

Abstract
Background
The existence of moderate to high levels of linkage disequilibrium (LD) between genetic markers and quantitative trait loci (QTL) affecting traits of interest is fundamental for the success of genome-wide association (GWAS) and genomic selection (GS) studies. Knowledge about the extent and the pattern of LD in livestock populations is essential to determine the density of single nucleotide polymorphisms (SNP) required for accurate GWAS and GS. Moreover, observed LD is related to historical effective population sizes (Ne), and can provide insights into the genetic diversity history of populations. Estimates of the consistency of linkage phase across breeds (RH,B) can be used to determine if there is sufficient relationship to use pooled reference populations in multi-breed GS programs. The objective of this study was to estimate LD levels, persistence of phase and effective population size in Hereford and Braford cattle populations sampled in Brazil.
Results
Mean LD estimates, measured using the squared correlation of alleles at two loci (r2), obtained between adjacent SNP across all chromosomes were 0.21 ± 0.27 for Herefords (391 samples with 41,241 SNP) and 0.16 ± 0.22 for Brafords (2044 samples and 41,207 SNP). Estimated r2 was > 0.2 and 0.3, respectively, for 34 and 25 % of adjacent markers in Herefords, and 26 and 17 % in Brafords. Estimated Ne for Brafords and Herefords at the current generation was 220 and 153 individuals, respectively. The two breeds demonstrated moderate to strong persistence of phase at all distances (RH,B = 0.53 to 0.97). The largest phase correlations were found in the 0 to 50 Kb bins (RH,B = 0.92 to 0.97). Estimated LD decreased rapidly with increasing distance between SNP, however, useful linkage for GWAS and GS (r2 > 0.2) was found spanning to ~50 Kb.
Conclusions
Panels containing about 50,000 and 150,000 SNP markers are necessary to detect minimal levels of LD between adjacent markers that would be useful for GWAS and GS studies to Hereford and Braford breeds, respectively. Markers are expected to be linked to the same QTL alleles in distances < 50 Kb in both populations due to observed high persistence of phase levels.
Keywords: Beef cattle, Genetic diversity, Linkage disequilibrium, Quantitative trait loci, Single nucleotide polymorphism
Background
The evolution of molecular biology tools and techniques occurred over the last decades helped unveiling underlying genetic factors and improving rates of genetic gains for economically important traits in livestock. The availability of high-density single nucleotide polymorphisms (SNP) genotyping assays for cattle coupled with advances in computational and statistical methods allowed the generation and use of large amounts of genomic data in genome-wide association (GWAS) and genomic selection (GS) studies for production-relevant traits [1]. These advancements created new opportunities to identify and select animals with superior genetic merit, while reducing generation intervals and overall associated costs [2, 3].
Existing genome-wide linkage disequilibrium (LD) levels are usually affected by genetic (selection, mutation, drift, migration, and non-random mating) and non-genetic (marker ascertainment bias) factors [4–6] and they can reflect historical effective population sizes and rates of recombination in a population [7]. Studies to understand LD levels and structure in livestock species are necessary for revealing diversity levels among breeds and detecting regions of genome that have been historically subjected to different selection pressures [8, 9]. Knowledge about historical effective population sizes is also important to determine optimal selection pressures [10] for achieving breeding goals while maintaining acceptable levels of genetic diversity in breeding populations [11]. Estimates of linkage phase consistency across breeds and populations are also essential for determining the potential success of using data from pooled reference populations for multi-breed genomic evaluation and selection programs [12].
Hereford and Braford cattle are important breeds for beef production in southern Brazil, where subtropical climates are observed with average low temperatures in winter months. Local climate conditions naturally control the incidence of tropical ectoparasites such as the bovine tick (Rhipicephalus (Boophilus) microplus) however, infestation peaks can be observed sporadically, raising risks of losses associated with tick-borne diseases [13–15]. Development of strategies to implement GS methods for genetic improvement of tick-resistance in these breeds are underway [16], in addition to studies focusing on unrevealing biological mechanisms underlying this trait [17]. Minimal levels of linkage disequilibrium between causative variants and genetic markers are fundamental for performing effective GWAS and GS studies, since these approaches rely on the non-random associations between markers and functional mutations affecting traits of interest [18, 19]. Successful experimental designs for performing GWAS and GS with Brazilian Hereford and Braford cattle will be highly dependent of the right choice of marker density, which in term is dependent on the estimated levels of LD across the genome [20].
The objective of this study was to estimate linkage disequilibrium levels at varying SNP densities, persistence of phase and effective population sizes in Brazilian Hereford and Braford cattle populations.
Methods
Animal welfare
All experimental procedures involving live animals were approved by the Committee for Ethics in Animal Experimentation from the Federal University of Pelotas (Comissão de Ética em Experimentação Animal, Pelotas, Rio Grande do Sul, Brazil; process number 6849).
Sample collection and genotyping
Samples from a total of 391 Hereford and 2079 Braford (with a breed composition ranging between ½ Hereford + ½ Zebu and ¾ Hereford + ¼ Zebu) cattle born between 2008 and 2010 in commercial farms associated with the Delta G Connection Genetic Improvement Consortium [21] were used in this study. DNA was extracted from blood samples from FTA cards or from cryopreserved semen. Genotyping of all samples was performed with Illumina BovineSNP50v2 (50 K) BeadChip (Illumina Inc., San Diego, USA). Additionally, data from 40 bulls (17 Hereford and 23 Braford) genotyped with Illumina High-Density (HD) Bovine BeadChip Array (Illumina Inc., San Diego, USA) were also included in the final dataset. Genotype calling and initial data quality control (QC) were performed using GenomeStudio software (Illumina Inc. San Diego, CA), according to manufacturer's protocols. Genotypes with a GenCall Score < 0.15 were set as missing genotypes.
Data quality control
Additional QC was performed with R snpStats package [22, 23]. Samples with call rates < 0.90, heterozygosity deviations > 3.0 standard deviations, conflicts between declared and genotype-based sex, and duplicated genotypes with different sample identification were removed from the final dataset. Only SNP located on autosomes (BTA) were considered in further analyses. SNP with call rates < 0.98, minor allele frequencies (MAF) < 0.03 and highly significant deviations (P < 10-6) from Hardy-Weinberg Equilibrium (HWE) were also excluded. Moreover, whenever multiple SNP were observed in the same physical map position, only one SNP with highest MAF was retained. The high-density (HD) panel was filtered to select only the SNP also present in the 50K panel.
