

Abstract
Cholinergic regulation of arterial luminal diameter involves intricate network of intercellular communication between the endothelial and smooth muscle cells that is highly dependent on the molecular mediators released by the endothelium. Albeit the well-recognized contribution of nitric oxide (NO) towards vasodilation, the identity of compensatory mechanisms that maintain vasomotor tone when NO synthesis is deranged remain largely unknown in the ophthalmic artery. This is the first study to identify the vasodilatory signalling mechanisms of the ophthalmic artery employing wild type mice. Acetylcholine (ACh)-induced vasodilation was only partially attenuated when NO synthesis was inhibited. Intriguingly, the combined blocking of cytochrome P450 oxygenase (CYP450) and lipoxygenase (LOX), as well as CYP450 and gap junctions, abolished vasodilation; demonstrating that the key compensatory mechanisms comprise arachidonic acid metabolites which, work in concert with gap junctions for downstream signal transmission. Furthermore, the voltage-gated potassium ion channel, Kv1.6, was functionally relevant in mediating vasodilation. Its localization was found exclusively in the smooth muscle. In conclusion, ACh-induced vasodilation of mouse ophthalmic artery is mediated in part by NO and predominantly via arachidonic acid metabolites, with active involvement of gap junctions. Particularly, the Kv1.6 channel represents an attractive therapeutic target in ophthalmopathologies when NO synthesis is compromised.
The magnitude of global visual impairment is estimated to reach an approximate 285 million people at present and this number is expected to escalate as a result of the aging population1. Among the many factors that contribute towards vision loss, there is accumulating evidence that emphasizes the dysregulation of ocular blood flow as one of the leading causes of various sight threatening ophthalmopathologies namely, glaucoma, diabetic retinopathy and nonarteritic anterior ischemic optic neuropathy (NAION)2,3,4,5,6. Modulation of the ocular blood flow is largely attributed to the vascular endothelium, which consists of a group of highly specialized cells that play indispensable physiological roles in the maintenance of vascular tone, especially by the release of various diffusible vasoactive substances. Although nitric oxide (NO) is the common messenger molecule implicated in the vasodilatory responses in ocular blood vessels7,8, other key mediators released by the endothelium include prostacyclin (PGI2) and the endothelium-derived hyperpolarizing factor (EDHF) that play crucial roles in maintaining the hemodynamic balance in ocular vasculature9.These mediators can be released by shear stress, autacoids or neurotransmitters from the autonomic nervous system10,11. One important substance that can act as an autacoid or neurotransmitter of the parasympathetic nervous system and has been shown to induce marked vasodilation in many blood vessels is acetylcholine (ACh). Previous studies reported the presence of nitric oxide synthase (NOS) in the endothelium of ocular arteries and demonstrated its contribution to ACh-induced vasodilation in a wide range of species including in humans12,13, dogs14, rats15, pigs16, cows17, and primates18,19 On the other hand, it has been shown in different ocular vascular beds that apart from NO, PGI2 and EDHFs may also contribute to endothelium-dependent vasodilation20,21. In recent years, with the use of gene-knockout mice and isoform-selective NOS inhibitors, we demonstrated that endothelium-dependent vasodilation induced by ACh is mediated predominantly by the activation of endothelial NOS (eNOS) in the retinal arterioles22. However, in the mouse ophthalmic artery, eNOS mediated only a part of the cholinergic vasodilation response while another, yet unknown, mechanism also substantially contributed towards ACh-induced vasodilation23.The mouse ophthalmic artery is a small vessel with an inner diameter between 80 and 150 μm that develops moderate myogenic tone24,25,26. Since the relative contribution of these three mediators to agonist-induced vasodilation varies among vascular beds, species and also the diameter of the blood vessels, we hypothesize that PGI2 and EDHFs may be of physiological relevance in the mouse ophthalmic circulation. Therefore, the purpose of the present study was to identify the mechanisms contributing to endothelium-dependent vasodilator responses in the mouse ophthalmic artery using in vitro vascular preparations and pharmacological inhibitors. For the first time, this experimental approach allowed for in-depth identification of post-receptor signalling pathways involved in the vasodilatory mechanisms of the mouse ophthalmic artery. Physiologically, the specific modulator molecules and potassium ion channels identified may be potential targets for therapeutic intervention to enhance the circulatory status of the eye in pathologic conditions when NO availability is compromised.
Results
Cholinergic vasodilation responses are endothelium-dependent
To investigate the role of endothelium in mediating cholinergic vasodilation of the OA, preconstricted arteries with intact and denuded endothelium were stimulated with ACh (10−4 M). The removal of endothelium in ophthalmic arteries resulted in marked attenuation of the vasodilator response (3.77 ± 4.20%, P < 0.0001) compared to the arteries with intact endothelium (77.41 ± 6.78%). Conversely, the vasodilator response in both endothelium-intact (82.00 ± 10.76%) and –denuded (70.53 ± 8.09%) ophthalmic arteries was almost similar following treatment with endothelium-independent, exogenous NO donor, sodium nitroprusside (SNP) (10−4 M), indicating that the smooth muscle reactivity remained unaffected after endothelium denudation (Fig. 1).
Figure 1. Responses of the wild-type mouse ophthalmic artery before and after removal of endothelium to ACh (10−4 M) and to the exogenous NO donor, SNP (10−4 M).
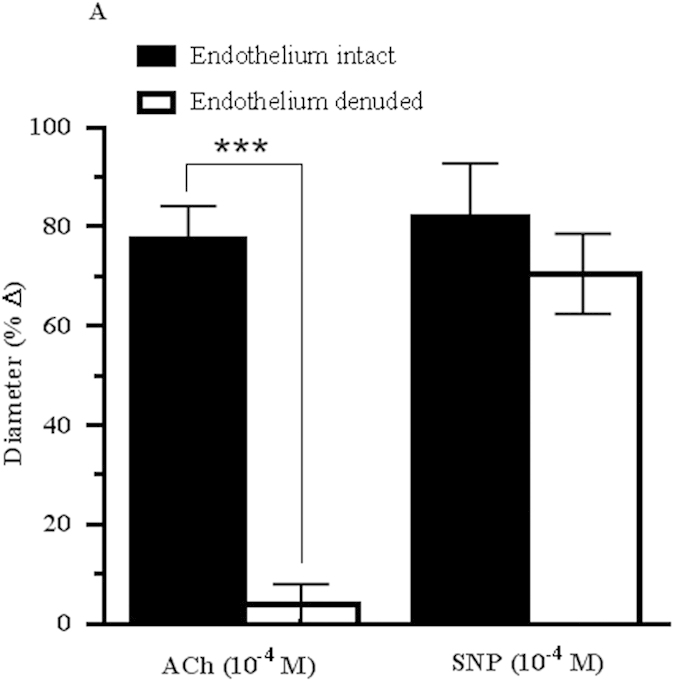
Vasodilatory responses to ACh were markedly attenuated in the endothelium-denuded vessels, whereas vasodilation responses to SNP were retained in both endothelium-denuded and –intact vessels. Values are expressed as mean ± standard error of the mean (s.e.m) (n = 6 per group; ***P < 0.0001, endothelium-denuded versus endothelium-intact).
