with osteosarcoma because of the use of chemotherapy, leading to an improved overall survival rate of up to 65%. Chemotherapy drugs usually include doxorubicin, methotrexate, ifosfamide, and cisplatin. Unfortunately, the efficacy of these agents is often hampered by the development of multidrug resistance (MDR). In osteosarcoma, 30% to 40% of patients will experience MDR associated with recurrence or metastasis despite improved multimodality therapy3. A number of patients will also develop resistance to multiple types of chemotherapy after prolonged periods of treatment. Drug resistance to various functionally and structurally unrelated chemotherapeutic drugs in sarcoma cells may be intrinsic or acquired, and subsequently limits the overall utility of chemotherapy. Improvements to the survival rate of sarcoma patients have reached a plateau in the past few decades. Almost one third of patients with localized sarcoma experience recurrent or progressive disease, and the average survival period after recurrence is about one year. The mechanism of MDR in sarcoma is not well understood. There is a wide range of mechanisms that contribute to drug sensitivity/resistance, including ATP-binding cassette transporter (ABC transporters) mediated drug efflux, alteration of apoptosis, cancer stem cells (CSC), alteration/mutation of specific targets of the drugs, aberrant activation cell signaling pathways, DNA damage and repair, autophagy induction, miRNA regulation, hypoxia induction, epigenetic regulation, tumor microenvironment, etc. Overexpression/activation of protein kinases in sarcoma has also been recently identified, which enables sarcoma cells to escape the cytotoxic effects of chemotherapeutic agents. Strategies to reverse MDR have been a high priority goal for clinical and investigational sarcoma oncologists. One promising approach is the specific targeting of protein kinases implicated in different types of sarcoma to reverse MDR4.
The human kinome contains at least 600 protein kinases that carry out the phosphorylaton of proteins at 250,000 or more sites. Generally, protein kinases are divided into tyrosine, serine/threonine, histidine, and mixed protein kinases based on their phosphorylated substrates. Protein kinases are vital to regulate many tumor processes, including cell growth, survival, angiogenesis, apoptosis, recurrence, and metastasis. Consequently, protein kinases have emerged as one of the most promising therapeutic target families for the treatment of cancers. To date, the Food and Drug Administration (FDA) has approved approximately 30 protein kinases inhibitors for clinical use5. Though the understanding of the functional roles in the kinome for MDR is not yet understood, studies of these protein kinases and their functions will contribute to the discovery and development of new therapeutic strategies. A number of protein kinases have been found to be highly expressed and activated in a variety of sarcomas, particularly in the late stage of drug resistant tumors6. Suppression of several protein kinases, such as mammalian target of rapamycin (mTOR), SRC non-receptor tyrosine kinase (SRC), insulin-like growth factor 1 (IGF-1) receptor (IGF-1R), epidermal growth factor receptor (EGFR), c-Jun N-terminal kinases (JNKs), Janus kinase (JAK), mitogen-activated protein kinase (MAPK), or extracellular signal-regulated kinase (ERK), enhances cell death in the presence of low concentrations of chemotherapeutic drug, implicating the potential utility of these protein kinases as drug targets7.
Here, we present an overview of the most recent targets within the protein kinases, which may reverse chemotherapeutic drugs resistance in sarcomas.
Targeting tyrosine kinases to reverse MDR in sarcomas
Tyrosine kinases (TKs) catalyze the transfer of a phosphate of ATP to tyrosine residues on protein substrates. Tyrosine kinases play a key role in signal transduction and regulating cell cycle, mitogenesis, proliferation, differentiation, adhesion, migration, and apoptosis8. The tyrosine kinase families include 90 tyrosine kinases in the human genome and are divided into two main subfamilies: receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (NRTKs). RTKs consist of an extracellular domain, a transmembrane domain, and an intracellular catalytic domain. A specific ligand (e.g., a particular growth factor or hormone) binds the extracellular domain and triggers a cascade of reactions; as a result, an extracellular signal is transduced to the nucleus through phosphorylation of intracellular substrates proteins, which consequently regulates the expression of gene and protein function. There are 58 types of RTKs, distributed into 20 subfamilies, including EGFR, vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), platelet-derived growth factor receptor (PDGFR), anaplastic lymphoma receptor tyrosine kinase (ALK), AXL receptor tyrosine kinase (AXL), MET proto-oncogene (MET, also known as Hepatocyte Growth Factor Receptor), and insulin receptor (INSR). NRTKs are phosphotransferase enzymes that are responsible for catalyzing the transfer of a phosphate group from ATP to tyrosine residues in proteins. NRTKs transfer extracellular signals to the nucleus, regulate cellular diverse processes, and are also vital to regulating the immune system. Unlike RTKs, most NRTKs are localized in the cytoplasm; however, there are few NRTKs anchored to the cell membrane through amino-terminal modification, whereas others lack an extracellular ligand-binding domain and transmembrane-spanning region. Thirty-two types of NRTKs have been identified and divided into 10 subfamilies, including SRC, ABL, JAK, and Focal adhesion kinase (FAK). Aberrant expression of certain tyrosine kinases and/or constitutive activation of downstream pathways may be responsible for the progression of sarcoma, including tumor cell survival, apoptosis, neovascularization, and invasion, particularly in MDR9. Accumulating evidence reveals that tyrosine kinases are highly expressed in sarcoma, such as overexpression of JAK1 in Ewing sarcoma, EGFR and PDGFRA in synovial sarcoma, and FGFR in osteosarcoma and rhabdomyosarcoma10. Targeting tyrosine kinases can not only lead to inhibition of these kinases and downregulation of downstream signal pathways to decrease tumor cell growth, but may also present a potential strategy to reverse MDR in sarcoma (Figure. 1, Table 1).
Figure 1. Targeting tyrosine kinases reverse MDR in sarcomas.
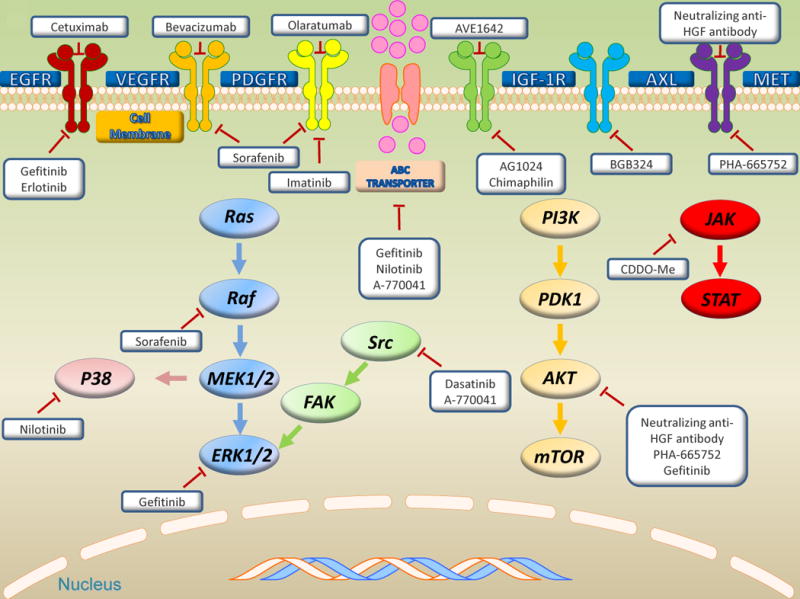
Intracellular signal transduction of tyrosine kinases and potential target sites in sarcomas, including receptor tyrosine kinases, non-receptor tyrosine kinases, ABC transporters, and three major downstream signaling pathways: PI3K/AKT/mTOR, MAPK, and JAK/STAT pathways. Bold represent target tyrosine kinases by small molecule kinase inhibitors and mAb.
Table1.
Targeting tyrosine kinase reverse MDR in sarcomas
| Target Kinase | Inhibition compound | functional mechanism | Effect on anticancer drug | Sarcoma | References |
|---|---|---|---|---|---|
| Gefitinib | apoptosis, cell-cycle arrest, ABCC1 | Vincristine | Leiomyosarcomas | 9 | |
| EGFR | Erlotinib | apoptosis, cell-cycle arrest | Cisplatin | Chondrosarcoma | 16 |
| Cetuximab | natural killer cells | Doxorubicin, methotrexate | Osteosarcoma | 17 | |
| AXL | BGB324 | PI3K/AKT, ERK pathways | Vincristine, doxorubicin | Ewing sarcoma | 24 |
| VEGF | Bevacizumab | VEGF | Gemcitabine, docetaxel | Leiomyosarcoma, angiosarcoma, liposarcoma | 41 |
| PDGFR | Imatinib | Reactive oxygen species accumulation | Doxorubicin | Osteosarcoma | 38 |
| Sorafenib | VEGFR,PDGFR,Raf | Topotecan | Osteosarcoma, Ewing sarcoma | 4 | |
| MET | Anti-HGF antibody | PI3K/AKT pathway | Cisplatin | Osteosarcoma | 45 |
| PHA-665752 | PI3K/AKT pathway | Cisplatin | Osteosarcoma | 45 | |
| Chimaphilin | apoptosis | Doxorubicin | Osteosarcoma | 34 | |
| IGF-1R | AG1024 | apoptosis, cell cycle arrest, cytotoxicity | Doxorubicin | Osteosarcoma | 32 |
| AVE1642 | apoptosis | Gemcitabine | Leiomyosarcoma | 31 | |
| SRC | Dasatinib | apoptosis | Doxorubicin, vincristine, doxorubicin, actinomycin D | Chondrosarcoma synovial sarcoma | 57,50 |
| A-770041 | apoptosis, ABCB1 | Paclitaxel, doxorubicin | Osteosarcoma | 56 | |
| BCR-ABL,c-KIT, PDGFR | Nilotinib | ABCB1, ABCC1 | Doxorubicin | Synovial sarcoma, leiomyosarcoma | 7 |
| JAK | CDDO-Me | apoptosis, JAK/STAT pathway | Doxorubicin | Osteosarcoma | 66 |
Targeting receptor tyrosine kinases (RTKs)
Targeting EGFR (epidermal growth factor receptor)
EGFR is a receptor tyrosine kinase of the ErbB family, which also includes ErbB2 (human epidermal growth factor receptor-2 (HER2)), ErbB3 (HER3), and ErbB4 (HER4). EGFR may form an activated heterodimer conjugate with another member of the ErbB receptor family, such as ErbB2. EGFR dimerization and the subsequent autophosphorylation of tyrosines induces various downstream signal transduction cascades and lysosomal degradation. The downstream signals predominantly activate the Ras/Raf/MEK/ERK, PI3K/AKT, JNK, signal transducer and activator of transcription (STAT), and PLCγ/PKC pathways, leading to DNA synthesis and regulation cell proliferation, adhesion, and differentiation11. EGFR is both overexpressed and activated in various sarcomas, including osteosarcoma, fibrosarcoma, rhabdomyosarcoma, and liposarcoma, which promotes tumor cell progression, activation of downstream signal transduction pathways, resistance to chemotherapeutic drugs, and is associated with poor prognosis12. Targeting EGFR with small molecule kinase inhibitors and monoclonal antibodies (mAb) has become a rational targeting strategy for the treatment of sarcoma. Recent studies have demonstrated in vitro and in vivo efficacy of the small molecule kinase inhibitor imatinib to treat osteosarcoma and the mAb cetuximab for the treatment of rhabdomyosarcoma13, 14.