Haplotype reconstruction and phasing
Haplotype reconstruction and imputation of sporadically missing genotypes (0.39 %) were carried out using FImpute 2.0 [24]. Expectation-Maximization algorithm employed initially estimates the most probable haplotypes considering observed genotypes, using pedigree relationship information. Subsequently, the program performs genotype imputation using a haplotype search based on a sliding window approach, walking along each chromosome and using overlapping windows to reconstruct haplotypes [25].
Linkage disequilibrium analysis
Linkage disequilibrium was calculated as pairwise r2 [26], which relies on the allele phase information at the gametic level. Considering two marker loci (A and B), each one with two alleles (A1, A2, B1 and B2), the frequency of alleles in the population can be denoted pA1, pA2, pB1 and pB2, and the frequency of haplotypes with allele 1 at marker locus A and allele 1 at locus B, for example, denoted pA1B1. Following Hill and Roberson [26],
| 1 |
For each population, LD values between all pairs of SNP of all chromosomes were binned according to pairwise physical distances into intervals of 100 Kb starting from 0 up to 10 Mb. Average values of r2 were calculated for each bin.
To evaluate the feasibility of successfully using sparser marker panels in GWAS and GS studies, average r2 between adjacent markers was calculated for different marker densities, sequentially removing SNP from the total dataset using every second, fourth, fifth, sixth, seventh and 14th marker (using, respectively, 50, 25, 20, 17, 14 and 7 % of available SNP). Linkage disequilibrium estimates were calculated according to Badke et al. [27], using R scripts [23] available at https://www.msu.edu/~steibelj/JP_files/LD_estimate.html.
Intra and inter chromosomal and breed heterogeneities
To investigate intra and inter chromosomal and breed variation in LD, two analysis of covariance with general linear models were fitted. The following linear model (Equation 2) was used to estimate the effects of physical distance (covariate), chromosome, breed and the chromosome x breed interaction on LD through a total of 82,356 adjacent breed-specific marker pairs:
| 2 |
were was the observed LD over marker distance on chromosome i of breed j, μ is the overall mean of across markers pairs, is the effect of chromosome i, is the effect of breed j, is the regression coefficient on marker distance, is the adjusted marker distance , dk is the observed physical distance for marker pair k, is the average physical distance between markers, and is the residual effect. Distances were log10 transformed in an attempt to linearize the relationship between LD and the log-transformed distance [28].
Additionally, a more complex linear model was fit to all 12,911,174 syntenic SNP pairs to investigate the adjusted mean r2 in a broader range of inter marker distances than that observed in Equation 2, where just adjacent markers were considered. This more comprehensive model can be represented by:
| 3 |
In addition to the variables already described above for Equation 2, here and are the regression coefficients (quadratic and cubic, respectively) on marker distance. Although the log10 transformation of physical distances to linearize the relationship between LD and the log-transformed distances, we decided to fit these higher order coefficients to consider possible deviations of the expected linearity and interaction with breed and chromosome effects (cβ1i, cβ2i, cβ3i, bβ1j, bβ2j, bβ3j, cbβ1ij, cbβ2ij and cbβ3ij terms).
The physical distances of interest to predict r2 using Equation 3 were related to the average inter marker distance values observed in some commercial panels available for cattle genomic selection. The density of the considered panels was 150K (GeneSeek Genomic Profiler HD-150K), 80K (GeneSeek Genomic Profiler HD-80K), 50K (Illumina BovineSNP50v2 BeadChip), 20K (GeneSeek Genomic Profiler LD v2), 8K (Illumina BovineLD v.2 BeadChip) and 3K (Illumina Bovine3k BeadChip). Assuming a bovine genome size of 3.000 Mb [29], the target distance values were calculated dividing the genome length in Mb by the number of markers in each panel. As example, for the 150K panel, we divided 3.000 Mb by 150.000 markers and obtained an average inter marker distance (dk) of 20 Kb. Accordingly, chromosome by breed predicted r2 values were obtained from estimated parameters of Equation 3 at physical inter marker distances of 20, 38, 60, 150, 375 and 1000Kb. All analysis was performed using R package [23].
Analysis of persistence of phase
The degree of phase concordance between the two breeds for pairs of SNP was calculated according to Badke et al. [27] with the following formula:
| 4 |
where RB,H is the correlation of phase between rij(B) in the Braford (B) population and rij(H) in the Hereford (H) population, S(B) and S(H) are the standard deviations of rij(B) and rij(H), respectively, and are the average rij across all SNP i and j within interval p for populations B and H, respectively.
Positive r values are expected when two markers are in LD and show equal linkage phase in both studied populations [30]. Marker pairs were binned according to marker distances (intervals of 100 Kb starting from 0 up to 10 Mb), and average values of RB,H were calculated for each bin, using markers common to both breeds.
Estimation of effective population size
Estimates of LD decay in relation to different SNP distances were used to infer past effective population sizes (Ne) of the two studied cattle breeds. The relationship between r2 and Ne can be expressed by the formula:
| 5 |
where c is the genetic distance between two markers expressed in Morgans [31]. Effective population size was estimated considering each SNP pair located within 100 Mb of the same chromosome, with physical distances between SNP converted to genetic distances, assuming 1 Mb = 1 cM [32, 33]. Because generations are discrete and distances between SNP are continuous, historical effective population size (Net) for a given generation t = 1/2c [34] in the past was assessed by selecting SNP pairs with map distance within corresponding ranges of c values. When applied in t = 1/2c, the resulting t value was rounded to the target generation. For example, r2 of all SNP pairs with distance between 33.3 cM (t = 1.5) and 1 M (t = 0.5) of the same autosomal chromosome were selected to calculate Ne at t = 1. Then, within each bin, the average values of r2 and c were obtained and applied in the formula:
| 6 |
for 0.0 < r2 < 1.0. Longer c ranges were used to define generation bins as we moved further in the past, because they correspond to shorter distances with fewer markers and we wanted to ensure sufficient numbers of SNP pairs for reliably estimating Net within each bin [35]. The actual bins were of one generation for t between 1 and 10, e.g. a range from 0.5 to 1.5 for the current generation; five generations for t between 15 and 100, and of 50 generations for t between 150 and 1000 (see Table 1 for additional details).