Role of NO, and PGI2 in ACh-induced vasodilation
Cumulative administration of ACh (10−9–10−4M) evoked concentration-dependent vasodilatory responses (76.44 ± 9.45%) that were markedly attenuated (49.18 ± 10.48%, P < 0.01) following incubation with the non-isoform-selective NOS inhibitor, L-NAME (10−4 M) (Fig. 2A). To test whether the NO receptor, sGC was involved in vasodilation, the ophthalmic arteries were stimulated with ACh (10−9–10−4 M) before and after addition of the sGC inhibitor, ODQ (10−5M). Responses to acetylcholine were markedly reduced (ACh reference: 83.61 ± 8.67% vs ODQ: 48.92 ± 8.71%, P < 0.001) after ODQ treatment (Fig. 2B), indicative of NO involvement. Conversely, responses to acetylcholine in the ophthalmic arteries cannot be ascribed to PGI2 because exposure of the arteries to the non-isoform-selective COX inhibitor, indomethacin (10−5M), did not significantly affect the vasodilation (ACh reference: 76.63 ± 5.93% vs indomethacin: 69.60 ± 9.84%), as shown in Supplementary Fig. S1. Additionally, combined incubation with L-NAME and indomethacin (48.91 ± 9.01%) did not alter the vasodilatory response compared to inhibition with L-NAME alone (49.18 ± 10.48%) (Fig. 2A,C), suggesting that COX metabolites did not contribute to the cholinergic vasodilation in the mouse ophthalmic artery. However, the residual dilatory response observed after the blocking of both NOS and COX with L-NAME and indomethacin, respectively, was abolished by the addition of 30 mM K+ solution (potassium chloride, KCl), as depicted in Fig. 2C. At this concentration, KCl acts as a partial depolarizing agent and antagonizes the action of EDHF. Therefore, this finding suggests the important involvement of EDHF in ACh-mediated vasodilation in this vascular bed.
Figure 2. Effect of NOS inhibition on the vasodilatory responses of ophthalmic artery from wild-type mice with intact endothelium.

(A) The non-subtype-selective NOS inhibitor, L-NAME (10−4 M, n = 5) partially attenuated vasodilation to ACh (**P < 0.01, ACh reference vs L-NAME). (B) The sGC inhibitor, ODQ (10−4 M, n = 6) evoked partial yet significant attenuation of the dilatory responses to ACh (***P < 0.001, ACh reference vs ODQ). (C) The residual dilatory responses in the presence of both L-NAME and indomethacin were abolished by 30 mM of potassium (K+) solution (***P < 0.001, ACh reference vs L-NAME + Indomethacin; ***P < 0.001, L-NAME + Indomethacin vs L-NAME + Indomethacin + KCl). Values are expressed as mean ± s.e.m.
Contribution of EDHF-mediated vasodilator responses to ACh
To assess the EDHF mechanisms involved in mediating ACh-induced vasodilation in the mouse OA, various pharmacological agents were employed to inhibit different factors implicated as the putative EDHF. Acetylcholine was previously reported to induce generation of vasoactive amounts of H2O2 both NOS-dependently and –independently27,28. To evaluate the contribution of H2O2 to endothelium-dependent dilatation, responses to ACh before and after incubation with catalase (1000 units/ml) were tested. Catalase, when applied either alone or in combination with both L-NAME and indomethacin, evoked negligible inhibitory effect on vasodilation responses elicited by ACh (Supplementary Fig. S2).
The inhibition of CYP450 and LOX with 17-ODYA and baicalein, respectively, elicited significant blunting of the relaxation (L-NAME and indomethacin: 53.48 ± 4.81% vs 17-ODYA: 33.13 ± 7.10%, P < 0.01 and L-NAME and indomethacin: 59.36 ± 8.76% vs baicalein: 17.98 ± 14.09%, P < 0.001, respectively) (Fig. 3A,B). Gap junction inhibitor, 18α-GA, also produced significant inhibitory responses to ACh-mediated dilatation (L-NAME and indomethacin: 63.80 ± 5.55% vs 18α-GA: 18.75 ± 7.03%, P < 0.001) (Fig. 3C). Interestingly, the combination blocking with 17-ODYA and baicalein almost abolished vasodilation to ACh (L-NAME and indomethacin: 61.28 ± 5.30% vs 17-ODYA and baicalein: 4.87 ± 2.86%, P < 0.001) (Fig. 3D). In order to investigate which of these two pathways (CYP450 and LOX) may activate the gap junction, ophthalmic arteries were incubated with combinations of 17-ODYA with 18α-GA and baicalein with 18α-GA. The vasodilation elicited by cumulative application of ACh in the presence of L-NAME and indomethacin was abolished by the CYP450 and gap junction blocker combination, as depicted in Fig. 3E (L-NAME and indomethacin: 70.54 ± 10.19% vs 17-ODYA and 18α-GA: 0.83 ± 0.55%, P < 0.001), whereas combination inhibition of both LOX and gap junction resulted in significant attenuation of vasodilatation (L-NAME and indomethacin: 56.03 ± 6.68% vs baicalein and 18α-GA: 19.48 ± 3.84%, P < 0.001) (Fig. 3F). These experiments were performed in the presence of both L-NAME and indomethacin to rule out the influence of NOS and COX.
Figure 3. The ACh-evoked vasodilatory responses of ophthalmic artery from wild-type mice mediated by EDHFs.

The inhibition of the arachidonic acid metabolites synthesized via the (A) CYP450 and (B) LOX pathways with 17-ODYA and baicalein, respectively, produced significant attenuation of the vasodilation. (C) The blocking of gap junctions with 18α-GA caused marked blunting of the dilatory responses. Vasodilation to ACh was almost abolished by the combination blocking with (D) 17-ODYA and baicalein and with (E) 17-ODYA and 18α-GA. (F) Combination blocking with baicalein and 18α-GA caused a partial but significant inhibition of vasodilation. Values are expressed as mean ± s.e.m [n = 5–6 per group; **P < 0.01, ***P < 0.001, L-NAME and Indomethacin vs L-NAME and Indomethacin and blocker(s)].
Effect of potassium ion channel blockers on endothelium-dependent vasodilation
To further characterize the EDHF-related dilatation, specifically the contribution fostered by KCa channels, all three channel subtypes, SKCa, IKCa and BKCa, were inhibited with combination of Apa and ChTX. Marked inhibition of cholinergic responses was observed with this combination blocking (L-NAME and indomethacin: 56.96 ± 7.40% vs Apa and ChTX: 4.07 ± 1.65%, P < 0.0001), as shown in Fig. 4A. To further validate this finding, each KCa channel subtype was blocked in combination with their respective, highly specific inhibitors consisting of Apa for SKCa, TRAM-34 for IKCa and IbTX for BKCa. Remarkably, this combination blocking demonstrated that the vasodilator response to ACh remained unchanged (L-NAME and indomethacin: 63.26 ± 9.07% vs Apa and TRAM-34 and IbTX: 66.06 ± 11.34%, P > 0.05) (Fig. 4B). Each of these three KCa channels was inhibited individually with Apa, TRAM-34, IbTX and ChTX and our findings showed that only the inhibition with ChTX displayed significant attenuation of the vasodilation (Supplementary Fig. S3), while the other blockers comprising of Apa, TRAM-34 and IbTX did not contribute to significant blunting of vasodilation in the OA (Supplementary Fig. S4- S6, respectively). Based on these results, we suspected that ChTX, which had also been reported to block several Kv channel subtypes, blocked one or more of the Kv channel subunits in the mouse ophthalmic artery.
Figure 4. Responses of ophthalmic artery from wild-type mice to ACh-evoked vasodilation in the presence of calcium-activated potassium ion channel blockers.
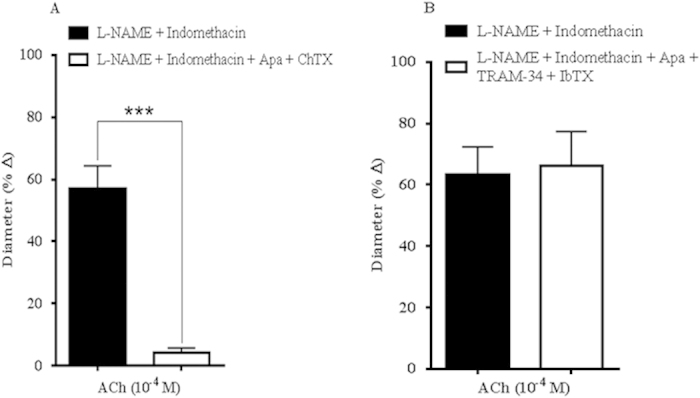
(A) Combined blocking with Apa and ChTX elicited total attenuation of endothelium-dependent vasodilation. (B) The combination blocking of the KCa channels with their respective specific blockers, Apa, TRAM-34 and IbTX, conferred negligible inhibitory effects on ACh-induced vasodilation. Values are expressed as mean ± s.e.m [n = 5–6 per group; ***P < 0.0001, L-NAME and Indomethacin vs L-NAME and Indomethacin and blocker(s)].
