

Abstract
It has been estimated that up to half of circulating Factor XIIIa (FXIIIa) is stored in platelets. The release of FXIIIa from platelets upon stimulation with ADP in patients with coronary artery disease treated with dual antiplatelet therapy has not been previously examined. Samples from 96 patients with established coronary artery disease treated with aspirin and clopidogrel were examined. Platelet aggregation was performed by light transmittance aggregometry (LTA) in platelet rich plasma (PRP) with platelet poor plasma (PPP) as reference and ADP 5μM as agonist. Kaolin activated TEG was performed in citrate PPP. PRP after aggregation was centrifuged and plasma supernatant (PSN) collected. FXIIIa was measured in PPP and PSN.
Platelet aggregation after stimulation with ADP 5μM resulted in 24% additional FXIIIa release in PSN as compared to PPP (99.3 ± 27 vs. 80.3 ± 24 %, p<0.0001). FXIIIa concentration in PSN correlated with maximal plasma clot strength (TEG-G) (r=0.48, p<0.0001), but not in PPP (r=0.15, p=0.14). Increasing quartiles of platelet derived FXIIIa were associated with incrementally higher TEG-G (p=0.012). FXIIIa release was similar between clopidogrel responders and non-responders (p=0.18). In summary, platelets treated with aspirin and clopidogrel release a significant amount of FXIIIa upon aggregation by ADP. Platelet derived FXIIIa may contribute to differences in plasma TEG-G, and thus in part provide a mechanistic explanation for high clot strength observed as a consequence of platelet activation. Variability in clopidogrel response does not significantly influence FXIIIa release from platelets.
Keywords: clopidogrel, Factor XIII, platelet aggregation, coagulation, thrombelastography
Introduction
Factor XIII (FXIII) is a transglutaminase consisting of 2 separate isoforms assembled into a tetramer of 2 FXIIIa active isomers and 2 FXIIIb isomers that bind the active FXIIIa [1]. Cleavage by thrombin frees FXIIIa with its primary role being cross stabilization of soluble fibrin strands [1,2]. Congenital FXIII deficiency leads to a bleeding diathesis, that if untreated can be fatal early in life [3]. Beyond its purpose of fibrin stabilization, other roles of FXIII have been identified in angiogenesis and wound healing [4,5].
FXIIIa is predominantly synthesized in cells of bone marrow origin and bound by the excess FXIIIb in plasma as an inactive tetramer (A2B2) [6]. In megacaryocytes, platelets, and leukocytes it is present in a cellular form (cFXIII) in a dimer structure of FXIIIa (A2) [6]. Megacaryocytes synthesize the majority of FXIIIa and package FXIIIa as well as encoding mRNA into platelets [7]. FXIIIa is highly abundant in platelets, and has been demonstrated predominantly in the cytoplasm [8,9]. It has been estimated that up to 50% of total FXIIIa is stored in platelets with a lesser amount found in macrophages/monocytes [1]. The role of FXIIIa derived from platelets in local dynamics of fibrin stabilization in platelet rich thrombus, such as found in high shear conditions of arterial thrombosis remains uncertain. Recently Kasahara et al. reported that platelet-dependent clot retraction requires factor XIII (FXIII), which covalently associates fibrin polymers with protein located within the platelet plasma membrane at lipid rafts [10]. High clot strength in whole blood assays measured by thrombelastography (TEG) appears to be a risk factor for increased risk of coronary thrombosis after coronary stenting and coronary artery bypass grafting (CABG) [11,12]. Antiplatelet therapy may affect local thrombus generation dynamics and fibrin stabilization by inhibiting FXIIIa activity on the surface of platelets or preventing release of FXIIIa into plasma [13]. FXIIIa release from platelets during platelet aggregation in patients with coronary artery disease treated with dual antiplatelet therapy has not been previously quantified.
We hypothesized that despite dual antiplatelet therapy with aspirin and clopidogrel, FXIII is being released from platelets and thus may contribute to fibrin stabilization in vivo in patients with coronary artery disease treated with standard antiplatelet therapy.
Methods
Patients
The study protocol was approved by the Indiana University institutional review board for research. Written informed consent was obtained from all subjects. Subjects with established coronary artery disease who were taking clopidogrel 75 mg and aspirin 81-325 mg daily for at least 14 days prior to enrollment were eligible for recruitment in the study. Subjects were excluded if they had a history of medication noncompliance, drug or alcohol abuse, bleeding disorder, platelet count less than 150,000/mm3, myelodysplastic or myeloproliferative disorders, if they were taking dipyridamole or warfarin, if they had chronic liver disease (hepatic transaminases greater than or equal to 3×ULN), or renal disease (serum creatinine greater than 2.0 mg/dl). Because the recruited subjects were requiring dual antiplatelet pharmacotherapy, baseline predrug platelet aggregation studies were not performed.
Blood samples
Blood was obtained from an antecubital or forearm vein using a 21 gauge needle with vacutainer collection tubing. The first 4 ml of blood drawn was discarded to avoid spontaneous platelet activation. Blood was collected in 3.2 % sodium citrate tubes to fill volume and a single Greiner partial fill (1.8 ml) blood collection tube containing 3.2% sodium citrate (Greiner Bio-One, NA, Inc., Monroe, NC, USA). VerifyNow was completed within 30 minutes after blood draw but no sooner than 10 minutes according to testing instructions, and LTA was completed within 2 hours of the blood draw.