Table 1.
Description of generation binning process
| Generation range applied to | Number of generations represented by each bin | Example for first bin | ||
|---|---|---|---|---|
| Generation | Generation range | Corresponding distance range [mid-point] (Morgans) | ||
| 1–10 | 1 | 1 | 0.5 to 1.5 | 0.33 to 1.0 [6.7 × 10-1] |
| 15–100 | 5 | 15 | 12.5 to 17.5 | 0.04 to 0.02857 [3.33 × 10-2] |
| 150–1000 | 50 | 150 | 125 to 175 | 0.004 to 0.00286 [3.33 × 10-3] |
Results
SNP quality control
Data QC excluded 43 samples with call rates < 0.90, 24 samples with heterozygosity deviations > 3.0 standard deviations, eight samples with incorrect sex assignment and eight duplicated genotypes with different sample identification. The final resulting dataset contained 2435 samples (391 Hereford and 2044 Braford). Some samples were excluded for not meeting more than one criteria of QC. Marker QC also removed 4232 SNP with call rates < 0.98, 5712 SNP with MAF < 0.03, and 1342 SNP with highly significant HWE deviations (P < 10-6). Estimated means for call rate, MAF and HWE were 0.998, 0.271, and 0.541, respectively. Additionally, 34 monomorphic markers in the subset of Hereford samples were excluded from subsequent analyses of this breed. The final resulting dataset contained a total of 41,241 autosomal SNP (75.52 %) in Brafords and 41,207 autosomal SNP (75.46 %) in Herefords.
Linkage disequilibrium
Average r2 ± SD between adjacent SNP across all chromosomes was 0.21 ± 0.27 for Hereford and 0.16 ± 0.22 for Braford. The analysis revealed a rapid decrease in LD in both populations with increasing physical distances (Fig. 1). Herefords showed higher LD than Brafords up to a distance of 1780 Kb. For larger distances, Brafords showed a mean of r2 slightly higher than Herefords. Average LD for markers at some distance intervals is presented in Table 2. At distances of 50 Kb, average LD was 0.25 ± 0.29 for Herefords and 0.18 ± 0.24 for Brafords. In Brafords, average r2 decayed faster than in Herefords with the increase of distance, declining to 50 % of its initial value at ~5 Kb whereas in Herefords the same decline was observed at ~50 Kb. Observed LD was > 0.2 and 0.3, respectively, for 34 % (14,010 SNP) and 25 % (10,302 SNP) of adjacent markers in Herefords, and 26 % (10,722 SNP) and 17 % (7011 SNP) in Brafords.
Fig. 1.
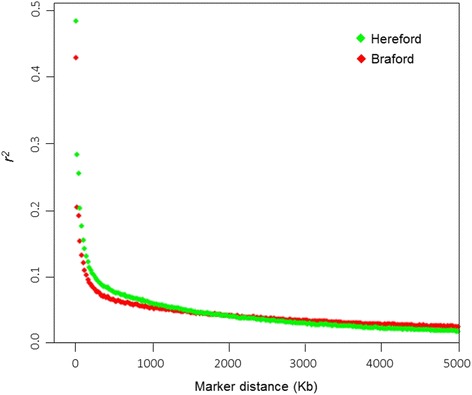
Extent of r 2 as a function of inter-marker distance in Hereford and Braford populations
Table 2.
Average r 2 ± SD between adjacent markers according to inter-marker distances
| Inter-marker distance | Hereford | Braford |
|---|---|---|
| 0–1 Kb | 0.49 ± 0.45 | 0.43 ± 0.39 |
| 1–5 Kb | 0.28 ± 0.35 | 0.20 ± 0.30 |
| 0–20 Kb | 0.29 ± 0.34 | 0.21 ± 0.27 |
| 20–40 Kb | 0.26 ± 0.30 | 0.19 ± 0.24 |
| 5–50 Kb | 0.25 ± 0.29 | 0.18 ± 0.24 |
| 40–60 Kb | 0.20 ± 0.26 | 0.15 ± 0.21 |
| 60–80 Kb | 0.18 ± 0.23 | 0.13 ± 0.19 |
| 50–100 Kb | 0.17 ± 0.23 | 0.13 ± 0.18 |
| 0.1–0.5 Mb | 0.10 ± 0.15 | 0.08 ± 0.12 |
| 0.5–1 Mb | 0.07 ± 0.11 | 0.06 ± 0.09 |
| 2–5 Mb | 0.03 ± 0.05 | 0.03 ± 0.05 |
| 5–10 Mb | 0.01 ± 0.02 | 0.02 ± 0.03 |
Estimated r2 values > 0.2 were observed in the 0 to 60 Kb bins in Herefords (range = 0.20 to 0.49), and in the 0 to 20 Kb bins in Brafords (range = 0.21 to 0.43). Average r2 values > 0.3 were observed in the 0 to 1 Kb bins in Herefords (0.49) and Brafords (0.43). Linkage disequilibrium estimates obtained from sparse maps of markers were low (Table 3). Considering only 50 % of available SNP (a panel with about 20K SNP), observed average r2 decreased from 0.21 to 0.15 in Herefords, and from 0.16 to 0.12 in Brafords. When 20 % of SNP were maintained in the map (using only every fifth SNP and a density similar to the 8K panel), average r2 decreased to 0.09 in Herefords and to 0.08 in Brafords. Similarly, as observed in LD values, the percentage of pairs of markers with r2 values > 0.2 and with average r2 > 0.3 decreased when sparse maps were analyzed (Table 4). Using only every fifth marker (20 % of available SNP), the percentage of markers with r2 > 0.2 decreased from 34 to 15 % in Herefords and from 26 to 10.1 % in Brafords, while the percentage of markers with r2 > 0.3 decreased from 25 to 8.3 % in Herefords, and from 17 to 4.9 % in Brafords.
Table 3.