Hence, to confirm this hypothesis, a number of agents that block Kv channels were employed to assess the nature of the Kv channel involved in mediating ACh-elicited vasodilation. In order to determine which of these ChTX-sensitive Kv channel subtype(s) is particularly involved in mediating the ACh-induced relaxations, vessels were incubated with MgTX, a selective blocker of the Kv1.3 and Kv1.6 channels in the presence of NOS and COX inhibitors. Incubation with MgTX completely blunted ACh-induced dilations (L-NAME and indomethacin: 64.67 ± 4.93% vs MgTX: 1.25 ± 1.25%, P < 0.001) (Fig. 5A), underscoring the probable role(s) of ChTX- and MgTX-sensitive Kv1.3 and Kv1.6 channels in the mouse ophthalmic arteries. Following incubation with MTX, psora-4 and β-DTX which, specifically blocks the Kv1.2, Kv1.3 and combinations of Kv1.1 and Kv1.2 channels, respectively, the ACh -induced vasodilatation remained unaffected ((Supplementary Figs S7–9) In contrast, incubation with α-DTX, a potent blocker of Kv1.1, Kv1.2 and Kv1.6 channels evoked complete attenuation of the vasodilatory responses to ACh, as demonstrated in Fig. 5B (L-NAME and indomethacin: 92.73 ± 3.48% vs α-DTX: 4.96 ± 2.34%, P < 0.001). Taken together, the results of the Kv channel inhibitions is attributed to the involvement of the Kv1.6 channel that is sensitive to the blocking effects of ChTX, MgTX as well as α-DTX.
Figure 5. Specific inhibition of the Kv channel family in the ophthalmic artery of wild type mice indicated an active participation of this channel subtype in mediating the agonist-evoked vasodilatory mechanisms.

(A) Blocking with MgTXalmost abolished dilatory responses of the arteries. (B)The inhibition of Kv1.1, Kv1.2 and Kv1.6 channels with α-DTX demonstrated a significantabolishment of vasodilation. Values are expressed as mean ± s.e.m [n = 5–6 per group; ***P < 0.001, L-NAME and Indomethacin vs L-NAME and Indomethacin and blocker(s)]. Absence of error bar indicates that the SEM was less than the size of the symbol.
Next, to evaluate the potential functional relevance of other potassium channels, arteries were treated with blockers of Kir and KATP channels, and Na+/K+-ATPase. Neither glibenclamide nor ouabain had any significant effect on the ACh-mediated vasodilation ((Supplementary Figs S10–11)), indicating the null involvement of the KATP channel and Na+/K+-ATPase, respectively. Conversely, Fig. 6 shows that BaCl2, a blocker of Kir channels, caused 38.07% inhibition (P < 0.001) of the ACh-elicited vasodilation in the presence of L-NAME and indomethacin (L-NAME and indomethacin: 48.64 ± 8.35%).
Figure 6. Cholinergic vasodilation of the mouse ophthalmic artery in the presence of Kir channels blocker.
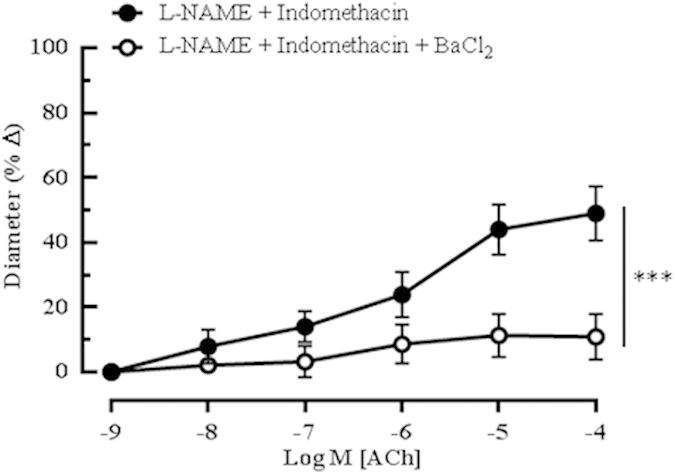
The inhibition of Kir channels with BaCl2 caused a significant inhibition of dilation. All experiments were carried out in the presence of both NOS and COX inhibitors. Values are expressed as mean ± s.e.m (n = 5 per group; ***P < 0.001, L-NAME and Indomethacin vs L-NAME and Indomethacin and blocker).
Localization of the Kv1.6 channel in the mouse ophthalmic artery
The Kv1.6 channel plays a central role in the regulation of the ophthalmic blood flow as demonstrated by the functional experiments in the present study. In order to determine the localization of this Kv channel subtype in the ophthalmic artery, immunostaining was carried out on the sagittal cryosections of ophthalmic artery. Localization of Kv1.6 was particularly restricted to the vascular smooth muscle cell layer but no expression was observed in the endothelial cells, as shown in Fig. 7A. The negative control of the same tissue in which the primary antibody was omitted, was not stained (Fig. 7B).
Figure 7. Photomicrographs representing the transverse cryosections of the mouse ophthalmic artery for Kv1.6 channel immunolocalization.

(A) The expression of the Kv1.6 channel is prominent in the smooth muscle cell layer compared to the endothelial cells. (B) The negative control demonstrates no staining in the absence of the primary antibody. EC, Endothelial cell; VSMC, Vascular smooth muscle cell; L, Lumen. Scale bars indicate 10 μm at 600× magnification.
Discussion
This is the first functional study reporting on the EDHF mechanisms mediating agonist-induced vasodilator response in the mouse ophthalmic artery. There are several key findings, including some novel aspects, emerging from the current study. First, in addition to the well-established observations of the role of endothelium in various vascular beds and species, we endeavoured to investigate the role of endothelium in vasodilator response to ACh particularly in the mouse ophthalmic artery. The use of ACh instead of other agonists e.g. bradykinin is advocated in this study to circumvent desensitization of the endothelial receptors due to tachyphylaxis10. Moreover, our previous study has clearly demonstrated that in the murine ophthalmic artery, endothelium-dependent vasodilator responses were mediated by the M3 muscarinic ACh receptor25,26. Mechanical denudation of endothelium abolished ACh-induced vasorelaxation, thereby demonstrating that the vascular endothelium plays an obligatory role in the cholinergic vasodilation of mouse ophthalmic artery. Our results also showed that the endothelium-dependent responses were partially mediated by a NOS- and sGC-dependent mechanism, supporting the involvement of NO. These findings are in contrast to the vasodilatory mechanism in the human ophthalmic artery, where the ACh- induced dilation is mediated predominantly by the NO pathway12. Conversely, the involvement of PGI2 was discounted in the mouse ophthalmic artery because indomethacin exerted no inhibitory effect on the dilatory responses to ACh. These results imply that prostanoid-dependent signalling pathway do not account for the ACh-mediated vasodilatory response in the mouse ophthalmic artery.
Secondly, the predominant involvement of EDHF accounts for the residual dilatory response observed in the mouse ophthalmic artery in the presence of both L-NAME and indomethacin, whereby the abolishment of dilation by concomitant addition of depolarizing concentration of potassium solution was observed29. It is widely recognized that the EDHF phenomenon evokes vasodilatation in the presence of COX and NOS inhibitors30,31 and its physiologic influence is deemed more prominent as the vessel diameter decreases. Since the smaller vessels have fundamental roles in vascular resistance, EDHF is suggested to be of major importance in the blood flow control in these vessels32,33. Consistent with this possibility, the involvement of these different factors implicated as EDHFs was tested and we found that endothelium-dependent vasodilation in the ophthalmic vasculature was mediated in part by CYP450 and predominantly by LOX metabolites, with a major involvement of the gap junctions. While the individual blockade of CYP450 and LOX only partially reduced vasodilation responses, combined blockade of CYP450 and LOX virtually abolished vasodilation suggesting that metabolites of both enzymes almost exclusively contribute to the EDHF-mediated responses in this vascular bed. The CYP450 pathway appears not to be completely dependent on the gap junctions because combined blockade of both CYP450 and gap junctions only resulted in additive response to the individual inhibitions. It is becoming increasingly well recognized that the arachidonic acid metabolites generated via the CYP450 pathway, most likely the four epoxyecosatrienoic acid regioisomers (EETs), have been implicated in the augmentation of gap junctional communications and to regulate active communications between endothelial cells34,35,36. This is especially relevant because EETs are highly lipophilic transferable factors that cannot pass through the gap junctions, which comprise of aqueous pores, but rather may act as modulators to hyperpolarize the VSMC via the gap junctions37,38,39.