In a study that demonstrated highly activated EGFR in leiomyosarcoma, the use of an EGFR inhibitor showed a potent ability to overcome MDR9. The group investigated the effect of EGFR inhibitor gefitinib on leiomyosarcoma stem-like cells. It is believed that stem-like cells specifically use their stem cell processes of self-renewal and asymmetric differentiation to develop into various cancer cell types and are characterized by their innate MDR to chemotherapy15. These leiomyosarcoma stem-like cells exhibit chemoresistance and show high activation of AKT and ERK pathways. Treatment with EGFR inhibitior gefitinib reduced PI3/AKT and MAPK/ERK pathway activation and chemosensitized leiomyosarcoma stem-like cells in vitro. Moreover, gefitinib used as single agent killed tumor cells in vitro, reduced tumor growth in vivo, and in combination with vincristine, showed additive effects by increasing the activity of vincristine. Furthermore, gefitinib decreased anti-apoptotic factor Bcl-2 levels and increased intracellular doxorubicin retention. The combination of gefitinib with gemcitabine also enhanced inhibition of tumor cell proliferation, increased tumor cell apoptosis, and induced G1 cell cycle arrest in vitro. In vivo, the combination of gefitinib and doxorubicin resulted in a markedly synergistic effect on tumor cell growth inhibition and apoptosis. In addition to STS such as leiomyosarcoma as described above, EGFR is also overexpressed and highly activated in chondrosarcoma cell lines. Accordingly, overexpression of EGFR contributes to cisplatin resistance in chondrosarcoma16. The combination of erlotinib (an inhibitor of EGFR that specifically targets the ATP binding site of the receptor in a reversible fashion) with cisplatin showed a sensitization effect in chemoresistant chondrosarcoma cells that induced apoptosis and cell cycle arrest16.
An anti-EGFR mAb is designed to specifically bind to the extracellular domain of EGFR, which creates a competitive ligand inhibition, and subsequently prevents receptor dimerization, inhibits receptor phosphorylation, and suppresses downstream signal pathways. Cetuximab, a recombinant chimeric human murine IgG1 type mAb, is the first anti-EGFR mAb approved by the FDA for clinical use. Cetuximab increases lysis in EGFR-expressing chemoresistant osteosarcoma cell lines by enhancing the cytotoxic function of resting natural killer (NK) cells17. Consequently, cetuximab-mediated anti-EGFR effects related to immunotherapy should be a novel subject for further research studies in the management of chemoresistant sarcomas. Collectively, targeting EGFR represents a promising therapeutic strategy for overcoming MDR in sarcoma.
Targeting AXL receptor tyrosine kinase
AXL (also known as ARK; UFO; JTK11; Tyro7) is a member of the receptor tyrosine kinase TAM subfamily, which also includes Tyro3 and Mer. AXL is ubiquitously expressed in epithelial cells, endothelial cells, mesenchymal, and hematopoietic origins throughout all tissues. Overexpression and activation of AXL was found to be correlated with poor survival in a variety of sarcomas, including osteosarcoma, liposarcoma, rhabdomyosarcoma, and leiomyosarcoma18–21. AXL plays a pivotal role in tumor cell differentiation, apoptosis, invasion, and metastasis through PI3K/AKT/S6K, MAPK/ERK, IL-6-STAT-3, NF-κB pathways, and the epithelial-to-mesenchymal transition (EMT) phenotype22. Targeting the 3′-UTR of AXL with miR-199a-3p downregulated the expression of the AXL gene and subsequently inhibited the progression in osteosarcoma cells through the AKT pathway23.
Recently, a study demonstrated that there was abundant expression of AXL and Gas6 in Ewing sarcoma patient samples24. Higher levels of AXL correlated with a worse prognosis compared with lower expression in primary patient tissues. In vitro, BGB324, an AXL kinase inhibitor, decreased viability and migration in Ewing sarcoma cell lines. More importantly, the Ewing sarcoma cell lines most resistant to vincristine and doxorubicin are associated with higher AXL and Gas6 expression levels. Furthermore, treatment of BGB324 sensitized chemoresistant cell lines to vincristine and doxorubicin. The study demonstrated that treatment of BGB324 reduced expression of pAKT and pERK levels and reversed MDR in Ewing sarcoma cells; however, the latter may be related to downregulation of PI3K/AKT and ERK pathways, which need further investigation25. From a clinical perspective, the combination of BGB324 with chemotherapeutic drugs may benefit Ewing sarcoma patients with chemoresistance.
In addition to EGFR and AXL as described above, there are several other RTKs that have also been identified to be involved in MDR sarcomas, such as IGF-1R, PDGFR, VEGFR, and MET.
Targeting other RTKs
The insulin-like growth factor 1 receptor (IGF-1R) binds IGF-1 and IGF-2, and subsequently activates two main downstream signaling pathways, Ras/Raf/MEK/ERK and PI3K/AKT pathways, which are implicated in tumor cell proliferation, survival, growth, neovascularization, migration, invasion, and metastasis. Expression of IGF-IR has been found in various sarcomas, including rhabdomyosarcoma, osteosarcoma, synovial sarcoma, leiomyosarcoma, and Ewing sarcoma, and represents a negative prognostic biomarker for patients26–31. AVE1642, a mAb directed against IGF-1R, showed synergistic effects in the treatment of leiomyosarcoma in combination with gemcitabine31. Targeting IGF-1R with AG1024, a commercially available small molecule kinase inhibitor, showed enhanced doxorubicin chemosensitivity in human osteosarcoma cell lines32. Targeting IGF-1R via RNAi also showed a marked chemosensitization effect in osteosarcoma cells33. In addition, a recent study demonstrated that chimaphilin, a naphthoquinone extracted from pyrola, could induce apoptosis and reverse MDR osteosarcoma cell lines by targeting IGF-IR signaling34.
The PDGFR includes two types of receptors: alpha-type and beta-type PDGFRs. When the receptor binds its ligands (PDGF-AA, -BB, -AB, -CC, -DD), downstream signal transduction pathways become activated, such as PI3K and STAT3 pathways. Overexpression and constitutive activation of PDGFR play a key role in regulation of cell growth, differentiation, proliferation, and migration, particularly in blood vessel formation (angiogenesis) in sarcomas35. Notably, in a multi-platform profiling study of 2539 sarcoma specimens covering 22 sarcoma subtypes, researchers demonstrated that PDGFRA was overexpressed in 22.1% of these sarcomas, including in angiosarcoma, liposarcoma, fibrosarcoma, Ewing sarcoma, chondrosarcoma, osteosarcoma, UPS, and leiomyosarcoma36. Olaratumab, a human monoclonal antibody that directly binds PDGFRA and blocks ligand binding, concurrent with doxorubicin, dramatically improved overall survival in advanced/metastatic STS in a randomized phase Ib/II trial; a phase III trial is currently underway37. Targeting PDGFR with the small molecule inhibitor imatinib exhibits a synergistic anti-proliferative effect on osteosarcoma when combined with doxorubicin38. Sorafenib is a small molecule multi-target kinase inhibitor of several protein kinases, including VEGFR, PDGFR, and Raf kinases, that showed a synergistic effect on osteosarcoma and Ewing sarcoma in combination with topotecan4.
VEGFR is vital in vasculogenesis and angiogenesis, and represents an attractive strategy to block the development of new blood supply for tumors by targeting VEGFR39. Overexpression and/or aberrant activation of VEGFR are associated with poor prognosis in sarcomas40. Recently, a phase II clinical trial demonstrated that bevacizumab, a humanized monoclonal antibody that targets VEGF-A, inhibited the binding of VEGFR and showed favorable activity in combination with chemotherapeutic drugs in STS41. In another multicentre, non-randomized phase II study conducted by the Italian Sarcoma Group, researchers demonstrated that the combination of sorafenib with the mTOR inhibitor everolimus showed favorable 6-month progression-free survival for 45% of patients with relapsed or unresectable high-grade osteosarcoma, who had progressed after failure of the standard chemotherapeutic drugs treatments42.