Average r 2 ± SD for adjacent SNP according to marker panel density
| % SNP kepta | Hereford | Braford | ||
|---|---|---|---|---|
| Average r 2 | Average distance (Kb) | Average r 2 | Average distance (Kb) | |
| 50 % | 0.15 ± 0.21 | 121.80 | 0.12 ± 0.17 | 121.83 |
| 25 % | 0.11 ± 0.16 | 243.57 | 0.09 ± 0.13 | 243.00 |
| 20 % | 0.09 ± 0.14 | 304.22 | 0.08 ± 0.11 | 303.78 |
| 17 % | 0.09 ± 0.14 | 364.97 | 0.07 ± 0.11 | 364.47 |
| 14 % | 0.08 ± 0.13 | 425.83 | 0.07 ± 0.10 | 425.30 |
| 7 % | 0.07 ± 0.10 | 850.22 | 0.06 ± 0.09 | 849.50 |
aIn relation to the total number of SNP obtained for each population after the quality control (41,207 SNP in Hereford and 41,241 SNP in Braford)
Table 4.
Percentage of adjacent SNP with average r 2 > 0.2 and > 0.3 according to marker panel density
| % SNP kepta | Hereford | Braford | ||
|---|---|---|---|---|
| % r 2 > 0.2 | % r 2 > 0.3 | % r 2 > 0.2 | % r 2 > 0.3 | |
| 50 % | 24.49 | 16.41 | 18.33 | 11.09 |
| 25 % | 17.35 | 10.59 | 12.52 | 6.69 |
| 20 % | 15.02 | 8.33 | 10.12 | 4.96 |
| 17 % | 14.80 | 8.03 | 9.98 | 4.58 |
| 14 % | 13.18 | 6.59 | 8.95 | 4.03 |
| 7 % | 8.37 | 3.83 | 6.76 | 2.70 |
aIn relation to the total number of SNP obtained for each population after the quality control (41,207 SNP in Hereford and 41,241 SNP in Braford)
Intra and inter chromosomal heterogeneity and differences between breed in r2
Average distances between SNP in different chromosomes were similar, and average physical distances between adjacent markers on Hereford and Braford autosomes was 61 Kb. For all chromosomes, average r2 between adjacent SNP was larger in Herefords than Brafords (Table 5). Estimated r2 values ranged from 0.16 (BTA23) to 0.26 (BTA6) in Herefords, and from 0.13 (BTA23, BTA28 and BTA29) to 0.20 (BTA6) in Brafords. Squared correlation estimates between adjacent SNP (Equation 2) revealed that physical distance, breed and chromosome have significant effects on r2 (P < 0.001), whereas the interaction between breed and chromosome was not significant. Therefore, predicted marginal (least square) means ± SE of r2 were obtained for each chromosome averaged across breeds, chromosome by breed interactions and adjacent SNP distances (Fig. 2).
Table 5.
Average r 2 ± SD and mean of length distances between adjacent markers in chromosomes
| Chra | Length (Mb) | Hereford | Braford | ||
|---|---|---|---|---|---|
| n SNPb | Average r 2 | n SNP | Average r 2 | ||
| 1 | 161.02 | 2697 | 0.24 ± 0.29 | 2699 | 0.17 ± 0.23 |
| 2 | 137.63 | 2197 | 0.23 ± 0.29 | 2200 | 0.18 ± 0.23 |
| 3 | 125.67 | 2000 | 0.21 ± 0.27 | 2001 | 0.17 ± 0.22 |
| 4 | 120.64 | 1987 | 0.23 ± 0.28 | 1988 | 0.16 ± 0.21 |
| 5 | 124.68 | 1732 | 0.20 ± 0.26 | 1734 | 0.16 ± 0.22 |
| 6 | 119.22 | 2066 | 0.26 ± 0.30 | 2069 | 0.20 ± 0.24 |
| 7 | 112.63 | 1824 | 0.25 ± 0.29 | 1824 | 0.19 ± 0.24 |
| 8 | 116.03 | 1958 | 0.23 ± 0.29 | 1959 | 0.18 ± 0.23 |
| 9 | 105.59 | 1670 | 0.23 ± 0.29 | 1671 | 0.17 ± 0.23 |
| 10 | 104.22 | 1760 | 0.21 ± 0.27 | 1761 | 0.16 ± 0.22 |
| 11 | 107.25 | 1805 | 0.20 ± 0.26 | 1806 | 0.16 ± 0.22 |
| 12 | 91.06 | 1342 | 0.21 ± 0.27 | 1344 | 0.15 ± 0.20 |
| 13 | 84.18 | 1449 | 0.19 ± 0.26 | 1449 | 0.15 ± 0.20 |
| 14 | 84.59 | 1471 | 0.23 ± 0.28 | 1471 | 0.18 ± 0.23 |
| 15 | 85.26 | 1374 | 0.19 ± 0.25 | 1374 | 0.15 ± 0.20 |
| 16 | 81.32 | 1314 | 0.21 ± 0.27 | 1314 | 0.16 ± 0.22 |
| 17 | 75.00 | 1249 | 0.20 ± 0.27 | 1250 | 0.15 ± 0.21 |
| 18 | 65.98 | 1036 | 0.21 ± 0.27 | 1038 | 0.16 ± 0.21 |
| 19 | 64.01 | 1058 | 0.17 ± 0.23 | 1059 | 0.14 ± 0.20 |
| 20 | 72.20 | 1264 | 0.19 ± 0.25 | 1264 | 0.15 ± 0.21 |
| 21 | 71.08 | 1124 | 0.20 ± 0.26 | 1124 | 0.16 ± 0.22 |
| 22 | 61.38 | 1021 | 0.20 ± 0.27 | 1025 | 0.15 ± 0.21 |
| 23 | 52.29 | 838 | 0.16 ± 0.23 | 838 | 0.13 ± 0.19 |
| 24 | 63.40 | 1014 | 0.21 ± 0.27 | 1018 | 0.16 ± 0.21 |
| 25 | 42.77 | 753 | 0.19 ± 0.25 | 755 | 0.15 ± 0.21 |
| 26 | 51.64 | 867 | 0.20 ± 0.27 | 867 | 0.14 ± 0.21 |
| 27 | 45.37 | 763 | 0.17 ± 0.24 | 764 | 0.14 ± 0.20 |
| 28 | 46.22 | 747 | 0.19 ± 0.24 | 747 | 0.13 ± 0.18 |
| 29 | 51.48 | 827 | 0.18 ± 0.25 | 828 | 0.13 ± 0.19 |
aChromosomes
bNumber of SNP
Fig. 2.