In contrast to the CYP450-mediated signalling, the LOX signalling mechanism(s) seemed to be highly dependent on the gap junctions, through which the downstream signals and/or molecule(s) that dilate the VSMC are transmitted, since combined blockade of LOX and gap junctions did not result in any further attenuation of the response. However, it should be remarked that the molecular weights of LOX- derived metabolites, namely 15-hydroxy-11, 12-epoxyeicosatrienoic acid (HEETA) and 11, 12, 15-trihydroxyeicosatrienoic acid (THETA,) are large and since the aqueous central pore of the gap junctions can only permit the passage of molecules < 1 kDa, it is unlikely that LOX and/or its metabolites are transferable via this channel to hyperpolarize the VSMC40,41,42. However, a plausible explanation can be that arachidonic acid metabolites generated via the LOX pathway may act as autocrine or intracellular modulators of gap junctions, as was previously proposed in the rat middle cerebral artery and rabbit arteries, where LOX metabolites directly stimulated the SKCa channels, instead of the gap junctions as observed in our study, to hyperpolarize the VSMC43,44,45. The murine LOX share a highly conserved sequence similarity with the human’s based on the phylogenetic classification and they belong to the same 12/15-LOX subfamily46. Intriguingly, 12/15-LOX was found to be associated with key regulation roles in pathologies of the central nervous system such as Parkinson’s disease and Alzheimer’s47,48. Looking at the pivotal roles of the LOX- derived metabolites in human pathologies and the high similarity between both mouse and human, these findings broadens the use of murine models for further in-depth investigations of the molecular mechanisms of LOX-related pathway in the next studies.
Despite the rapid progress made in the past decade in elucidating the physiological roles of the arachidonic acid metabolites in various biological systems, many important questions still remain unanswered. For example, in our study, the existence of putative receptor(s) of the downstream signalling cascade of CYP450, especially for the EETs, require further investigation to extend our current hypothesis beyond the present findings. Likewise, studies involving the CYP450 metabolites in cardioprotection are also seeking to identify the precise molecular receptor(s) target(s) of EETs for potential development of new therapeutic strategies39,49. Additionally, it is important to define the precise identity of the arachidonic acid metabolites generated via the CYP450 and LOX mechanisms responsible for the observed vasodilatory phenomenon, as emphasized by Thollon et al.50.
Thirdly, our data strongly suggest the important involvement of the Kir and Kv1.6 channels in mediating endothelium-dependent dilation to ACh. Previous studies have shown that K+ released from the endothelium can act as an EDHF by activating KCa and stimulating Na+/K+-ATPase and Kir channel in guinea pig choroidal arterioles and rat hepatic arteries, respectively51,52. Therefore, we examined the possible role of potassium channels in endothelium-dependent vasodilation of the mouse ophthalmic artery. Consistent with the finding that KATP channels are usually not involved in EDHF-mediated vasodilation53, our results indicated that the inhibitory effect of glibenclamide on KATP channel had negligible influence on the vasodilatation of mouse ophthalmic artery induced by ACh. The application of ouabain also failed to inhibit dilation, suggesting the lack of Na+/K+-ATPase involvement in mediating responses to ACh. However, it is of interest that the blockade of Kir channels caused significant attenuation of vasodilation in the ophthalmic artery. The Kir, channel localized on the SMCs, is one the major targets of external K+ions, which activate the channel conductance to lower intracellular Ca2+ and leads to vasodilatation54,55.
Accumulating evidences imply that the action of EDHF is generally inhibited by combination blockade of the SKCa with Apa and, IKCa and BKCa with ChTX32,56. Correspondingly, our study demonstrated that the combined inhibition with these blockers virtually abolished cholinergic responses in the mouse ophthalmic artery. However, it is important to highlight here that the combined blockade of all three KCa channels with their respective specific blockers, and not ChTX, had no significant effect on the ACh-mediated vasodilatory responses in the current study as hypothesized. Of note, an interesting phenomenon was observed in mice where the expression of IKCa and SKCa in the endothelial cells was relatively low as the size of the artery decreased57 which, was in sharp contrast to the increased expression of both channel subtypes in the rat artery as the vessel size decreased58,59. On the basis of our results, these observations support the hypothesis of our study that the expression of the KCa channels in the mouse ophthalmic artery may be low or null and are unlikely to account for the attenuation of the vasodilation when blocked with Apa and ChTX. Taken together, this confounding finding can be extrapolated to the multi-channel blocking properties of ChTX, which not only blocks the IKCa and BKCa channels, but also inhibits the Shaker-related voltage-gated K+ channels Kv1.1, 1.2, 1.3, and 1.6 with high affinity60,61.
An ongoing, unresolved restriction to study the post- receptor mechanisms is the use of most characterized pharmacological blockers and inhibitors with unspecific nature that may be affecting another alternative EDHF signalling cascade with similar affinity, as demonstrated in the current study. Therefore, to confirm the involvement of the Kv channels and in particular to dissect which of these is/are involved in the vasodilation of the mouse ophthalmic artery, several highly specific Kv channel inhibitors were employed. Complete attenuation in vasodilation was observed in the presence of MgTX and α- DTX. MgTX is widely used as a potent inhibitor of the Kv1.3 in ion channel investigations62. However, a recent study by Bartok et al. provided critical evidence that MgTX is not a highly specific Kv1.3 inhibitor as had been assumed in many previous studies63 and this toxin has also been shown to inhibit other Kv channels, namely Kv1.1, 1.2 and 1.6, with high potency61,64,65,66,67. Our results support a possible participation of other channel subtype(s) considering the potential overlap in blocking selectivity exhibited by ChTX and MgTX, as summarized in the Venn diagram (Fig. 8). Therefore, with the use of several other toxins, this study unravelled that the Kv1.6 channel is functionally relevant in mediating vasodilatory responses. Additionally, immunostaining confirmed the localization of this voltage-gated channel subtype in the VSMC of the mouse ophthalmic artery.
Figure 8. Venn diagram representing the various pharmacological blockers employed in this study to dissect the contribution of the Kv channels to ACh-induced vasodilation of the mouse ophthalmic artery.
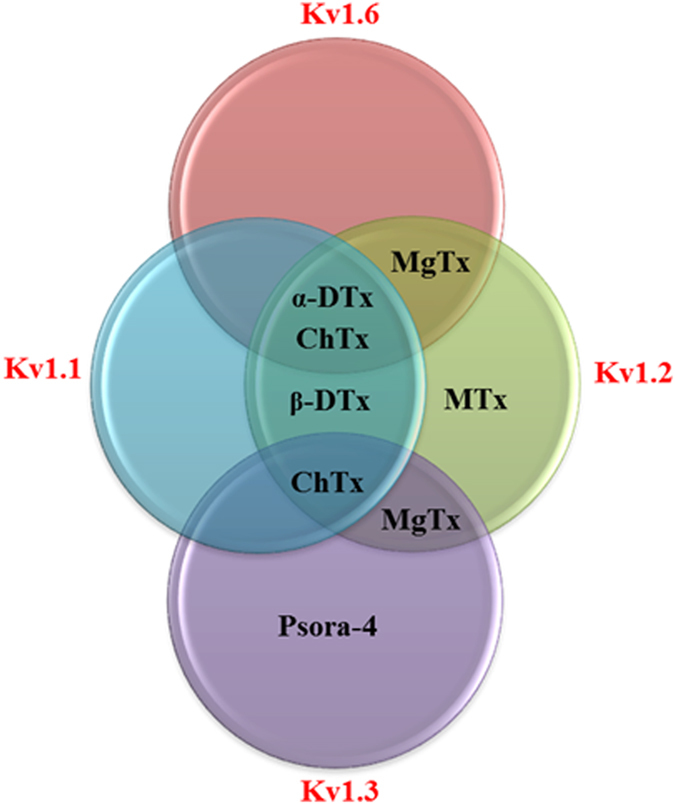
There are overlaps in the affinity of the blockers for one or more Kv channels. MgTX, Margatoxin; MTX, Maurotoxin; ChTX, Charybdotoxin; α-DTX, alpha Dendrotoxin; β- DTX, beta Dendrotoxin; Psora-4, [5-(4-Phenylbutoxy)psoralen].