MET, also called c-Met and hepatocyte growth factor receptor (HGFR), is a membrane receptor tyrosine kinase encoded by the MET oncogene, which binds HGF and subsequently transduces signal through MAPK, PI3K, STAT, Notch, and Wnt signaling pathways. Overexpression and aberrantly activated MET triggers cell growth, angiogenesis, invasion, and metastasis correlated with poor prognosis in various sarcomas43, 44. Targeting MET using the small molecule kinase inhibitor PHA-665752 and the neutralizing anti-HGF antibody sensitized osteosarcoma cells to cisplatin through suppression of the PI3K/AKT signaling45. PHA-665752 and neutralizing anti-HGF antibody substantially decreased the levels of pAKT with no changes on total AKT levels.
Targeting non-receptor tyrosine kinases (NRTKs)
Targeting SRC Kinase
The SRC family kinases (SFKs) have nine members, including SRC, Lck, Yes, Fyn, Fgr, Yrk, Lyn, Blk, and Hck. In humans, SRC, Fyn, and Yes are ubiquitously expressed in all tissues, while the others are generally expressed in hematopoietic cells, with high levels in platelets, neural tissue, and osteoclasts with SRC. SRC can be activated by many different proteins, including RTKs, adhesion receptors, integrins, G-protein coupled receptors, cytokine receptors, as well as immune response receptors. The SRC kinase plays a crucial role in promoting tumor cell proliferation, survival, adhesion, migration, invasion, angiogenesis, and reducing apoptosis by interacting with other tyrosine kinase receptors and intracellular signaling pathways, such as PDGFR, EGFR, VEGFR, PI3K/AKT, and Ras/Raf/MEK/MAPK signal pathways46.
There is increasing evidence that demonstrates that SFKs are expressed and hyperactivated in most sarcomas, including in osteosarcoma, Ewing sarcoma, liposarcoma, leiomyosarcoma, synovial sarcoma, fibrosarcoma, and rhabdomyosarcoma47–53. Therefore, targeting SFKs represents a promising therapeutic target. Several small molecule kinase inhibitors of SFKs have demonstrated efficacy in sarcoma cells, such as SI-83 in osteosarcoma; AP23994 in Ewing sarcoma; dasatinib in osteosarcoma, Ewing sarcoma, liposarcoma and synovial sarcoma; SI221 in rhabdomyosarcoma; and saracatinib in fibrosarcoma54, 55
A-770041 is a potent SFK (Lck and SRC) inhibitor, which not only showed inhibition of SRC, but also reversed MDR ability in osteosarcoma56. The combination of A-770041 with paclitaxel and doxorubicin sensitized and reversed chemotherapeutic drugs resistance in MDR human osteosarcoma cell lines. Moreover, lentiviral shRNA targeting SRC induced downregulation of SRC expression levels and reversed MDR. A-770041 significantly increased intracellular accumulation of drug concentration in MDR osteosarcoma cell lines that overexpressed ABCB1 (also known as P-gp, MDR1) in a dose-dependent manner without altering the expression of ABCB1 protein levels. These results indicated that A-770041 inhibited ABCB1 efflux function via interaction with ABCB1 and contributed to the reversal of MDR. In addition, A-770041 in combination with doxorubicin increased apoptosis to further enhance chemosensitization in MDR osteosarcoma cell lines. Dasatinib also showed a potent sensitization effect with doxorubicin treatment in TP53 mutant chemoresistant chondrosarcoma cells exhibited by increased doxorubicin induced-apoptosis57. In synovial sarcoma, dasatinib inhibited tumor cell growth and induced apoptosis in vitro and in vivo, leading to decreased phosphorylation of FAK, STAT3, IGF-IR, and AKT. Concurrent administration of dasatinib with chemotherapeutic agents also displayed additive effects in synovial sarcoma50.
Targeting other non-receptor tyrosine kinases (NRTKs)
Nilotinib, a small molecule multi-target tyrosine kinase inhibitor, inhibits the BCR-ABL, c-KIT, PDGFRB, and MAPK11 kinases. Recently, a study demonstrated that nilotinib plays an important role in inhibiting tumor cell growth and overcoming MDR in STS7. The group showed that the combination of nilotinib with doxorubicin exhibited synergistic antitumor effects and apoptosis in synovial sarcoma and leiomyosarcoma cells. Moreover, nilotinib inhibited ABCB1 and ABCC1 (also known as MRP1) activity, which resulted in increased intracellular doxorubicin accumulation. Furthermore, nilotinib decreased basal ABCB1 expression levels and p38 MAPK phosphorylation, as well as fully inhibited overexpression of ABCB1 levels and p38 MAPK phosphorylation induced by doxorubicin in synovial sarcoma cell lines. In addition, the study demonstrated that p38 MAPK activation also results in the development of MDR by modulating the expression and activity of ABCB1. Given the ABC transporters significant roles in MDR, nilotinib presents a novel potential strategy for overcoming MDR in sarcoma. In another impressive prospective phase III trial performed by the Italian Sarcoma Group in collaboration with the Spanish Sarcoma Group, researchers enrolled 328 high-risk localized STS patients and randomized each to receive either three or five cycles of epirubicin plus ifosfamide58. The group demonstrated that expression of ABCC1 (MRP1) was the only independent prognostic factor for both relapse-free survival and overall survival as compared with expression of ABCB1 and glutathione S-transferase pi.
FAK is vital to cell progression, including adhesion, growth, and survival. Overexpression and activity FAK was demonstrated in Ewing sarcoma59. Targeting FAK with the kinase inhibitor PF-562271 and shRNA inhibited Ewing sarcoma cell growth in vitro and in vivo. High expression of FAK in osteosarcoma cells was associated with a stronger migration ability, which could be suppressed by targeting FAK with selective siRNA60. The combination of targeting FAK by shRNA with cisplatin showed enhanced apoptosis in osteosarcoma cells61.
JAK is located intracellularly and is activated by receptor tyrosine kinases and transduce cytokine-mediated signals, which subsequently activate STAT transcription factors62. High levels of activated JAK/STAT3 were found in rhabdomyosarcoma, Ewing sarcoma, and osteosarcoma tumor samples and cell lines associated with poorer prognosis63, 64. Targeting JAK by small molecule kinase inhibitor AZD1480 inhibited rhabdomyosarcoma and Ewing sarcoma growth in vitro and xenografts in vivo63. Moreover, targeting JAK with siRNA sensitized FGF-2-mediated cisplatin resistance and inhibited cell proliferation in osteosarcoma cells65. In addition, inhibitor of STAT3 CDDO-Me reduced resistance to doxorubicin in MDR osteosarcoma through down-regulate STAT3-mediated antiapoptotic proteins in both dose and time dependent manner66.
Targeting serine/threonine protein kinases to reverse MDR in sarcoma
Serine/threonine kinases (STKs) are enzymes that phosphorylation the OH group of serine or threonine, which have similar side-chains. There are at least 125 kinds of STKs, which can be divided into several groups, including the TKL group, AGC group, CMGC group, CAMK group, CK1 group, RGC group, PKL group, atypical group, and Other group. There is increasing evidence that shows that STKs play a paramount role in maintaining cellular homeostasis and cell signaling transduction, and regulation of cell proliferation, differentiation, apoptosis, and embryonic development67. DNA damage and reactive oxygen species (ROS) stress, such as cAMP/cGMP and Ca2+/calmodulin, can regulate activity of STKs. Dysregulation of STKs has been implicated to be associated with tumor growth, metastasis, recurrence, and chemoresistance in a variety of sarcomas64. Representative intracellular signal transduction of serine/threonine kinases and potential target sites in sarcomas are shown in Figure. 2. Functional mechanisms reverse MDR by targeting serine/threonine kinases in sarcomas are summarized in Table 2.
Figure 2. Targeting serine/threonine kinases reverse MDR in sarcomas.
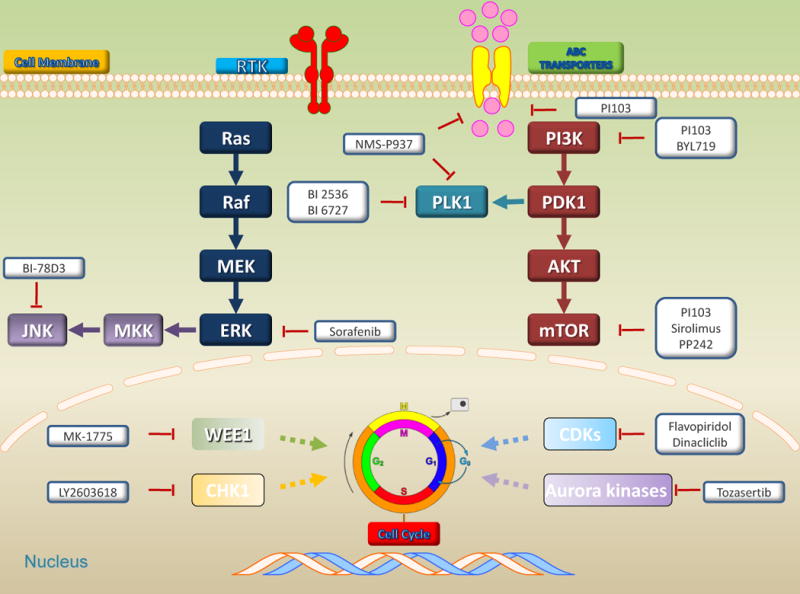
Intracellular signal transduction of serine/threonine kinases and potential target sites in sarcomas, including serine/threonine kinases, ABC transporters, cell cycle, and two major downstream signaling pathways: PI3K/AKT/mTOR and MAPK pathways. Bold represent target serine/threonine kinases by small molecule kinase inhibitors.