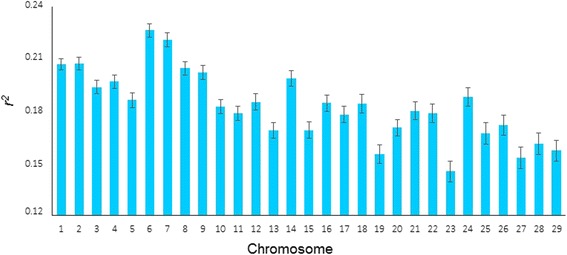
Extent of r 2 ± standard errors by chromosomes in Hereford and Braford populations
Squared correlation estimates considering all SNP pairs (Equation 3) revealed significant effects of linear, quadratic and cubic physical distance coefficients, breed, chromosome, and all interactions (P < 0.001). Predicted r2 values at specific distances for Herefords and Brafords are shown in Fig. 3 for chromosomes with the greatest, lowest and intermediate LD averages (BTA6, BTA23 and BTA10, respectively).
Fig. 3.
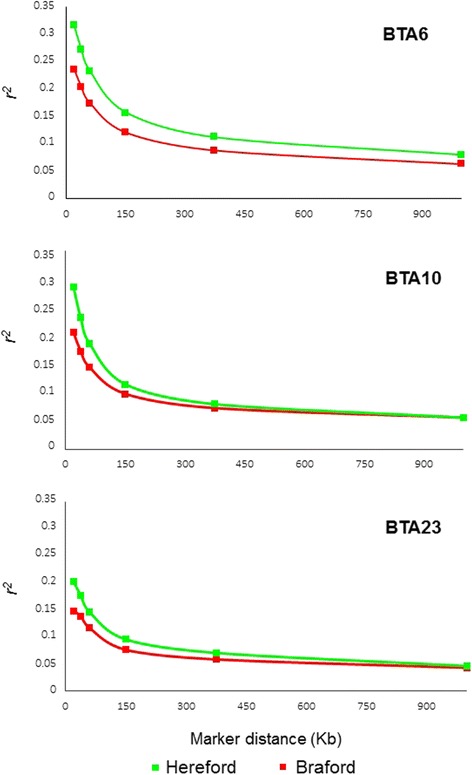
Predicted r 2 as a function of inter-marker distance considering different panels densities. Square symbols pinpoint predicted r 2 at distances of 20, 38, 60, 150, 375 and 1000 Kb corresponding, respectively, to average physical distances expected for panels of 150K, 80K, 50K, 20K, 8K, and 3K equally-spaced markers in chromosomes 6 (BTA6), 10 (BTA10) and 23 (BTA23) for Hereford and Braford populations
Effective population size
Although current estimated effective population size (Fig. 4) for Brafords (Ne = 220) is larger than for Herefords (Ne = 153), different recent Ne trends were observed for the two studied populations. Braford´s decreasing historical Ne trend was reversed and sharply increased in the last two generations, while Herefords had an accelerated Ne decline starting four generations ago. Consequently, the relative positions of the breed were changed, restoring the larger past effective size of Braford compared to Hereford, which was observed up to about thirty generations ago, when respective values were 315 and 309 for the two populations.
Fig. 4.
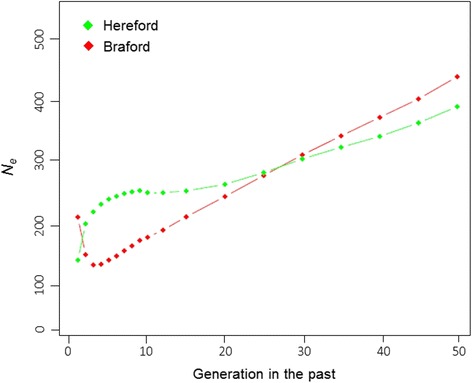
Estimated N e as a function of generation in the past in Hereford and Braford populations
Persistence of phase
Moderate to strong persistence of phase at all distances (RB,H = 0.53 to 0.97) were observed for both breeds (Fig. 5). Phase correlations decreased rapidly with increasing distances between SNP, as was similarly observed for average r2. For markers < 50 Kb apart mean estimated RB,H was 0.92, decreasing to 0.74 and 0.53 at marker distances of 1 and 5 Mb, respectively. Marker phase correlation values of RB,H > 0.9 were found in the 0 to 50 Kb bins (RB,H = 0.92 to 0.97), and the proportion of SNP with reversed r signs was lower, ranging from 5 % at 1 Kb to 34 % at 10 Mb distance.
Fig. 5.
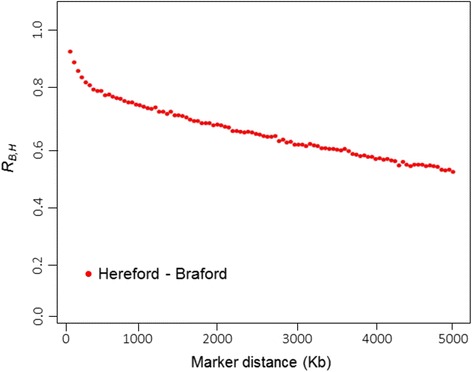
Correlation of phase (R B,H) between Hereford and Braford populations for SNP pairs at varying distances
Discussion
Numerous studies have been conducted to estimate the pattern and extent of LD in different domestic animal species [8, 27, 28, 36]. Obtained results are essential for fine-tuning experimental designs to increase GWAS and GS efficiency and accuracies in studied populations, and can therefore have great impact in realized rates of genetic progress in economically important traits. Linkage disequilibrium patterns and scale within and between populations/breeds can be influenced by several factors such as marker allele frequencies, selection history, population structure and effective size, marker type and density, as well as which LD measure is used [9, 32, 37, 38]. Therefore, these factors should be considered when conclusions are drawn from comparisons between different studies. The choice of r2 [23] to estimate LD in Hereford and Braford data was based on the parameter´s lower sensitivity to variations in allele frequency [38] and sample size [39, 40], when compared to D and D´.
Robust ascertainment bias towards informativeness in taurine breeds has been observed in the 50K panel [41] and has to be considered when interpreting population genetics inferences based on LD estimates. Using a high density marker panel (446,985 SNP) and 795 genotyped Nelore steers, Espigolan et al. [42] observed an overall average r2 of 0.17.