The novelty of the present investigation lies in the identification of the Kv1.6 channel in mediating vasodilation that has never been reported hitherto. It is well recognized that the altered Kv1.6 channel expression is associated with neurodegenerative diseases such as Amyotrophic Lateral Sclerosis (ALS) that affects the duration of action potential of motor neurons68. On the other hand, study by Carrisoza-Gaytan et al. emphasized the importance of the Kv1.6 channel in K+ reabsorption in the thick ascending limb of the rat nephron69. This channel is also implicated in the pulmonary artery smooth muscle cells as one of the crucial hypoxia-sensitive Kv channels that regulate membrane potential and intracellular Ca2+ homeostasis during hypoxia70,71,72. As our understanding of the Kv channels continues to evolve, it is therefore tempting to conjecture that the specific identification of the Kv1.6 channel in this study may represent an innovative molecular target in the ophthalmic circulation to enhance vasodilation in conditions of channelopathy, albeit the exact function of this channel subtype in the ophthalmic circulation warrants further investigation. Our current investigation provides a plausible hypothesis as to how ACh-induced vasodilatation may occur in the mouse ophthalmic artery and based on our results, the hypothesized signalling pathways involved in the vasodilator mechanisms are as depicted in Fig. 9.
Figure 9. The proposed hypothetical endothelial cell-dependent signalling pathways of conducted vasodilation in response to ACh in the mouse ophthalmic artery.

The first proposed mechanism involves the NO/cGMP pathway: Activation of the endothelial M3 receptor by ACh induces an influx of [Ca2+]i. Following interaction with CaM (calmodulin), Ca2+ activates eNOS and release of NO. NO causes relaxation by interacting with the haem group of the enzyme, sGC, which then mediates the formation of cyclic guanosine monophosphate (cGMP) and activation of protein kinase G (PKG) that relaxes the VSMC. The second vasodilator mechanism is via the arachidonic acid (AA) metabolites synthesized through the CYP450 oxygenase pathway: The increase in [Ca2+]i elicits translocation of phospholipase A2 (PLA2) to the membrane and its major hydrolysis product is AA, which can be metabolized by CYP450 oxygenase to EETs. The EETs function as messenger molecules that modulate gap junctions (GJ) to spread the conductance to the VSMC. EETs may also directly activate a channel/receptor on the VSMC to hyperpolarize and dilate the vessel. The third signalling pathway involves the AA metabolites generated via the LOX pathway: It is hypothesized that LOX and/or its metabolites, namely THETA and HEETA do not pass the GJ as EDHF per se but activate GJ to hyperpolarize the VSMC. The forth key players are the gap junctions. The fifth proposed mechanism involves the active participation of the Kir and Kv1.6 channels: It is hypothesized that both Kir and Kv1.6 channels on the VSMC are activated by the increase in extracellular K+ resulting in hyperpolarization and vasodilation. The precise identity of the putative channel(s) on the endothelial cells that is activated and opened for K+ efflux for hyperpolarization to occur is unknown. Question marks represent unknown receptors that are yet to be identified. Blockers and inhibitors are indicated in red boxes. Green arrows indicate the activation of the gap junctions and downstream receptor(s). Blue solid arrows show potential pathways for transfer of hyperpolarization from the endothelium to the smooth muscle cells. Blue quadrangular point arrow indicates the hypothesized transfer of hyperpolarization via an unknown receptor on the VSMC.
In conclusion, the hallmark of this study was the identification of the major signalling cascades that mediate endothelium-dependent vasodilation in the mouse ophthalmic artery which, were previously uncharacterized. Although the findings emerging from this experimental study do not fully account for the precise molecular mechanisms underlying the observed vasodilation in vitro, the current elucidation of EDHF mechanisms in mouse ophthalmic artery assigns a pivotal platform for the use of mice to further explore the functional relevance of specific CYP450 and LOX metabolites in mediating ACh-induced vasodilation, as well as the existence of a potential putative, ‘novel’ receptor on the endothelial cells that mediates the efflux of K+ remains to be determined in this vascular bed. This study also addressed the potential therapeutic target(s) for future translational applications in human ocular diseases. It will be interesting to determine whether the contribution of the specific potassium ion channels outlined here in the mice ophthalmic artery could also play similar roles in the human ophthalmic circulation, particularly in pathological conditions when NO synthesis is impaired.
Materials and Methods
Experimental animals
This study was approved by the Animal Care Committee of Rhineland-Palatinate, Germany, and animal care conformed to the institutional guidelines and ‘The Association for Research in Vision and Ophthalmology’ (ARVO) statement for the use of animals in ophthalmic and vision research. Mice were treated according to the EU Directive 2010/63/EU for animal experiments. Male C57BL/6J mice (The Jackson Laboratory, Bar Harbour, ME, USA) aged 3 to 7 months old were used for the experiments. Animals were housed under standard conditions (temperature 23 ± 2 °C, humidity range 55 ± 10% and 12 h light/dark cycles), and had access to standard mouse chow and water ad libitum.
Drugs
The following drugs were used in this experiment: Nω-nitro L-arginine methyl ester (L-NAME), indomethacin, acetylcholine hydrochloride (ACh), phenylephrine, 1H-(1, 2, 4) oxadiazole (4, 3-alpha) quinoxaline-1-one (ODQ), catalase, baicalein 18 alpha-glycyrrhetinic acid (18α-GA), ouabain, glibenclamide, barium chloride (BaCl2), and psora-4 [5-(4-Phenylbutoxy)psoralen] (all purchased from Sigma-Aldrich Chemie GmbH, Steinheim, Germany), 17-octadecynoic acid (17-ODYA) and 1-[(2-chlorophenyl) Fdiphenylmethyl]-1H-pyrazole (TRAM-34) (Tocris Bioscience, Bristol, UK), iberiotoxin (IbTX), charybdotoxin (ChTX) and apamin (AnaSpec Inc., Fremont, CA, USA), margatoxin (MgTX), maurotoxin (MTX), α- and β-dendrotoxin (α- and β- DTX) (Alomone Labs, Jerusalem, Israel). Indomethacin, ODQ, 17-ODYA, baicalein, glibenclamide and TRAM-34 were dissolved in dimethyl sulfoxide (DMSO). DMSO at ≤ 0.2% (v/v) did not influence vascular reactivity to agonists and antagonists tested, as described elsewhere73. 18α-GA was dissolved in chloroform: ethanol (2:3) according to the manufacturer’s instructions and this solvent mixture did not affect the vasoreactivity (personal observation). Apamin, charybdotoxin, iberiotoxin and L-NAME were dissolved in phosphate buffer saline (PBS), whereas all other drugs were dissolved in distilled water.