Table2.
Targeting Serine/threonine kinase reverse MDR in sarcomas
| Target Kinase | Inhibition compound | functional mechanism | Effect on anticancer drug |
|---|---|---|---|
| BI 6727 | apoptosis, cell cycle arrest | Vincristine | |
| Polo-like kinase 1 | BI 2563 | apoptosis, cell cycle arrest | Vincristine, Eribulin |
| NMS-P937 | apoptosis, cell cycle arrest, ABCB1 | Doxorubicin | |
| JNK | BI-78D3 | apoptosis | Doxorubicin |
| Flavopiridol | apoptosis, cell cycle arrest | Doxorubicin | |
| CDK | Dinacliclib | apoptosis | Doxorubicin |
| PI3K | BYL719 | apoptosis | Mafosfamide, ifosamide |
| Dual PI3K/mTOR | PI103 | ABCB1, ABCC1 | Doxorubicin |
| mTOR | PP242 | apoptosis | Cisplatin |
| Sirolimus | apoptosis | Gemcitabine Etoposide, doxorubicin |
|
| Aurora kinase | Tozasertib | apoptosis, cell cycle arrest | Cisplatin, doxorubicin, methotrexate |
| ERK | Sorafenib | ERK | Cisplatin |
| WEE1 | MK-1775 | apoptosis | Gemcitabine |
| CHK1 | LY2603618 | apoptosis, cell cycle arrest | Cisplatin |
Targeting polo-like kinases
Polo-like kinases (PLKs) are a group of serine/threonine kinases, including PLK1, PLK2, PLK3, PLK4 and PLK5. PLKs play important roles in the regulation of cell cycle progression through the G2/M transition and mitosis. Among PLKs, PLK1 (Polo-like kinase 1) is the most comprehensively characterized member of the PLK family. PLK1 is highly expressed in proliferative tissues, such as embryonic tissue, testis, and bone marrow. Overexpression of PLK1 increases centrosome size and/or number and aneuploidy, which contributes to tumorigenesis68. PLK1 is overexpressed in several sarcomas, such as osteosarcoma, liposarcoma, Ewing sarcoma, and rhabdomyosarcoma, and is associated with poor prognosis69–72. Targeting PLK1 with the small molecule kinase inhibitor BI 2536 significantly decreased osteosarcoma growth in vitro and in vivo70.
PLK1 expression was significantly higher in osteosarcoma cell lines, as well as clinical specimens compared with normal muscle tissues, which was associated with a worse clinical prognosis73. Targeting PLK1 with small molecule kinase inhibitor NMS-P937 reduced osteosarcoma cell growth, as well as decreased cloning efficiency and migration ability. Furthermore, combination of NMS-P937 with doxorubicin exhibited a synergistic effect in chemoresistant osteosarcoma cell lines through increased apoptosis and cell cycle arrest. More importantly, NMS-P937 showed reversal effects on doxorubicin-resistant osteosarcoma cell lines with overexpression of ABCB1 by interfering with ABCB1 mediated efflux activity, which resulted in intracellular accumulation of chemotherapeutic drugs. Recently, another study demonstrated efficacy of using RNAi to target PLK1 in the treatment of osteosarcoma. The combination of PLK1 shRNA/PEI-Lys with doxorubicin also showed marked synergistic effects on osteosarcoma cells via induced apoptosis and cell cycle arrest in vitro and in vivo74. The combination of another PLK1 small molecule kinase inhibitor BI 6727 with vincristine showed synergistic inhibition effects in Ewing sarcoma through the induction of mitochondrial pathway apoptosis and triggered mitotic arrest72. In rhabdomyosarcoma, BI 2536 and BI 6727 also synergistically induced apoptosis and mitotic arrest with co-treatment with vincristine in vitro and in vivo75. The group also confirmed that the combination of BI 2536 with eribulin showed synergistic inhibition effects in rhabdomyosarcoma71. These findings present targeting PLK1 as a promising therapeutic strategy to reverse MDR in sarcoma.
Targeting JNK kinase
JNKs were originally identified as serine/threonine protein kinases that phosphorylate c-Jun on serines 63 and 73 in the amino-terminal activation domain. JNKs consist of JNK1, JNK2, and JNK3 belonging to the MAPK family, encoded by the MAP8, MAP9, and MAP10 genes, respectively. As the final kinases of the MAPK cascade, JNKs are responsible for integrating multiple signals and regulating cell proliferation, differentiation, transcription, and development processes. They also play a role in T-cell differentiation and the cellular apoptosis pathway76. JNK was found to be expressed in various sarcomas, including Ewing sarcoma, osteosarcoma, chondrosarcoma, synovial sarcoma, and fibrosarcoma77–81. Targeting JNK with arsenic exhibited a potential inhibitive effect on Ewing sarcoma77. Moreover, the JNK signaling pathway is correlated with autophagy in rhabdomyosarcoma82. To date, two types of JNK inhibitors have been developed: peptide-based and small molecule ATP-non-competitive inhibitors, which target the upstream kinases or the downstream substrates and scaffolding proteins76. JNK-interacting protein-1 (JIP1) is a scaffolding protein and belongs to a larger group of scaffold proteins, which also includes JIP2, JIP3, and JIP4. JIP1 selectively binds MKK4/7 (MAP kinase kinase 4, 7), MLK3 (mixed-lineage kinase 3), and JNK, which subsequently forms a functional JNK-signaling complex and regulates JNK activation, as well as JNK phosphorylation of c-Jun83.
Recently, a study demonstrated that BI-78D3, an ATP-non-competitive JNK inhibitor, blocked JIP1-JNK binding and sensitized osteosarcoma cells to doxorubicin78. The group investigated 71 osteosarcoma specimens, of which 55 samples were JIP1 positive and associated with poor prognosis. Furthermore, they found that JIP1 was more highly expressed in human osteosarcoma cell lines than in primary osteoblasts, and the combination of BI-78D3 with doxorubicin decreased JNK and JNK-JIP1 complex expression levels. In addition, BI-78D3 enhanced doxorubicin-induced apoptosis by inhibiting the anti-apoptotic JNK signaling pathway. Overexpression of c-Jun in chemoresistant tumor cells and activated c-Jun promoted the expression of ABCB1, which suggested that JNK inhibition may be related to ABCB1 in overcoming MDR84.
Targeting cyclin-dependent kinases
Cyclin-dependent kinases (CDKs) are a family of STKs, which belong to the CMGC group and include 21 CDKs (CDK1-21) and five CDK-like (CDKL) kinases based on sequence similarity85. CDKs play a crucial role in regulating the progression of the cell cycle. When a CDK binds a cyclin, a regulatory subunit protein, a CDK-cyclin complex is formed and the presence of CDK-activating kinase (CAK) activates kinases. CDK-cyclin complexes tightly control and monitor four sequential phases of cell cycle namely, G1, S, G2, and M phase, and dysregulation in any phase results in uncontrolled cell growth and tumorigenesis86.
Cyclin D-dependent CDK4 and CDK6 are found to be expressed and active in rhabdomyosarcoma87. Targeting with the small molecule kinase inhibitor PD0332991 decreased rhabdomyosarcoma in vitro and in vivo through cell cycle G1 arrest87. CDK4 was also overexpressed in liposarcoma and associated with significantly poorer prognosis compared with the low-CDK4 group88. A Phase II trial reported that PD0332991 could inhibit tumor cell growth and improve progression-free survival rates in patients with a CDK4 amplification and Rb expression in liposarcoma89. In another notable report, flavopiridol, a pan-CDK inhibitor that specifically targets CDK2, CDK4, CDK6, and CDK9, inhibited liposarcoma growth90. More importantly, flavopiridol exhibited significant enhancement of doxorubicin effects in vitro and in vivo through cell-cycle arrest and subsequent apoptosis. Furthermore, the group conducted a Phase I clinical trial of the combination of flavopiridol with doxorubicin for the treatment of advanced sarcomas, including leiomyosarcoma, fibrosarcoma, and rhabdomyosarcoma. The results also showed synergistic effects on STS. Another study demonstrated that targeting CDK1/CDK2 with the small molecule kinase inhibitor dinacliclib or siRNA induced apoptosis in osteosarcoma, fibrosarcoma, rhabdomyosarcoma, Ewing sarcoma, and liposarcoma, even in doxorubicin-resistant osteosarcoma91.sarcomas.