Lower mean LD estimates were also reported for B. indicus breeds by Villa-Angulo et al. [43] when analyzing a dataset with 31,857 SNP derived from the 50K panel from 487 animals of seventeen taurine and indicine breeds. Reports of estimated larger historical effective population sizes for B. indicus breeds may be representative of differences which occurred during the domestication and selection processes of B. indicus and B. taurus cattle, and offers a plausible explanation for the lower LD levels observed in indicine populations [8, 44]. Considering an expected average of 3/8 Zebu contribution to Braford breed composition, the lower average r2 observed in this population can be a result of those differences reported for B. indicus and B. taurus breeds in terms of past effective population sizes and selection processes during breed formation.
Both studied populations presented an inverse relationship between LD and inter-marker distances, and this decline of LD as a function of distance agrees with other studies based on r2 estimates in cattle [8, 38, 42, 45]. The Bovine HapMap Consortium [46] reported that, in general, B. indicus breeds had lower r2 values at short distances and higher r2 values at longer distances between markers when compared to B. taurus breeds. As the extent of LD at short inter-marker distances reflect the historic effective population size [44]. Rapid decline in average r2 of Braford compared to the decrease of r2 in Hereford can be associated to differences in effective population size of the breeds.
Decreases in effective population sizes have been widely observed within the last ~50 years in several cattle breeds, mostly due to the advent of artificial insemination which allowed the intense use of fewer males and high selection pressures for specific traits [47]. As demonstrated in Fig. 4, effective population sizes for both breeds have declined over time as reflection of the historical process of domestication and breed formation [46]. Brazilian Brafords were essentially formed with a limited pool of Nelore sires and the increased Ne trend observed in the last two generations (Fig. 1) could have resulted from recent efforts to increase diversity of this population through the introduction of foreign lineages formed with Brahman zebu. Conversely, the decreasing Ne trend observed in Brazilian Herefords is supported by the known restricted use of few purebred sires observed in most recent generations (Lopa TBP, personal communication).
Moreover, selection can lead to increased interchromosomal LD heterogeneity [48]. Thus, higher LD values observed in BTA6 in Brafords and Herefords in comparison to other chromosomes can be indicative of the presence of QTL affecting traits that have been under intense selection in both breeds. Evidence of QTL in BTA6 affecting growth traits such as birth weight [49], carcass weight [50] and ribeye area [49], and also feed intake and body weight gain [51, 52], have been reported in different cattle breeds. Additionally, the “spotted” locus (S) [53] is also located at BTA6 in the region containing the KIT gene. The SH allele is responsible for Hereford pattern of coat colour (white face, belly, feet and tail, often with a white stripe over the shoulder when homozygous) [53, 54]. Since the breed is fixed for this phenotype, it is expected that a historical selection occurred during breed formation fixed the spotted allele at this locus. The absence of breed × chromosome interaction effects obtained by Equation 2 indicates that even though high levels of interchromosomal LD heterogeneity were observed, similar trends were observed in Brafords and Herefords between adjacent markers at the 50K panel distances. This is in agreement with the common selection objectives applied to both populations analysed in this study [21] and to fact that Herefords are expected to have contributed 5/8 of the Braford genome. Nevertheless, as we considered a wider range of distances and had additional statistical power by including all pairs of syntenic markers in Equation 3, we were able to capture significant differences between breeds that were specific for chromosome and distance.
Knowledge about breed and chromosome-specific variation in LD levels could be used to determine optimal marker density for performing GWAS and GS studies [39, 48]. This information could be used to design customized marker panels for Hereford and Braford cattle with different chromosome-specific densities or to choose commercially available panels that would yield the desired LD levels across the whole genome. For example, if a minimal LD value of r2 = 0.2 is established as a threshold and the 80K panel will be used for genotyping, only BTA6 and BTA10 in Hereford and BTA6 in Braford would be expected to meet the target LD threshold (Fig. 3). To reach average r2 > 0.2 in all chromosomes, genotyping with 150K is required for Herefords, while for Brafords even higher densities would be required, such as available with HD commercial panels (as Illumina High-Density Bovine BeadChip Array Illumina 777K or Affymetrix Axiom Genome-Wide BOS 1 Array 650K). Alternatively, animals genotyped with panels below the desirable density could be imputed to higher densities, provided that reference haplotype panels are available for the respective populations [55].
The across-population, or across-breed, accuracy of GS estimates based on prediction equations derived from a specific reference population depends basically on the persistence of LD phase across populations, which reflects their genetic relationships [56]. Even if r2 values close to 1 are observed across populations, if a large proportion of SNP are in reversed phase the result of selection for these markers will lead to antagonistic responses [12]. Our estimates of phase correlation indicated that markers in LD at distances lower than 50 Kb in Herefords show similar levels of LD in Brafords, and a high proportion of these SNP share the same linkage phase. These results could be expected, since Brafords are composites with a contribution of 62.5 % of the Hereford breed. The correlation of phase values between populations proportionally decreases with the extent of divergence between the populations. To find markers that are in LD with QTL across diverging breeds, such as Australian Angus and New Zealand Jersey populations, de Roos et al. [12] concluded that a panel of approximately 300,000 marker would be required. In our case, due to the genetic similarity of Hereford and Braford breeds, 50K panel data can be pooled into a single reference population for performing GS and GWAS studies and results can subsequently be applied to either breed.
Conclusions
Our results indicate that at least 50K and 150K of evenly spaced SNP are necessary to effectively perform GWAS and GS studies, in Hereford and Braford respectively. For distances < 50 Kb, SNP are expected to be linked to the same QTL alleles in both breeds, due to high persistence of phase; therefore, Hereford and Braford data can be pooled into a single reference population for multibreed GS and GWAS studies performed with the above mentioned marker densities.
Ethics approval and consent to participate
Biological samples used for genotyping were collected according the Brazilian Guidelines for Use of Animals for Scientific and Educational Purposes, available at http://www.mct.gov.br/upd_blob/0234/234054.pdf.
There was a written informed consent from cattle owners (Embrapa SAIC 10200.10/01623) to cover the use of their animals and sample collection.
Acknowledgements
Research supported by CNPq – National Council for Scientific and Technological Development grant 478992/2012-2, Embrapa – Brazilian Agricultural Research Corporation grants 02.09.07.004 and 01.11.07.002, and CAPES – Coordination for the Improvement of Higher Level Personnel grant PNPD 02645/09-2. Authors acknowledge the Delta G Connection and Gensys Associated Consulting for providing the data for this research.