Vascular preparation and reactivity studies
Mice were sacrificed by CO2 inhalation, and the eyes were rapidly removed and placed in cold Krebs-Henseleit buffer composed of 118.3 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 11 mM glucose (Carl Roth GmbH, Karlsruhe, Germany). The ophthalmic arteries were carefully isolated and cleaned of surrounding connective tissues using fine-point tweezers under a dissecting microscope. Arterial segments were then placed in an organ bath with ice-cold Krebs-Henseleit buffer, cannulated onto two glass micropipettes and secured with 10–0 nylon monofilament suture. Vessels were pressurized via these micropipettes to 50 mm Hg under no-flow conditions using two reservoirs filled with Krebs-Henseleit buffer. The ophthalmic artery was equilibrated for 30–40 minutes before the commencement of the experiments. During this equilibration period, the vessels developed a stable spontaneous myogenic tone by constricting to ~86 to 81% of the initial arterial luminal diameter measured immediately after pressurization to 50 mmHg, as described elsewhere24. Video sequences were captured to a personal computer using a video camera mounted on an inverted microscope for off-line analysis. The organ bath was continuously circulated with Krebs solution maintained at 37 °C and pH 7.4 and, aerated with 95% O2 and 5% CO2. A minimum 50% vasoconstriction from the resting diameter in response to 100 mM K+ solution was used as a criterion to assess vessel viability26.
In some experiments, the endothelium was mechanically removed by rubbing the luminal surface of the arteries with a human hair, as described previously26. Next, arteries were preconstricted to 70–50% of the initial vessel diameter by titrating the α1-adrenoceptor agonist phenylephrine, and concentration-response curves to ACh (10−9–10−4 M) was obtained by cumulative application of ACh to the circulating bath solution. The pre-treatment of the arteries with L-NAME slightly constricted the vessels and in this circumstance, the phenylephrine concentration was adjusted to reach a similar preconstriction level in all experiments. All reported drug concentrations refer to final molar concentrations in the organ bath.
Experimental Protocols
Protocol 1: The role of endothelium in acetylcholine-induced vasodilation
To test whether ACh-induced responses were completely endothelium-dependent in preconstricted ophthalmic arteries of the C57BL/6J genotype, endothelium-intact and endothelium-denuded arteries were stimulated with ACh (10−4 M) and with the endothelium-independent NO donor, sodium nitroprusside (SNP, 10−4 M) to ensure that smooth muscle reactivity was not affected by endothelium removal74.
Protocol 2: Contribution of NO and cyclooxygenase (COX) metabolites to ACh- induced vasodilation
To assess the role of NO and prostanoids in mediating ophthalmic artery vasodilation, responses of arteries to cumulative application of ACh (10−9–10−4 M) were tested before and after incubation (30 min) with the non-isoform selective NOS inhibitor, L-NAME (10−4 M) or COX inhibitor, indomethacin (10−5 M). Similarly, responses of arteries to cumulative application of ACh were tested before and after treatment (30 min) with soluble guanylate cyclase (sGC) inhibitor, ODQ (10−5 M). Arteries were preconstricted with phenylephrine after the incubation with blockers.
Protocol 3: Contribution of EDHFs- mediated vasodilator response to ACh
To investigate the contribution of putative EDHFs to cholinergic vasodilation, responses of ophthalmic artery to ACh (10−9–10−4 M) were tested before and after 30 minutes of incubation with the following inhibitors alone or in combinations: 17-ODYA (10−4 M), a suicide substrate inhibitor of both ω- hydroxylation and epoxygenation of AA via the CYP450 pathway; baicalein (10−5 M), a specific inhibitor of 12/15-lipoxygenase (12/15-LOX); catalase (1000 units/ml), a hydrogen peroxide (H2O2) inhibitor; and 18α-GA (3 × 10−5 M), a gap junction uncoupler. Both L-NAME (10−4 M) and indomethacin (10−5 M) were present in the organ bath in addition to the inhibitors to prevent the formation of NO and prostanoids, respectively.
Protocol 4: Contribution of calcium-activated potassium channels (K Ca ) and voltage-gated potassium channels (K v ) to ACh-induced vasodilation
To characterize the KCa that mediate ACh-induced dilator reactivity, ophthalmic arteries were pre-treated with the combination of following agents: Apamin (10−7 M), a specific blocker of the small conductance KCa (SKCa) and ChTX (10−7 M), an inhibitor of both intermediate conductance KCa (IKCa) and big conductance KCa (BKCa). Due to the limited specificity of ChTX, which also blocks some of the voltage-gated channels34, highly selective KCa blocker combinations were employed, as follows: IbTX (10−7 M), a selective BKCa blocker (Maxi KCa) and TRAM-34 (10−6 M), a specific blocker of the IKCa60. The role of specific Shaker-related type 1 Kv channels was evaluated employing blockers with varying sensitivity and specificity for the different Kv channels: MgTX (10−8 M), α- and β-DTX (5 × 10−8M) and MTX (5 × 10−8M).
Protocol 5: Contribution of potassium channels (Kir, and KATP) and sodium-potassium pump (Na+/K+− ATPase) to ACh- induced vasodilation
Depending on the channel, pump or enzyme targeted, vasodilatory responses of the vessels to ACh (10−9–10−4M) were tested before and after 30 minutes incubation with the following blockers: BaCl2 (10−5 M), an inward rectifier (Kir) channel blocker; ouabain (10−4 M) a Na+/K+- ATPase inhibitor and glibenclamide (10−5 M), a KATP channel inhibitor.
Statistical analysis
Data are expressed as mean ± SEM, with n representing the number of animals per group. Changes in vascular responses to various reagents tested are presented as percentage of diameter change from the initial precontraction levels or the percent vasodilator responses as compared to maximal vasodilator response induced by ACh. Statistical comparisons of concentration- response curves were made using the two-way ANOVA for repeated measures followed by Bonferroni post-hoc test. Unpaired two-tailed t-test was used for single-dose responses. The level of significance α was set at 0.05. Graph Pad Prism 6 software (GraphPad Inc., San Diego, USA) was used for statistical analyses.
Immunohistochemistry
To determine the localization of the Kv1.6 channel in the mouse ophthalmic artery, segments of the ophthalmic artery were subjected to immunohistochemistry. The blood vessels were carefully isolated, rinsed in cold Krebs-Henseleit buffer and cryopreserved in Tissue-TEK OCT media (Sakura FineTek Europe, Alphen aan den Rijn, Netherlands) and immediately frozen at −20 °C in a freezer. Transverse cryosections of the arterial rings (8 μm thick) were thaw mounted onto Superfrost Plus slides (Thermo Scientific, Gerhard Menzel GmbH, Braunschweig, Germany), air-dried and stored at −20 °C until use. Prior to immunolabelling, the sections were fixed in 4% paraformaldehyde for 20 min, followed by permeabilization in PBS (0.05 M Na2HPO4, 0.14 M NaCl, pH 7.40) containing 0.1% Triton X-100 (TX). Sections were then blocked with PBS-TX containing 1% BSA and 10% normal goat serum for 30 min followed by overnight incubation with the primary antibody diluted at 1:50 at 4 °C. The rabbit polyclonal Kv1.6 antibody (APC-003, Alomone Labs, Jerusalem, Israel) was generated against a glutathione S-transferase (GST) fusion protein corresponding to residues 463–530 of the rat Kv1.6 protein. After overnight incubation, slides were rinsed in PBS and incubated with peroxidase conjugated polyclonal goat anti-rabbit IgG, H & L chain specific secondary antibody (Calbiochem, San Diego, CA, USA) at 1:200 for 1 h at room temperature. Negative control sections were incubated with blocking media and the secondary antibody. Sections were extensively rinsed to remove unbound antibody and the detection of antibody binding was carried out with Vector® NovaRED™ Substrate Kit for peroxidase (Vector Laboratories, Burlingame, CA, USA). Finally, slides were mounted and cover-slipped.
Additional Information
How to cite this article: Manicam, C. et al. The Gatekeepers in the Mouse Ophthalmic Artery: Endothelium-Dependent Mechanisms of Cholinergic Vasodilation. Sci. Rep. 6, 20322; doi: 10.1038/srep20322 (2016).
Supplementary Material
Acknowledgments
The authors thank Dr. Stephen Peters, M.D. the Director of Surgical Pathology, University Hospital Rutgers, New Jersey School of Medicine and Mr. Natarajan Perumal, M.Sc from the Department of Ophthalmology, University Medical Center of the Johannes Gutenberg University Mainz for their critical appraisal of the manuscript. C.M. was funded by the Center for Translational Vascular Biology (CTVB) of the University Medical Center Mainz.