Targeting PI3K/AKT/mTOR
PI3K belongs to a family of lipid kinases, which phosphorylates the hydroxyl group at position 3 in the inositol ring to produce phosphatidylinositol 3-phosphate (PIP), phosphatidylinositol (3,4)-bisphosphate (PIP2), and phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Tyrosine kinase receptors (e.g. PDGFR, EGFR, IGF-IR) and G protein-coupled receptors activate PI3K as a second messenger that regulates diverse cellular functions, such as cell growth, proliferation, differentiation, membrane transport, cytoskeletal remodeling, and survival92. AKT (also known as Protein kinase B, PKB) is a serine/threonine protein kinase and plays a key role in regulating cell glucose metabolism, apoptosis, cell proliferation, survival, transcription, migration, and angiogenesis. AKT is a downstream mediator of the PI3K pathway, and is fully activated through simultaneous phosphorylation at two key sites: Thr308 (phosphorylated by PDK1) and Ser473 (phosphorylated by mTORC2)93. There is a broad range of substrates phosphorylated by AKT, such as forkhead box O1 (FOXO1), glycogen synthase kinase 3 (GSK3), Raf, ASK, and MDM2. mTOR is also a serine/threonine protein kinase and presents a catalytic subunit of two structurally and functionally distinct signaling complexes: mTORC1 and mTORC294. mTOR is a central component of cellular processes that regulates cellular metabolism, growth, and survival through input from upstream pathways and in response to diverse external stimuli, including growth factors (such as IGF-1 and IGF-2), hormones, amino acids, nutrients, oxygen, energy, and stress signals. There are more than 800 proteins phosphorylated directly or indirectly by mTOR. Mutations in PTEN and dysregulation of upstream effectors (e.g. PI3K, AKT, EGFR, FGFR, KRas, MET) and downstream effectors (e.g. 4EBP1, S6K) contribute to constitutive activation of mTOR signaling, which is involved in the initiation and development of sarcomas95. To date, mutation or constitutive activation of PI3K/AKT/mTOR has been found in most types of sarcomas, including osteosarcoma, rhabdomyosarcoma, Ewing sarcoma, synovial sarcoma, and liposarcoma96. As a convergence point of many signaling pathways for many sarcomas, as well as a poor prognostic factor, especially relating to MDR, targeting PI3K/AKT/mTOR represents an attractive therapeutic strategy.
PI103, a dual PI3K/mTOR kinase inhibitor, can reverse MDR in fibrosarcoma and rhabdomyosarcoma97. A study demonstrated that PI103 increased intracellular doxorubicin accumulation through decreased expression of ABCB1 and ABCC1. Furthermore, a specific inhibitor targeting the PI3K kinase, GDC-0941, showed inhibition of rhabdomyosarcoma cell growth. However, the combination of GDC-0941 with doxorubicin did not show synergistic effects. Another dual PI3K/mTOR kinase inhibitor NVP-BEZ235 did, on the other hand, show synergistic effects with chloroquine to induce apoptosis in rhabdomyosarcoma at subtoxic concentrations98. BYL719, a novel α-specific PI3K inhibitor, combined with mafosfamide and ifosamide demonstrated a striking synergistic inhibition effect in osteosarcoma by inducing apoptosis99. Sirolimus, which targets the mTOR pathway by directly binding to mTORC1, combined with gemcitabine enhanced antitumor growth activity in leiomyosarcoma compared with treatment by the individual single-agent drugs in vitro and in vivo100, 101. Moreover, the same group conducted a Phase I study that demonstrated favorable results using the combination treatment in patients with advanced solid tumors, including chondrosarcoma. Another study demonstrated that specifically targeting mTORC2 using the kinase inhibitor PP242 or siRNA markedly promoted cisplatin-induced apoptosis and inhibited osteosarcoma cell migration ability102. Besides small molecule kinase inhibitor and siRNA, complementary targeting of the 3′-untranslated region (UTR) of mTOR by miRNA-100 also sensitized chondrosarcoma cells to cisplatin103. In all, targeting PI3K/AKT/mTOR emerges as a promising strategy for overcoming MDR in sarcomas.
Targeting other serine/threonine protein kinases
Aurora kinases are classified between aurora kinase A, aurora kinase B, and aurora kinase C, and play a pivotal role in regulating cell mitosis and cell proliferation. Aurora kinase A and B are overexpressed in various sarcomas, including chondrosarcoma, osteosarcoma, synovial sarcoma, and Ewing sarcoma, and are associated with poor prognosis and metastasis104–107. Targeting aurora kinase A and B with small molecule kinase inhibitors, shRNA, and siRNA exhibited inhibition of tumor cells105, 107. Moreover, a study demonstrated that the combination of aurora kinase A and B kinase inhibitor VX-680 (Tozasertib) with etoposide and doxorubicin showed synergistic effects in Ewing sarcoma107. Recently, another report also confirmed that the combination of VX-680 with cisplatin, doxorubicin, and methotrexate exhibited a synthetic lethal interaction in osteosarcoma cell lines. In addition, the synergistic effect of the combination of VX-680 with cisplatin was demonstrated in cisplatin-resistant osteosarcoma cells108. The combination of aurora kinase inhibitor MSC1992371A with gemcitabine was investigated in a Phase I study in patients with solid tumors, including in sarcoma patients109.
ERK belongs to the MAPK family, which is a downstream component of the MAPK cascade, known as Ras/Raf/MEK/ERK. The pathway relays signals from the cell surface for growth, stress, and other responses, and is implicated in cell survival, growth, and apoptosis in response to DNA damaging stress. Aberrant activation and/or overexpression of upstream signaling components constitutively activate ERK, which is often exhibited in different types of sarcomas, and targeting this pathway demonstrates an inhibition effect on tumors110. Sorafenib, a multitarget kinase inhibitor, in combination with cisplatin showed a synergistic effect in osteosarcoma cells through downregulation of ERK111. Targeting ERK signaling by inorganic phosphate (Pi) also exhibited sensitization to doxorubicin in osteosarcoma cells112.
Checkpoint kinase 1 (CHK1) is associated with DNA damage repair, cell cycle checkpoint, and cycle arrest. The WEE1 G2 checkpoint kinase (WEE1) is also a key regulator of cell cycle progression involved in regulating the cell G2/M checkpoint entry into mitosis, cell size checkpoint, and DNA damage checkpoint. Constitutive activation and/or overexpression of CHK1 and/or WEE1 in sarcomas are implicated in tumor cell proliferation, apoptosis, and especially contribute to anticancer therapy resistance due to DNA damage repair ability113. Targeting WEE1 by kinase inhibitor MK-1775 inhibited the growth of various sarcomas, including osteosarcoma, Ewing sarcoma, and fibrosarcoma114. Moreover, the combination of MK-1775 with gemcitabine showed a synergistic effect in vitro and in vivo through increased apoptosis115. In addition, targeting CHK1 by kinase inhibitor LY2603618 increased cisplatin sensitivity in osteosarcoma116.
Conclusions
Sarcomas are rare malignancies associated with poor prognosis. Chemotherapy has been implemented for the last several decades and has achieved modest benefits in patients with STS. However, the development of MDR in tumor cells limits the curative potential of chemotherapeutic agents and presents challenges to the patients and the oncology team. Advances in the understanding of the molecular mechanisms of sarcomas over the past years have revealed broader roles for protein kinases in oncogenesis and the development of sarcomas, particularly in regard to MDR. Tremendous research and clinical trials have shown that targeting protein kinases by small molecule agents, antibodies, RNAi, and in combination with conventional chemotherapy represents a promising therapeutic approach to overcoming MDR in sarcoma.
Highlights.
MDR attenuates the efficacy of anticancer drugs and results in treatment failure for sarcomas.
Certain protein kinases demonstrate aberrant expression and/or activity in sarcoma cells, which have been found to be involved in the MDR of sarcoma.
Targeting protein kinases in combination with conventional chemotherapy represents a promising therapeutic approach to overcoming MDR in sarcoma.
Acknowledgments
This work was supported, in part, by the Gattegno and Wechsler funds, the Kenneth Stanton Fund, and the Jennifer Hunter Yates Foundation. Dr. Duan is supported, in part, through a grant from Sarcoma Foundation of America (SFA), a grant from National Cancer Institute (NCI)/National Institutes of Health (NIH), UO1, CA 151452-01, a pilot grant from Sarcoma SPORE/NIH, and a grant from an Academic Enrichment Fund of MGH Orthopaedics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest
EC has received consulting fees from Amgen, EMD Serono, Pfizer, and Bayer.