Abbreviations
- B
Braford
- BTA
Bos taurus autosome
- GS
Genomic selection
- GWAS
Genome-wide association study
- H
Hereford
- HD
High density
- HWE
Hardy-Weinberg equilibrium
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- QC
Quality control
- QTL
Quantitative trait loci
- SD
Standard deviation
- SE
Standard error
- SNP
Single nucleotide polymorphisms
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PB: data analysis, interpretation, and primary author of the manuscript. CCGG: experimental design, data collection and manuscript revision. ARC: data analysis and manuscript revision. JPS: helped with data analysis and manuscript revision. FFC: N e R script development, data analysis, interpretation and manuscript revision. All authors read and approved the final manuscript.
Contributor Information
Patrícia Biegelmeyer, Email: patriciabiegel@gmail.com.
Claudia C. Gulias-Gomes, Email: claudia.gulias@embrapa.br
Alexandre R. Caetano, Email: alexandre.caetano@embrapa.br
Juan P. Steibel, Email: steibelj@msu.edu
Fernando F. Cardoso, Email: fernando.cardoso@embrapa.br
References
- 1.Dekkers JCM. Application of genomics tools to animal breeding. Curr Genomics. 2012;13:207–212. doi: 10.2174/138920212800543057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaeffer LR. Strategy for applying genome-wide selection in dairy cattle. J Anim Breed Genet. 2006;123:218–223. doi: 10.1111/j.1439-0388.2006.00595.x. [DOI] [PubMed] [Google Scholar]
- 3.Konig S, Simianer H, Willam A. Economic evaluation of genomic breeding programs. J Dairy Sci. 2009;92:382–391. doi: 10.3168/jds.2008-1310. [DOI] [PubMed] [Google Scholar]
- 4.Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Gen. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith LP, Kuhner MK. The limits of fine-scale mapping. Genet Epidemiol. 2009;33:344–356. doi: 10.1002/gepi.20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218. doi: 10.1146/annurev.genet.39.073003.112420. [DOI] [PubMed] [Google Scholar]
- 8.McKay SD, Schnabel RD, Murdoch BM, Matukumalli LK, Aerts J, Coppieters W, et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007;8:74. doi: 10.1186/1471-2156-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bohmanova J, Sargolzaei M, Schenkel FS. Characteristics of linkage disequilibrium in North American Holsteins. BMC Genomics. 2010;11:421. doi: 10.1186/1471-2164-11-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rexroad CE, Vallejo RL. Estimates of linkage disequilibrium and effective population size in rainbow trout. BMC Genet. 2009;10:83. doi: 10.1186/1471-2156-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scraggs E, Zanella R, Wojtowicz A, Taylor JF, Gaskins CT, Reeves JJ, et al. Estimation of inbreeding and effective population size of fullblood wagyu cattle registered with the American Wagyu Cattle Association. J Anim Breed Genet. 2014;131:3–10. doi: 10.1111/jbg.12066. [DOI] [PubMed] [Google Scholar]
- 12.de Roos APW, Hayes BJ, Spelman RJ, Goddard ME. Linkage disequilibrium and persistence of phase in Holstein–Friesian, Jersey and Angus Cattle. Genetics. 2008;179(3):1503–1512. doi: 10.1534/genetics.107.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guglielmone AA. Epidemiology of babesiosis and anaplasmosis in South and Central America. Vet Parasitol. 1995;57:109–119. doi: 10.1016/0304-4017(94)03115-D. [DOI] [PubMed] [Google Scholar]
- 14.Nari A. Strategies for the control of one-host ticks and relationship with tick-borne diseases in South America. Vet Parasitol. 1995;57:153–165. doi: 10.1016/0304-4017(94)03117-F. [DOI] [PubMed] [Google Scholar]
- 15.Alves-Branco FPJ, Pinheiro AC, Sapper MFM. Controle dos principais ectoparasitos e endoparasitos em bovinos de corte no Rio Grande do Sul. Série Documentos, Embrapa Pecuária Sul, n.18, 2000.
- 16.Cardoso FF, Gomes CCG, Sollero BP, Oliveira MM, Roso VM, Picolli ML, et al. Genomic prediction for tick resistance in Braford and Hereford cattle. J Anim Sci. 2015;93:1–13. doi: 10.2527/jas.2014-8832. [DOI] [PubMed] [Google Scholar]
- 17.Biegelmeyer P, Nizolli LQ, Silva SS, Santos TRB, Dionello NJL, Gulias-Gomes CC, et al. Bovine genetic resistance effects on biological traits of Rhipicephalus (Boophilus) microplus. Vet Parasitol. 2015;208(3-4):231–237. doi: 10.1016/j.vetpar.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Goddard ME, Hayes BJ. Proceedings of the 18th Conference of the Association for the Advancement of Animal Breeding and Genetics. Barossa Valley, Australia: Association for the Advancement of Animal Breeding and Genetics; 2009. Genomic selection based on dense genotypes inferred from sparse genotypes; pp. 26–29. [Google Scholar]
- 19.Hayes BJ, Lewin HA, Goddard ME. The future of livestock breeding: genomic selection for efficiency, reduced emissions intensity, and adaptation. Trends Genet. 2013;29(4):206–214. doi: 10.1016/j.tig.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Khatkar MS, Nicholas FW, Collins AR, Zenger KR, Cavanagh JA, Barris W, et al. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics. 2008;9:187. doi: 10.1186/1471-2164-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sumário Conexão Delta G. 2015. http://www.gensys.com.br/home/show_page.php?id=703. Accessed 27 Jan 2016.
- 22.Clayton D. snpStats: SnpMatrix and XSnpMatrix classes and methods. R package version 1.20.0, 2015.