Footnotes
Author Contributions C.M. participated in the design of the study, performed the experiments, analysed and interpreted the data, and wrote the manuscript. J.S. carried out the experiments and analysed the data. J.S. will use the data of the experiments to which she contributed as a basis of her doctoral thesis in human medicine at the Johannes Gutenberg University Mainz. C.B. helped with the immunostaining and helped draft the manuscript. F.H.G. contributed essential materials, reagents and analysis tools and helped draft the manuscript. N.P. contributed essential materials, reagents and analysis tools and helped draft the manuscript. A.G. designed the study, analysed and interpreted the data and helped draft the manuscript. All authors read and approved the final manuscript.
References
- Pascolini D. & Mariotti S. P. Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 96, 614–618 (2012). [DOI] [PubMed] [Google Scholar]
- Behl T., Kaur I., Goel H. & Pandey R. K. Diabetic nephropathy and diabetic retinopathy as major health burdens in modern era. WJPPS 3, 370–387 (2014). [Google Scholar]
- Moore D., Harris A., Wudunn D., Kheradiya N. & Siesky B. Dysfunctional regulation of ocular blood flow: A risk factor for glaucoma? Clin. Ophthalmol. 2, 849–861 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir E. R., Renteria R. C. & Duong T. Q. Reduced ocular blood flow as an early indicator of diabetic retinopathy in a mouse model of diabetes. Invest. Ophthalmol. Vis. Sci. 53, 6488–6494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshani Y. et al. Impaired ocular blood flow regulation in patients with open-angle glaucoma and diabetes. Clin. Experiment. Ophthalmol. 40, 697–705 (2012). [DOI] [PubMed] [Google Scholar]
- Campion E. W., Biousse V. & Newman N. J. Ischemic Optic Neuropathies. N. Engl. J. Med. 372, 2428–2436 (2015). [DOI] [PubMed] [Google Scholar]
- Salomone S. et al. Regulation of vascular tone in rabbit ophthalmic artery: cross talk of endogenous and exogenous gas mediators. Biochem. Pharmacol. 92, 661–668 (2014). [DOI] [PubMed] [Google Scholar]
- Schmetterer L. & Polak K. Role of nitric oxide in the control of ocular blood flow. Prog. Retin. Eye Res. 20, 823–847 (2001). [DOI] [PubMed] [Google Scholar]
- Koss M. C. Functional role of nitric oxide in regulation of ocular blood flow. Eur. J. Pharmacol. 374, 161–174 (1999). [DOI] [PubMed] [Google Scholar]
- Brandes R. P. et al. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. P. Natl. Acad. Sci. USA 97, 9747–9752 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K. et al. Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br. J. Pharmacol. 161, 1722–1733 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haefliger I. O., Flammer J. & Luscher T. F. Nitric oxide and endothelin-1 are important regulators of human ophthalmic artery. Invest. Ophthalmol. Vis. Sci. 33, 2340–2343 (1992). [PubMed] [Google Scholar]
- Roufail E., Stringer M. & Rees S. Nitric oxide synthase immunoreactivity and NADPH diaphorase staining are co-localised in neurons closely associated with the vasculature in rat and human retina. Brain Res. 684, 36–46 (1995). [DOI] [PubMed] [Google Scholar]
- Wang Y., Okamura T. & Toda N. Mechanisms of acetylcholine-induced relaxation in dog external and internal ophthalmic arteries. Exp. Eye Res. 57, 275–281 (1993). [DOI] [PubMed] [Google Scholar]
- Yamamoto R., Bredt D. S., Snyder S. H. & Stone R. A. The localization of nitric oxide synthase in the rat eye and related cranial ganglia. Neuroscience 54, 189–200 (1993). [DOI] [PubMed] [Google Scholar]
- Yao K., Tschudi M., Flammer J. & Luscher T. F. Endothelium-dependent regulation of vascular tone of the porcine ophthalmic artery. Invest. Ophthalmol. Vis. Sci. 32, 1791–1798 (1991). [PubMed] [Google Scholar]
- Benedito S., Prieto D., Nielsen P. J. & Nyborg N. C. Role of the endothelium in acetylcholine-induced relaxation and spontaneous tone of bovine isolated retinal small arteries. Exp. Eye Res. 52, 575–579 (1991). [DOI] [PubMed] [Google Scholar]
- Chen Z., Gu Q., Kaufman P. L. & Cynader M. S. Histochemical mapping of NADPH-diaphorase in monkey and human eyes. Curr. Eye Res. 17, 370–379 (1998). [DOI] [PubMed] [Google Scholar]
- Toda N., Toda M., Ayajiki K. & Okamura T. Cholinergic nerve function in monkey ciliary arteries innervated by nitroxidergic nerve. Am. J. Physiol. 274, H1582–1589 (1998). [DOI] [PubMed] [Google Scholar]
- Mori A. et al. Stimulation of prostanoid IP and EP(2) receptors dilates retinal arterioles and increases retinal and choroidal blood flow in rats. Eur. J. Pharmacol. 570, 135–141 (2007). [DOI] [PubMed] [Google Scholar]
- Tare M., Parkington H. C. & Coleman H. A. EDHF, NO and a prostanoid: hyperpolarization-dependent and -independent relaxation in guinea-pig arteries. Br. J. Pharmacol. 130, 605–618 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gericke A. et al. Contribution of nitric oxide synthase isoforms to cholinergic vasodilation in murine retinal arterioles. Exp. Eye Res. 109, 60–66 (2013). [DOI] [PubMed] [Google Scholar]
- Laspas P. et al. Role of nitric oxide synthase isoforms for ophthalmic artery reactivity in mice. Exp. Eye Res. 127, 1–8 (2014). [DOI] [PubMed] [Google Scholar]
- Gericke A. et al. Functional role of alpha1-adrenoceptor subtypes in murine ophthalmic arteries. Invest. Ophthalmol. Vis. Sci. 52, 4795–4799 (2011). [DOI] [PubMed] [Google Scholar]
- Gericke A. et al. Cholinergic responses of ophthalmic arteries in M3 and M5 muscarinic acetylcholine receptor knockout mice. Invest. Ophthalmol. Vis. Sci. 50, 4822–4827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gericke A. et al. Role of the M3 muscarinic acetylcholine receptor subtype in murine ophthalmic arteries after endothelial removal. Invest. Ophthalmol. Vis. Sci. 55, 625–631 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iesaki T., Gupte S. A., Kaminski P. M. & Wolin M. S. Inhibition of guanylate cyclase stimulation by NO and bovine arterial relaxation to peroxynitrite and H2O2. Am. J. Physiol. Heart Circ. Physiol. 277, H978–H985 (1999). [DOI] [PubMed] [Google Scholar]
- Park S. W. et al. Hydrogen peroxide induces vasorelaxation by enhancing 4-aminopyridine-sensitive Kv currents through S-glutathionylation. Pflug. Arch. Eur. J Phy. 467, 285–297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanheel B. & Van de Voorde J. EDHF and residual NO: different factors. Cardiovasc. Res. 46, 370–375 (2000). [DOI] [PubMed] [Google Scholar]
- Garland C. J., Hiley C. R. & Dora K. A. EDHF: spreading the influence of the endothelium. Br. J. Pharmacol. 164, 839–852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow S. L. Factors, fiction and endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 31, 563–570 (2004). [DOI] [PubMed] [Google Scholar]
- Bryan R. M. Jr, You J., Golding E. M. & Marrelli S. P. Endothelium-derived hyperpolarizing factor: a cousin to nitric oxide and prostacyclin. Anesthesiology 102, 1261–1277 (2005). [DOI] [PubMed] [Google Scholar]
- Shimokawa H. et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc. Pharm. 28, 703–711 (1996). [DOI] [PubMed] [Google Scholar]
- Krummen S., Falck J. R. & Thorin E. Two distinct pathways account for EDHF-dependent dilatation in the gracilis artery of dyslipidaemic hApoB +/ +mice. Br. J. Pharmacol. 145, 264–270 (2005). [Google Scholar]