References
- 1.Borden EC, Baker LH, Bell RS, Bramwell V, Demetri GD, Eisenberg BL, et al. Soft tissue sarcomas of adults: state of the translational science. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:1941–56. [PubMed] [Google Scholar]
- 2.Ng VY, Scharschmidt TJ, Mayerson JL, Fisher JL. Incidence and survival in sarcoma in the United States: a focus on musculoskeletal lesions. Anticancer research. 2013;33:2597–604. [PubMed] [Google Scholar]
- 3.Li S, Sun W, Wang H, Zuo D, Hua Y, Cai Z. Research progress on the multidrug resistance mechanisms of osteosarcoma chemotherapy and reversal. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015;36:1329–38. doi: 10.1007/s13277-015-3181-0. [DOI] [PubMed] [Google Scholar]
- 4.Cubitt CL, Menth J, Dawson J, Martinez GV, Foroutan P, Morse DL, et al. Rapid screening of novel agents for combination therapy in sarcomas. Sarcoma. 2013;2013:365723. doi: 10.1155/2013/365723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends in pharmacological sciences. 2015;36:422–39. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Hannay JA, Liu J, Das P, Zhan M, Nguyen T, et al. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: implications for tumor growth, metastasis, and chemoresistance. Cancer research. 2006;66:8770–8. doi: 10.1158/0008-5472.CAN-06-1217. [DOI] [PubMed] [Google Scholar]
- 7.Villar VH, Vogler O, Martinez-Serra J, Ramos R, Calabuig-Farinas S, Gutierrez A, et al. Nilotinib counteracts P-glycoprotein-mediated multidrug resistance and synergizes the antitumoral effect of doxorubicin in soft tissue sarcomas. PloS one. 2012;7:e37735. doi: 10.1371/journal.pone.0037735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roskoski R., Jr A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacological research : the official journal of the Italian Pharmacological Society. 2015 doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Sette G, Salvati V, Memeo L, Fecchi K, Colarossi C, Di Matteo P, et al. EGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potential. PloS one. 2012;7:e46891. doi: 10.1371/journal.pone.0046891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baird K, Davis S, Antonescu CR, Harper UL, Walker RL, Chen Y, et al. Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer research. 2005;65:9226–35. doi: 10.1158/0008-5472.CAN-05-1699. [DOI] [PubMed] [Google Scholar]
- 11.Herbst RS. Review of epidermal growth factor receptor biology. International journal of radiation oncology, biology, physics. 2004;59:21–6. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 12.Ren W, Korchin B, Zhu QS, Wei C, Dicker A, Heymach J, et al. Epidermal growth factor receptor blockade in combination with conventional chemotherapy inhibits soft tissue sarcoma cell growth in vitro and in vivo. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:2785–95. doi: 10.1158/1078-0432.CCR-07-4471. [DOI] [PubMed] [Google Scholar]
- 13.Gobin B, Moriceau G, Ory B, Charrier C, Brion R, Blanchard F, et al. Imatinib mesylate exerts anti-proliferative effects on osteosarcoma cells and inhibits the tumour growth in immunocompetent murine models. PloS one. 2014;9:e90795. doi: 10.1371/journal.pone.0090795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto Y, Fukuda K, Fuchimoto Y, Matsuzaki Y, Saikawa Y, Kitagawa Y, et al. Cetuximab promotes anticancer drug toxicity in rhabdomyosarcomas with EGFR amplification in vitro. Oncology reports. 2013;30:1081–6. doi: 10.3892/or.2013.2588. [DOI] [PubMed] [Google Scholar]
- 15.Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Seminars in oncology. 2015;42(Suppl 1):S3–17. doi: 10.1053/j.seminoncol.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Song YD, Zhang KF, Liu D, Guo YQ, Wang DY, Cui MY, et al. Inhibition of EGFR-induced glucose metabolism sensitizes chondrosarcoma cells to cisplatin. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:7017–24. doi: 10.1007/s13277-014-1902-4. [DOI] [PubMed] [Google Scholar]
- 17.Pahl JH, Ruslan SE, Buddingh EP, Santos SJ, Szuhai K, Serra M, et al. Anti-EGFR antibody cetuximab enhances the cytolytic activity of natural killer cells toward osteosarcoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:432–41. doi: 10.1158/1078-0432.CCR-11-2277. [DOI] [PubMed] [Google Scholar]
- 18.Han J, Tian R, Yong B, Luo C, Tan P, Shen J, et al. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochemical and biophysical research communications. 2013;435:493–500. doi: 10.1016/j.bbrc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 19.Hoffman A, Ghadimi MP, Demicco EG, Creighton CJ, Torres K, Colombo C, et al. Localized and metastatic myxoid/round cell liposarcoma: clinical and molecular observations. Cancer. 2013;119:1868–77. doi: 10.1002/cncr.27847. [DOI] [PubMed] [Google Scholar]
- 20.el Sayadi H, Pissaloux D, Alberti L, Tabone-Eglinger S, Ranchere D, Decouvelaere AV, et al. Autocrine role for Gas6 with Tyro3 and Axl in leiomyosarcomas. Targeted oncology. 2013;8:261–9. doi: 10.1007/s11523-012-0249-2. [DOI] [PubMed] [Google Scholar]
- 21.Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, et al. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer research. 2010;70:7221–31. doi: 10.1158/0008-5472.CAN-10-0391. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546–63. doi: 10.18632/oncotarget.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian R, Xie X, Han J, Luo C, Yong B, Peng H, et al. miR-199a-3p negatively regulates the progression of osteosarcoma through targeting AXL. American journal of cancer research. 2014;4:738–50. [PMC free article] [PubMed] [Google Scholar]
- 24.Fleuren ED, Hillebrandt-Roeffen MH, Flucke UE, Te Loo DM, Boerman OC, van der Graaf WT, et al. The role of AXL and the in vitro activity of the receptor tyrosine kinase inhibitor BGB324 in Ewing sarcoma. Oncotarget. 2014;5:12753–68. doi: 10.18632/oncotarget.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto T, Ohno T, Wakahara K, Nagano A, Kawai G, Saitou M, et al. Simultaneous inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways augment the sensitivity to actinomycin D in Ewing sarcoma. Journal of cancer research and clinical oncology. 2009;135:1125–36. doi: 10.1007/s00432-009-0554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aslam MI, Hettmer S, Abraham J, Latocha D, Soundararajan A, Huang ET, et al. Dynamic and nuclear expression of PDGFRalpha and IGF-1R in alveolar Rhabdomyosarcoma. Molecular cancer research: MCR. 2013;11:1303–13. doi: 10.1158/1541-7786.MCR-12-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacEwen EG, Pastor J, Kutzke J, Tsan R, Kurzman ID, Thamm DH, et al. IGF-1 receptor contributes to the malignant phenotype in human and canine osteosarcoma. Journal of cellular biochemistry. 2004;92:77–91. doi: 10.1002/jcb.20046. [DOI] [PubMed] [Google Scholar]
- 28.Friedrichs N, Kuchler J, Endl E, Koch A, Czerwitzki J, Wurst P, et al. Insulin-like growth factor-1 receptor acts as a growth regulator in synovial sarcoma. The Journal of pathology. 2008;216:428–39. doi: 10.1002/path.2438. [DOI] [PubMed] [Google Scholar]
- 29.Scotlandi K, Manara MC, Serra M, Marino MT, Ventura S, Garofalo C, et al. Expression of insulin-like growth factor system components in Ewing’s sarcoma and their association with survival. European journal of cancer (Oxford, England : 1990) 2011;47:1258–66. doi: 10.1016/j.ejca.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Liang J, Li B, Yuan L, Ye Z. Prognostic value of IGF-1R expression in bone and soft tissue sarcomas: a meta-analysis. OncoTargets and therapy. 2015;8:1949–55. doi: 10.2147/OTT.S88293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macaulay VM, Middleton MR, Protheroe AS, Tolcher A, Dieras V, Sessa C, et al. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2013;24:784–91. doi: 10.1093/annonc/mds511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luk F, Yu Y, Walsh WR, Yang JL. IGF1R-targeted therapy and its enhancement of doxorubicin chemosensitivity in human osteosarcoma cell lines. Cancer investigation. 2011;29:521–32. doi: 10.3109/07357907.2011.606252. [DOI] [PubMed] [Google Scholar]
- 33.Wang YH, Xiong J, Wang SF, Yu Y, Wang B, Chen YX, et al. Lentivirus-mediated shRNA targeting insulin-like growth factor-1 receptor (IGF-1R) enhances chemosensitivity of osteosarcoma cells in vitro and in vivo. Molecular and cellular biochemistry. 2010;341:225–33. doi: 10.1007/s11010-010-0453-2. [DOI] [PubMed] [Google Scholar]
- 34.Daqian W, Chuandong W, Xinhua Q, Songtao A, Kerong D. Chimaphilin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines through insulin-like growth factor-I receptor (IGF-IR) signaling. Chemico-biological interactions. 2015;237:25–30. doi: 10.1016/j.cbi.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 35.Rivera-Valentin RK, Zhu L, Hughes DP. Bone Sarcomas in Pediatrics: Progress in Our Understanding of Tumor Biology and Implications for Therapy. Paediatric drugs. 2015;17:257–71. doi: 10.1007/s40272-015-0134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Movva S, Wen W, Chen W, Millis SZ, Gatalica Z, Reddy S, et al. Multi-platform profiling of over 2000 sarcomas: identification of biomarkers and novel therapeutic targets. Oncotarget. 2015;6:12234–47. doi: 10.18632/oncotarget.3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tap WD, Jones RL, Chmielowski B, Elias AD, Adkins D, Van Tine BA, et al. A randomized phase Ib/II study evaluating the safety and efficacy of olaratumab (IMC-3G3), a human anti-platelet-derived growth factor {alpha}(PDGFR {alpha}) monoclonal antibody, with or without doxorubicin (Dox), in advanced soft tissue sarcoma (STS) ASCO Annual Meeting Proceedings. 2015:10501. [Google Scholar]