- 23.R Development Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 24.Sargolzaei M, Chesnais JP, Schenkel FS. FImpute - an efficient imputation algorithm for dairy cattle populations. J Dairy Sci. 2011;94:421. [Google Scholar]
- 25.Sun C, Wu X-L, Weigel KA, Rosa GJM, Bauck S, Woodward BW, et al. An ensemble-based approach to imputation of moderate-density genotypes for genomic selection with application to Angus cattle. Genet Res. 2012;94:33–150. doi: 10.1017/S001667231200033X. [DOI] [PubMed] [Google Scholar]
- 26.Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- 27.Badke YM, Bates RO, Ernst CW, Schwab C, Steibel JP. Estimation of linkage disequilibrium in four US pig breeds. BMC Genomics. 2012;13:24. doi: 10.1186/1471-2164-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McRae AF, McEwan JC, Dodds KG, Wilson T, Crawford AM, Slate J. Linkage disequilibrium in domestic sheep. Genetics. 2002;160:1113–1122. doi: 10.1093/genetics/160.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snelling WM, Chiu R, Schein JE, Hobbs M, Abbey CA, Adelson DL, et al. A physical map of the bovine genome. Genome Biol. 2007;8:R165. doi: 10.1186/gb-2007-8-8-r165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uimari P, Tapio M. Extent of linkage disequilibrium and effective population size in Finnish Landrace and Finnish Yorkshire pig breeds. J Anim Sci. 2011;89(3):609–614. doi: 10.2527/jas.2010-3249. [DOI] [PubMed] [Google Scholar]
- 31.Sved JA. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol. 1971;2:125–151. doi: 10.1016/0040-5809(71)90011-6. [DOI] [PubMed] [Google Scholar]
- 32.Qanbari S, Pimentel ECG, Tetens J, Thaller G, Lichtner P, Sharifi AR, et al. The pattern of linkage disequilibrium in German Holstein cattle. Anim Genet. 2010;41(4):346–356. doi: 10.1111/j.1365-2052.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Q, Wang Z, Moore SS, Yang RC. Genome-wide analysis of zygotic linkage disequilibrium and its components in crossbred cattle. BMC Genet. 2012;13:65. doi: 10.1186/1471-2156-13-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayes BJ, Visscher PM, McPartlan HC, Goddard ME. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003;13:635–643. doi: 10.1101/gr.387103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbin LJ, Blott SC, Swinburne JE, Vaudin M, Bishop SC, Woolliams JA. Linkage disequilibrium and historical effective population size in the Thoroughbred horse. Anim Genet. 2010;41(Suppl.2):8–15. doi: 10.1111/j.1365-2052.2010.02092.x. [DOI] [PubMed] [Google Scholar]
- 36.Aerts J, Megens HJ, Veenendaal T, Ovcharenko I, Crooijmans R, Gordon L, et al. Extent of linkage disequilibrium in chicken. Cytogenet Genome Res. 2007;117:338–345. doi: 10.1159/000103196. [DOI] [PubMed] [Google Scholar]
- 37.Hill WG. Estimation of effective population size from data on linkage disequilibrium. Genet Res. 1981;38:209–216. doi: 10.1017/S0016672300020553. [DOI] [Google Scholar]
- 38.Beghain J, Boitard S, Weiss B, Boussaha M, Gut I, Rocha D. Genome-wide linkage disequilibrium in the Blonde d'Aquitaine cattle breed. J Anim Breed Gen. 2012;130:294–302. doi: 10.1111/j.1439-0388.2012.01020.x. [DOI] [PubMed] [Google Scholar]
- 39.Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- 40.Du FX, Clutter AC, Lohuis MM. Characterizing linkage disequilibrium in pig populations. Int J Biol Sci. 2007;3:166–178. doi: 10.7150/ijbs.3.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Utsunomiya YT, Bomba L, Lucente G, Colli L, Negrini R, Lenstra JA, et al. Revisiting AFLP fingerprinting for an unbiased assessment of genetic structure and differentiation of taurine and zebu cattle. BMC Genet. 2014;15:47. doi: 10.1186/1471-2156-15-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espigolan R, Baldi F, Boligon AA, Souza FRP, Gordo DGM, Tonussi RL, et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genomics. 2013;14:305. doi: 10.1186/1471-2164-14-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villa-Angulo R, Matukumalli L, Gill C, Choi J, Van Tassell C, Grefenstette J. High-resolution haplotype block structure in the cattle genome. BMC Genet. 2009;10:19–31. doi: 10.1186/1471-2156-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME, et al. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007;17(4):520–526. doi: 10.1101/gr.6023607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farnir F, Coppieters W, Arranz JJ, Berzi P, Cambisano N, Grisart B, et al. Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 2000;10:220–227. doi: 10.1101/gr.10.2.220. [DOI] [PubMed] [Google Scholar]
- 46.Bovine HapMap Consortium Genome-Wide Survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324(5926):528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayes BJ, Lien S, Nilsen H, Olsen HG, Berg P, Maceachern S, et al. The origin of selection signatures on bovine chromosome 6. Anim Genet. 2008;39(2):105–111. doi: 10.1111/j.1365-2052.2007.01683.x. [DOI] [PubMed] [Google Scholar]
- 48.Sargolzaei M, Schenkel FS, Jansen GB, Schaeffer LR. Extent of linkage disequilibrium in Holstein cattle in North America. J Dairy Sci. 2008;91:2106–2117. doi: 10.3168/jds.2007-0553. [DOI] [PubMed] [Google Scholar]
- 49.McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41:597–607. doi: 10.1111/j.1365-2052.2010.02063.x. [DOI] [PubMed] [Google Scholar]
- 50.Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, et al. Cross-breed comparisons identified a critical 591-kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genet. 2009;10:43. doi: 10.1186/1471-2156-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, et al. Association, effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011;12:103. doi: 10.1186/1471-2156-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snelling WM, Allan MF, Keele JW, Kuehn LA, Thallman RM, Bennett GL, et al. Partial-genome evaluation of postweaning feed intake and efficiency of crossbred beef cattle. J Anim Sci. 2011;89:1731–1741. doi: 10.2527/jas.2010-3526. [DOI] [PubMed] [Google Scholar]
- 53.Grosz MD, MacNeil MD. The “spotted” locus maps to bovine chromosome 6 in a Hereford-cross population. J Hered. 1999;90:233–236. doi: 10.1093/jhered/90.1.233. [DOI] [PubMed] [Google Scholar]
- 54.Olson TA. Genetics of colour variation. In: Fries R, Ruvinsky A, editors. The Genetics of Cattle. Wallingford: CAB International; 1999. pp. 33–53. [Google Scholar]
- 55.Piccoli ML, Braccini J, Cardoso FF, Sargolzaei M, Larmer SG, Schenkel FS. Accuracy of genome-wide imputation in Braford and Hereford beef cattle. BMC Genet. 2014;15:157. doi: 10.1186/s12863-014-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA. The impact of genetic architecture on genome-wide evaluation methods. Genetics. 2010;185(3):1021–1031. doi: 10.1534/genetics.110.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]