- Popp R., Brandes R. P., Ott G., Busse R. & Fleming I. Dynamic modulation of interendothelial gap junctional communication by 11,12-epoxyeicosatrienoic acid. Circ. Res. 90, 800–806 (2002). [DOI] [PubMed] [Google Scholar]
- Sandow S. L. et al. What’s Where and Why at a Vascular Myoendothelial Microdomain Signalling Complex. Clin. Exp. Pharmacol. Physiol. 36, 67–76 (2009). [DOI] [PubMed] [Google Scholar]
- Campbell W. B. & Harder D. R. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ. Res. 84, 484–488 (1999). [DOI] [PubMed] [Google Scholar]
- Fleming I. Myoendothelial Gap Junctions The Gap Is There, but Does EDHF Go Through It? Circ. Res. 86, 249–250 (2000). [DOI] [PubMed] [Google Scholar]
- Imig J. D. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol. Rev. 92, 101–130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit C. & Griffith T. M. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers. Arch. Eur. J. Phy. 459, 897–914 (2010). [DOI] [PubMed] [Google Scholar]
- Griffith T. M., Chaytor A. T. & Edwards D. H. The obligatory link: role of gap junctional communication in endothelium-dependent smooth muscle hyperpolarization. Pharmacol. Res. 49, 551–564 (2004). [DOI] [PubMed] [Google Scholar]
- Needleman P., Turk J., Jakschik B. A., Morrison A. R. & Lefkowith J. B. Arachidonic acid metabolism. Annu. Rev. Biochem. 55, 69–102 (1986). [DOI] [PubMed] [Google Scholar]
- Campbell W. B. & Falck J. R. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension 49, 590–596 (2007). [DOI] [PubMed] [Google Scholar]
- Campbell W. B. & Gauthier K. M. Inducible endothelium-derived hyperpolarizing factor (iEDHF): Role of the 15-lipoxygenase-EDHF pathway. J Cardiovasc. Pharmacol. 61, 176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G., Feletou M. & Weston A. H. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers. Arch. Eur. J. Phy. 459, 863–879 (2010). [DOI] [PubMed] [Google Scholar]
- Xu J. et al. Inhibition of 12/15-lipoxygenase by baicalein induces microglia PPARβ/δ: a potential therapeutic role for CNS autoimmune disease. Cell Death Dis. 4, e569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Maher P. & Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463 (1997). [DOI] [PubMed] [Google Scholar]
- Yang H., Zhuo J. M., Chu J., Chinnici C. & Pratico D. Amelioration of the Alzheimer’s disease phenotype by absence of 12/15-lipoxygenase. Biol. Psychiatry 68, 922–929 (2010). [DOI] [PubMed] [Google Scholar]
- Xu X., Zhang X. A. & Wang D. W. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv. Drug. Deliv. Rev. 63, 597–609 (2011). [DOI] [PubMed] [Google Scholar]
- Thollon C. et al. Alteration of endothelium-dependent hyperpolarizations in porcine coronary arteries with regenerated endothelium. Circ. Res. 84, 371–377 (1999). [DOI] [PubMed] [Google Scholar]
- Edwards G., Dora K., Gardener M., Garland C. & Weston A. K+is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396, 269–272 (1998). [DOI] [PubMed] [Google Scholar]
- Tamai K., Suzuki H., Hashitani H., Shirai S. & Ogura Y. Effects of K+ Channel Blockers on Acetylcholine-Induced Vasodilation in Guinea-pig Choroid. Exp. Eye Res. 69, 85–90 (1999). [DOI] [PubMed] [Google Scholar]
- Corriu C., Feletou M., Canet E. & Vanhoutte P. M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 119, 959–964 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman D. M. & Nelson M. T. Potassium ions as vasodilators - Role of inward rectifier potassium channels. Circ. Res. 88, 132–133 (2001). [DOI] [PubMed] [Google Scholar]
- Filosa J. A. et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 9, 1397–1403 (2006). [DOI] [PubMed] [Google Scholar]
- McNeish A. J., Wilson W. S. & Martin W. Dominant role of an endothelium-derived hyperpolarizing factor (EDHF)-like vasodilator in the ciliary vascular bed of the bovine isolated perfused eye. Br. J. Pharmacol. 134, 912–920 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br. J. Pharmacol. 156, 545–562 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers R. H., Todd J. Jr. & Webb R. C. Regional heterogeneity in acetylcholine-induced relaxation in rat vascular bed: role of calcium-activated K+ channels. Am. J. Physiol. Heart Circ. Physiol. 291, H216–222 (2006). [DOI] [PubMed] [Google Scholar]
- Sandow S. L. & Hill C. E. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 86, 341–346 (2000). [DOI] [PubMed] [Google Scholar]
- Eichler I. et al. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+−channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 138, 594–601 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Kurtz G., Vianna-Jorge R., Pereira B. F., Garcia M. L. & Kaczorowski G. J. Peptidyl inhibitors of Shaker-type Kv1 channels elicit twitches in guinea pig ileum by blocking Kv1.1 at enteric nervous system and enhancing acetylcholine release. J. Pharmacol.Exp. Ther. 289, 1517–1522 (1999). [PubMed] [Google Scholar]
- Garcia-Calvo M. et al. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 268, 18866–18874 (1993). [PubMed] [Google Scholar]
- Bartok A. et al. Margatoxin is a non-selective inhibitor of human Kv1.3 K+ channels. Toxicon 87, 6–16 (2014). [DOI] [PubMed] [Google Scholar]
- Garcia M. L., Knaus H. G., Munujos P., Slaughter R. S. & Kaczorowski G. J. Charybdotoxin and its effects on potassium channels. Am. J. Physiol. 269, C1–10 (1995). [DOI] [PubMed] [Google Scholar]
- Joseph B. & George J. Scorpion toxins and its applications. IJTPR 4, 57–61 (2012). [Google Scholar]
- Kiernan M. F., Barrie A., Szkolar J., Mills T. A. & Wareing M. Functional evidence for oxygen-sensitive voltage-gated potassium channels in human placental vasculature. Placenta 31, 553–555 (2010). [DOI] [PubMed] [Google Scholar]
- Novakovic A. et al. The mechanism of endothelium-independent relaxation induced by the wine polyphenol resveratrol in human internal mammary artery. J. Pharmacol. Sci. 101, 85–90 (2006). [DOI] [PubMed] [Google Scholar]
- Gunasekaran R. et al. Exposure to cerebrospinal fluid of sporadic amyotrophic lateral sclerosis patients alters Nav1.6 and Kv1.6 channel expression in rat spinal motor neurons. Brain Res. 1255, 170–179 (2009). [DOI] [PubMed] [Google Scholar]
- Carrisoza-Gaytan R., Salvador C., Diaz-Bello B. & Escobar L. I. Differential expression of the Kv1 voltage-gated potassium channel family in the rat nephron. J Mol Histol 45, 583–597 (2014). [DOI] [PubMed] [Google Scholar]
- Chu X. et al. Hypoxia suppresses Kv1.5 channel expression through endogenous 15-HETE in rat pulmonary artery. Prostaglandins Other Lipid Mediat. 88, 42–50 (2009). [DOI] [PubMed] [Google Scholar]
- Firth A. L. et al. Hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Ann. N. Y. Acad. Sci. 1177, 101–111 (2009). [DOI] [PubMed] [Google Scholar]
- Wang J., Weigand L., Wang W., Sylvester J. T. & Shimoda L. A. Chronic hypoxia inhibits Kv channel gene expression in rat distal pulmonary artery. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L1049–1058 (2005). [DOI] [PubMed] [Google Scholar]
- Minareci E. & Sadan G. An evaluation of vardenafil as a calcium channel blocker in pulmonary artery in rats. Indian J. Pharmacol. 46, 185–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka T., Hein T. W., Yoshida A. & Kuo L. Resveratrol, a component of red wine, elicits dilation of isolated porcine retinal arterioles: role of nitric oxide and potassium channels. Invest. Ophthalmol. Vis. Sci. 48, 4232–4239 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.