- 38.Yamaguchi SI, Ueki A, Sugihara E, Onishi N, Yaguchi T, Kawakami Y, et al. Synergistic antiproliferative effect of imatinib and adriamycin in platelet-derived growth factor receptor-expressing osteosarcoma cells. Cancer science. 2015;106:875–82. doi: 10.1111/cas.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaumann AK, Drexler HC, Lang SA, Stoeltzing O, Diermeier-Daucher S, Buchdunger E, et al. The inhibition of tyrosine kinase receptor signalling in leiomyosarcoma cells using the small molecule kinase inhibitor PTK787/ZK222584 (Vatalanib(R)) International journal of oncology. 2014;45:2267–77. doi: 10.3892/ijo.2014.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilvaer TK, Valkov A, Sorbye S, Smeland E, Bremnes RM, Busund LT, et al. Profiling of VEGFs and VEGFRs as prognostic factors in soft tissue sarcoma: VEGFR-3 is an independent predictor of poor prognosis. PloS one. 2010;5:e15368. doi: 10.1371/journal.pone.0015368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dickson MA, D’Adamo DR, Keohan ML, D’Angelo SP, Carvajal RD, Gounder MM, et al. Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma. 2015;2015:532478. doi: 10.1155/2015/532478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grignani G, Palmerini E, Ferraresi V, D’Ambrosio L, Bertulli R, Asaftei SD, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. The Lancet Oncology. 2015;16:98–107. doi: 10.1016/S1470-2045(14)71136-2. [DOI] [PubMed] [Google Scholar]
- 43.Fleuren ED, Roeffen MH, Leenders WP, Flucke UE, Vlenterie M, Schreuder HW, et al. Expression and clinical relevance of MET and ALK in Ewing sarcomas. International journal of cancer Journal international du cancer. 2013;133:427–36. doi: 10.1002/ijc.28047. [DOI] [PubMed] [Google Scholar]
- 44.Tsou HK, Chen HT, Hung YH, Chang CH, Li TM, Fong YC, et al. HGF and c-Met interaction promotes migration in human chondrosarcoma cells. PloS one. 2013;8:e53974. doi: 10.1371/journal.pone.0053974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Zhuang Y, Liu C, Li Y. Inhibition of c-Met activation sensitizes osteosarcoma cells to cisplatin via suppression of the PI3K-Akt signaling. Archives of biochemistry and biophysics. 2012;526:38–43. doi: 10.1016/j.abb.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Roskoski R., Jr Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacological research: the official journal of the Italian Pharmacological Society. 2015;94:9–25. doi: 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Bernardini G, Laschi M, Serchi T, Spreafico A, Botta M, Schenone S, et al. Proteomics and phosphoproteomics provide insights into the mechanism of action of a novel pyrazolo[3,4-d]pyrimidine Src inhibitor in human osteosarcoma. Molecular bioSystems. 2014;10:1305–12. doi: 10.1039/c3mb70328b. [DOI] [PubMed] [Google Scholar]
- 48.Casini N, Forte IM, Mastrogiovanni G, Pentimalli F, Angelucci A, Festuccia C, et al. SRC family kinase (SFK) inhibition reduces rhabdomyosarcoma cell growth in vitro and in vivo and triggers p38 MAP kinase-mediated differentiation. Oncotarget. 2015;6:12421–35. doi: 10.18632/oncotarget.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guan H, Zhou Z, Gallick GE, Jia SF, Morales J, Sood AK, et al. Targeting Lyn inhibits tumor growth and metastasis in Ewing’s sarcoma. Molecular cancer therapeutics. 2008;7:1807–16. doi: 10.1158/1535-7163.MCT-08-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Michels S, Trautmann M, Sievers E, Kindler D, Huss S, Renner M, et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer research. 2013;73:2518–28. doi: 10.1158/0008-5472.CAN-12-3023. [DOI] [PubMed] [Google Scholar]
- 51.Sievers E, Trautmann M, Kindler D, Huss S, Gruenewald I, Dirksen U, et al. SRC inhibition represents a potential therapeutic strategy in liposarcoma. International journal of cancer Journal international du cancer. 2015 doi: 10.1002/ijc.29645. [DOI] [PubMed] [Google Scholar]
- 52.Villacis RA, Silveira SM, Barros-Filho MC, Marchi FA, Domingues MA, Scapulatempo-Neto C, et al. Gene expression profiling in leiomyosarcomas and undifferentiated pleomorphic sarcomas: SRC as a new diagnostic marker. PloS one. 2014;9:e102281. doi: 10.1371/journal.pone.0102281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castoria G, Giovannelli P, Di Donato M, Hayashi R, Arra C, Appella E, et al. Targeting androgen receptor/Src complex impairs the aggressive phenotype of human fibrosarcoma cells. PloS one. 2013;8:e76899. doi: 10.1371/journal.pone.0076899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong M, Rice L, Lepler S, Pampo C, Siemann DW. Impact of the Src inhibitor saracatinib on the metastatic phenotype of a fibrosarcoma (KHT) tumor model. Anticancer research. 2010;30:4405–13. [PubMed] [Google Scholar]
- 55.Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer research. 2007;67:2800–8. doi: 10.1158/0008-5472.CAN-06-3469. [DOI] [PubMed] [Google Scholar]
- 56.Duan Z, Zhang J, Ye S, Shen J, Choy E, Cote G, et al. A-770041 reverses paclitaxel and doxorubicin resistance in osteosarcoma cells. BMC cancer. 2014;14:681. doi: 10.1186/1471-2407-14-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Oosterwijk JG, van Ruler MA, Briaire-de Bruijn IH, Herpers B, Gelderblom H, van de Water B, et al. Src kinases in chondrosarcoma chemoresistance and migration: dasatinib sensitises to doxorubicin in TP53 mutant cells. British journal of cancer. 2013;109:1214–22. doi: 10.1038/bjc.2013.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin-Broto J, Gutierrez AM, Ramos RF, Lopez-Guerrero JA, Ferrari S, Stacchiotti S, et al. MRP1 overexpression determines poor prognosis in prospectively treated patients with localized high-risk soft tissue sarcoma of limbs and trunk wall: an ISG/GEIS study. Molecular cancer therapeutics. 2014;13:249–59. doi: 10.1158/1535-7163.MCT-13-0406. [DOI] [PubMed] [Google Scholar]
- 59.Crompton BD, Carlton AL, Thorner AR, Christie AL, Du J, Calicchio ML, et al. High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer research. 2013;73:2873–83. doi: 10.1158/0008-5472.CAN-12-1944. [DOI] [PubMed] [Google Scholar]
- 60.Feng S, Shi X, Ren KE, Wu S, Sun X. Focal adhesion kinase is involved in the migration of human osteosarcoma cells. Oncology letters. 2015;9:2670–4. doi: 10.3892/ol.2015.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Zu J, Xu G, Zhao W, Jinglong Y. Inhibition of focal adhesion kinase induces apoptosis in human osteosarcoma SAOS-2 cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:1551–6. doi: 10.1007/s13277-013-1214-0. [DOI] [PubMed] [Google Scholar]
- 62.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 63.Yan S, Li Z, Thiele CJ. Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in Neuroblastoma and Pediatric Sarcomas In vitro and In vivo. Oncotarget. 2013;4:433–45. doi: 10.18632/oncotarget.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salas S, Jiguet-Jiglaire C, Campion L, Bartoli C, Frassineti F, Deville JL, et al. Correlation between ERK1 and STAT3 expression and chemoresistance in patients with conventional osteosarcoma. BMC cancer. 2014;14:606. doi: 10.1186/1471-2407-14-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carmo CR, Lyons-Lewis J, Seckl MJ, Costa-Pereira AP. A novel requirement for Janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PloS one. 2011;6:e19861. doi: 10.1371/journal.pone.0019861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryu K, Susa M, Choy E, Yang C, Hornicek FJ, Mankin HJ, et al. Oleanane triterpenoid CDDO-Me induces apoptosis in multidrug resistant osteosarcoma cells through inhibition of Stat3 pathway. BMC cancer. 2010;10:187. doi: 10.1186/1471-2407-10-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hunter T. A thousand and one protein kinases. Cell. 1987;50:823–9. doi: 10.1016/0092-8674(87)90509-5. [DOI] [PubMed] [Google Scholar]
- 68.de Carcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell cycle (Georgetown, Tex) 2011;10:2255–62. doi: 10.4161/cc.10.14.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ugras S, Brill E, Jacobsen A, Hafner M, Socci ND, Decarolis PL, et al. Small RNA sequencing and functional characterization reveals MicroRNA-143 tumor suppressor activity in liposarcoma. Cancer research. 2011;71:5659–69. doi: 10.1158/0008-5472.CAN-11-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu X, Choy E, Harmon D, Yang S, Yang C, Mankin H, et al. Inhibition of polo-like kinase 1 leads to the suppression of osteosarcoma cell growth in vitro and in vivo. Anti-cancer drugs. 2011;22:444–53. doi: 10.1097/CAD.0b013e32834513f4. [DOI] [PubMed] [Google Scholar]
- 71.Stehle A, Hugle M, Fulda S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer letters. 2015;365:37–46. doi: 10.1016/j.canlet.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 72.Weiss LM, Hugle M, Romero S, Fulda S. Synergistic induction of apoptosis by a polo-like kinase 1 inhibitor and microtubule-interfering drugs in Ewing sarcoma cells. International journal of cancer Journal international du cancer. 2015 doi: 10.1002/ijc.29725. [DOI] [PubMed] [Google Scholar]
- 73.Sero V, Tavanti E, Vella S, Hattinger CM, Fanelli M, Michelacci F, et al. Targeting polo-like kinase 1 by NMS-P937 in osteosarcoma cell lines inhibits tumor cell growth and partially overcomes drug resistance. Investigational new drugs. 2014;32:1167–80. doi: 10.1007/s10637-014-0158-6. [DOI] [PubMed] [Google Scholar]
- 74.Ma H, He C, Cheng Y, Li D, Gong Y, Liu J, et al. PLK1shRNA and doxorubicin co-loaded thermosensitive PLGA-PEG-PLGA hydrogels for osteosarcoma treatment. Biomaterials. 2014;35:8723–34. doi: 10.1016/j.biomaterials.2014.06.045. [DOI] [PubMed] [Google Scholar]
- 75.Hugle M, Belz K, Fulda S. Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs. Cell death and differentiation. 2015 doi: 10.1038/cdd.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koch P, Gehringer M, Laufer SA. Inhibitors of c-Jun N-terminal kinases: an update. Journal of medicinal chemistry. 2015;58:72–95. doi: 10.1021/jm501212r. [DOI] [PubMed] [Google Scholar]
- 77.Zhang S, Guo W, Ren TT, Lu XC, Tang GQ, Zhao FL. Arsenic trioxide inhibits Ewing’s sarcoma cell invasiveness by targeting p38(MAPK) and c-Jun N-terminal kinase. Anti-cancer drugs. 2012;23:108–18. doi: 10.1097/CAD.0b013e32834bfd68. [DOI] [PubMed] [Google Scholar]
- 78.Posthumadeboer J, van Egmond PW, Helder MN, de Menezes RX, Cleton-Jansen AM, Belien JA, et al. Targeting JNK-interacting-protein-1 (JIP1) sensitises osteosarcoma to doxorubicin. Oncotarget. 2012;3:1169–81. doi: 10.18632/oncotarget.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gweon EJ, Kim SJ. Resveratrol attenuates matrix metalloproteinase-9 and -2-regulated differentiation of HTB94 chondrosarcoma cells through the p38 kinase and JNK pathways. Oncology reports. 2014;32:71–8. doi: 10.3892/or.2014.3192. [DOI] [PubMed] [Google Scholar]
- 80.Fukukawa C, Nagayama S, Tsunoda T, Toguchida J, Nakamura Y, Katagiri T. Activation of the non-canonical Dvl-Rac1-JNK pathway by Frizzled homologue 10 in human synovial sarcoma. Oncogene. 2009;28:1110–20. doi: 10.1038/onc.2008.467. [DOI] [PubMed] [Google Scholar]
- 81.Kim A, Im M, Yim NH, Kim T, Ma JY. A novel herbal medicine, KIOM-C, induces autophagic and apoptotic cell death mediated by activation of JNK and reactive oxygen species in HT1080 human fibrosarcoma cells. PloS one. 2014;9:e98703. doi: 10.1371/journal.pone.0098703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou H, Shen T, Shang C, Luo Y, Liu L, Yan J, et al. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. Oncotarget. 2014;5:10140–50. doi: 10.18632/oncotarget.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Molecular and cellular biology. 1999;19:7245–54. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xia Y, Yang W, Bu W, Ji H, Zhao X, Zheng Y, et al. Differential regulation of c-Jun protein plays an instrumental role in chemoresistance of cancer cells. The Journal of biological chemistry. 2013;288:19321–9. doi: 10.1074/jbc.M113.475442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, et al. Cyclin-dependent kinases: a family portrait. Nature cell biology. 2009;11:1275–6. doi: 10.1038/ncb1109-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nature reviews Drug discovery. 2015;14:130–46. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saab R, Bills JL, Miceli AP, Anderson CM, Khoury JD, Fry DW, et al. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Molecular cancer therapeutics. 2006;5:1299–308. doi: 10.1158/1535-7163.MCT-05-0383. [DOI] [PubMed] [Google Scholar]
- 88.Lee SE, Kim YJ, Kwon MJ, Choi DI, Lee J, Cho J, et al. High level of CDK4 amplification is a poor prognostic factor in well-differentiated and dedifferentiated liposarcoma. Histology and histopathology. 2014;29:127–38. doi: 10.14670/HH-29.127. [DOI] [PubMed] [Google Scholar]
- 89.Dickson MA, Tap WD, Keohan ML, D’Angelo SP, Gounder MM, Antonescu CR, et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:2024–8. doi: 10.1200/JCO.2012.46.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luke JJ, D’Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Maki RG, et al. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: preclinical investigations and results of a phase I dose-escalation clinical trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:2638–47. doi: 10.1158/1078-0432.CCR-11-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer J, et al. The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. Molecular cancer therapeutics. 2011;10:1018–27. doi: 10.1158/1535-7163.MCT-11-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cantley LC. The phosphoinositide 3-kinase pathway. Science (New York, NY) 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 93.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 94.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E5564–73. doi: 10.1073/pnas.1419260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Molecular cancer therapeutics. 2015;14:395–406. doi: 10.1158/1535-7163.MCT-14-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marklein D, Graab U, Naumann I, Yan T, Ridzewski R, Nitzki F, et al. PI3K inhibition enhances doxorubicin-induced apoptosis in sarcoma cells. PloS one. 2012;7:e52898. doi: 10.1371/journal.pone.0052898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hugle M, Fulda S. Dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 synergizes with chloroquine to induce apoptosis in embryonal rhabdomyosarcoma. Cancer letters. 2015;360:1–9. doi: 10.1016/j.canlet.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 99.Gobin B, Huin MB, Lamoureux F, Ory B, Charrier C, Lanel R, et al. BYL719, a new alpha-specific PI3K inhibitor: single administration and in combination with conventional chemotherapy for the treatment of osteosarcoma. International journal of cancer Journal international du cancer. 2015;136:784–96. doi: 10.1002/ijc.29040. [DOI] [PubMed] [Google Scholar]
- 100.Martin-Liberal J, Gil-Martin M, Sainz-Jaspeado M, Gonzalo N, Rigo R, Colom H, et al. Phase I study and preclinical efficacy evaluation of the mTOR inhibitor sirolimus plus gemcitabine in patients with advanced solid tumours. British journal of cancer. 2014;111:858–65. doi: 10.1038/bjc.2014.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martin-Liberal J, Tirado OM, Garcia del Muro X. Sirolimus plus gemcitabine: a new therapeutic combination for resistant sarcomas? Expert review of anticancer therapy. 2015;15:257–9. doi: 10.1586/14737140.2015.1003045. [DOI] [PubMed] [Google Scholar]
- 102.Wang X, Lai P, Zhang Z, Huang M, Wang L, Yin M, et al. Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncology reports. 2014;32:382–8. doi: 10.3892/or.2014.3182. [DOI] [PubMed] [Google Scholar]
- 103.Zhu Z, Wang CP, Zhang YF, Nie L. MicroRNA-100 resensitizes resistant chondrosarcoma cells to cisplatin through direct targeting of mTOR. Asian Pacific journal of cancer prevention: APJCP. 2014;15:917–23. doi: 10.7314/apjcp.2014.15.2.917. [DOI] [PubMed] [Google Scholar]
- 104.Liang X, Wang D, Wang Y, Zhou Z, Zhang J, Li J. Expression of aurora kinase A and B in chondrosarcoma and its relationship with the prognosis. Diagnostic pathology. 2012;7:84. doi: 10.1186/1746-1596-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiang Z, Jiang J, Yang H, Ge Z, Wang Q, Zhang L, et al. Silencing of Aurora kinase A by RNA interference inhibits tumor growth in human osteosarcoma cells by inducing apoptosis and G2/M cell cycle arrest. Oncology reports. 2014;31:1249–54. doi: 10.3892/or.2014.2986. [DOI] [PubMed] [Google Scholar]
- 106.Przybyl J, Sciot R, Wozniak A, Schoffski P, Vanspauwen V, Samson I, et al. Metastatic potential is determined early in synovial sarcoma development and reflected by tumor molecular features. The international journal of biochemistry & cell biology. 2014;53:505–13. doi: 10.1016/j.biocel.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 107.Winter GE, Rix U, Lissat A, Stukalov A, Mullner MK, Bennett KL, et al. An integrated chemical biology approach identifies specific vulnerability of Ewing’s sarcoma to combined inhibition of Aurora kinases A and B. Molecular cancer therapeutics. 2011;10:1846–56. doi: 10.1158/1535-7163.MCT-11-0100. [DOI] [PubMed] [Google Scholar]
- 108.Tavanti E, Sero V, Vella S, Fanelli M, Michelacci F, Landuzzi L, et al. Preclinical validation of Aurora kinases-targeting drugs in osteosarcoma. British journal of cancer. 2013;109:2607–18. doi: 10.1038/bjc.2013.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raymond E, Alexandre J, Faivre S, Goldwasser F, Besse-Hammer T, Gianella-Borradori A, et al. A phase I schedule dependency study of the aurora kinase inhibitor MSC1992371A in combination with gemcitabine in patients with solid tumors. Investigational new drugs. 2014;32:94–103. doi: 10.1007/s10637-013-9950-y. [DOI] [PubMed] [Google Scholar]
- 110.Chandhanayingyong C, Kim Y, Staples JR, Hahn C, Lee FY. MAPK/ERK Signaling in Osteosarcomas, Ewing Sarcomas and Chondrosarcomas: Therapeutic Implications and Future Directions. Sarcoma. 2012;2012:404810. doi: 10.1155/2012/404810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang Q, Zhang S, Kang M, Dong R, Zhao J. Synergistic growth inhibition by sorafenib and cisplatin in human osteosarcoma cells. Oncology reports. 2015;33:2537–44. doi: 10.3892/or.2015.3832. [DOI] [PubMed] [Google Scholar]
- 112.Spina A, Sorvillo L, Di Maiolo F, Esposito A, D’Auria R, Di Gesto D, et al. Inorganic phosphate enhances sensitivity of human osteosarcoma U2OS cells to doxorubicin via a p53-dependent pathway. Journal of cellular physiology. 2013;228:198–206. doi: 10.1002/jcp.24124. [DOI] [PubMed] [Google Scholar]
- 113.Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell cycle (Georgetown, Tex) 2013;12:3159–64. doi: 10.4161/cc.26062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Molecular cancer therapeutics. 2012;11:174–82. doi: 10.1158/1535-7163.MCT-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kreahling JM, Foroutan P, Reed D, Martinez G, Razabdouski T, Bui MM, et al. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PloS one. 2013;8:e57523. doi: 10.1371/journal.pone.0057523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Duan L, Perez RE, Hansen M, Gitelis S, Maki CG. Increasing cisplatin sensitivity by schedule-dependent inhibition of AKT and Chk1. Cancer biology & therapy. 2014;15:1600–12. doi: 10.4161/15384047.2014.961876. [DOI] [PMC free article] [PubMed] [Google Scholar]