Platelet aggregation studies
Ex vivo platelet function was assessed by light transmittance aggregometry (LTA) at 37°C with an Optical Lumi-Aggregometer (Model 700 with AggroLink 8 software, Chrono-Log Corporation, Havertown, PA, USA). Platelet rich plasma (PRP) and platelet poor plasma (PPP) were obtained by differential centrifugation as previously described [12,14,15]. In short, PRP was obtained from citrate blood after centrifugation at 180 g for 5 minutes. After recovering the PRP, the blood samples were subjected to further centrifugation at 2000 g for 15 minutes to recover platelet poor plasma (PPP). The resulting PRP and PPP were kept at room temperature for use within one hour. Platelet aggregation in PRP was induced with adenosine diphosphate (ADP) at 5μmol/L. The resultant PRP after platelet aggregation was collected and centrifuged at 2000 × g for 15 minutes to separate the plasma from the platelet pellet and collect the platelet supernatant plasma (PSN). Platelet count was measured (ACT 10, Beckman Coulter Inc., Brea, CA, USA) in whole blood and PRP. PPP and PSN were stored frozen at -80° Celsius.
VerifyNow™(VN) P2Y12 (Accumetrics Inc., San Diego, CA, USA) point-of-care assay was used to assess platelet inhibition in whole blood in subjects during maintenance clopidogrel dosing according to the manufacturer's instructions. Clopidogrel non-response was defined as PRU>208 [16].
ELISA
Factor XIIIa concentration (FXIII-subunit A) was measured in both PPP and PSN from PRP previously aggregated with 5μM ADP stimulation by enzyme linked immunoassay according to the manufacturer's instructions (Aniara, West Chester, OH, USA). In the assay the FXIIIa concentration is expressed as (%), standardized to a normal human citrated plasma pool. The average normal concentration of FXIII tetramer in plasma is ∼25 μg/ml according to the manufacturer of the assay.
Thrombelastography
Thrombelastography was performed in citrate PPP according to the manufacturer's instructions (Haemoscope, USA) [17]. Plasma was mixed with kaolin, inverted five times, and then loaded in a cup containing 20μl of CaCl2. Thrombelastography was stopped after maximal fibrin clot strength was recorded. Time to fibrin formation (R), angle constant (K) , angle (α) and maximal clot strength (MA, mm and G, dyn/cm2) were recorded. TEG – G was calculated from TEG – MA by use of the following formula: G = (5000*MA)/(100-MA)
Statistical Analysis
Analysis was performed with SPSS software (version 21, IBM, USA). Statistical significance was defined as p < 0.05. Tests were conducted 2-sided and values are represented as mean ± SD unless otherwise specified. Categorical variables were compared using the χ2 test. Normal distribution of continuous data was assessed by the Kolmogorov–Smirnov test. Two-sided Student's t-test was used to compare normally distributed continuous data between two groups and by analysis of variance for multiple groups with Bonferroni correction for pairwise comparisons. Pearson's correlation coefficient was used to calculate correlation between measures of platelet reactivity, TEG, and FXIIIa.
Results
Clinical variables of subjects enrolled in the study are summarized in Table 1. Factor XIIIa concentration in PPP was within the expected range. FXIIIa concentration in plasma supernatant (PSN) after stimulation of PRP with ADP 5μM was 24% higher than in PPP (99.3 ± 27 vs. 80.3 ± 24 %, p<0.0001) (Fig 1). Mean platelet count in whole blood was 264 ± 73 (*103/μl) and 375 ± 117 (*103/μl) in PRP. The amount of platelet derived FXIIIa (Δ FXIIIa) was calculated by subtracting FXIIIa PPP from FXIIIa PSN concentrations. Mean Δ FXIIIa was 19 ± 29 %. The ratio of FXIIIa released in relation to platelet count was calculated by dividing Δ FXIIIa by platelet count in PRP and was 0.056 ± 0.09 %*μl/103 platelets. Maximal platelet aggregation by light transmittance aggregometry (ADP5 μM) was 32.2 ± 12 % and 206 ± 77 by VerifyNow P2Y12 assay. Mean TEG measures are listed in Table 2.
Table 1.
Demographics and clinical variables of subjects. Values represent mean ± SD.
| Clinical Variables | Frequency (%) |
|---|---|
| Age (mean ± SD) | 56 ± 10 |
| Male Gender (%) | 51/96 (63) |
| African American (%) | 28/96 (29) |
| Diabetes mellitus(%) | 34/96 (35) |
| Body Mass Index(kg/m2, mean ± SD) | 32.4 ± 7 |
| Prior percutaneous coronary intervention (%) | 86/96 (90) |
| Coronary Artery Bypass Grafting (%) | 15/96 (16) |
| Peripheral Vascular Disease (%) | 19/96 (20) |
| Hypertension (%) | 92/96 (96) |
| Hyperlipidemia (%) | 96/96 (100) |
| Current smoking (%) | 49/96 (51) |
| Beta-Blocker | 93/96 (97) |
| Angiotensin converting enzyme-inhibitor | 82/96 (85) |
| Calcium Channel Blocker | 18/96 (19) |
| Statin | 88/96 (92) |
Figure 1.
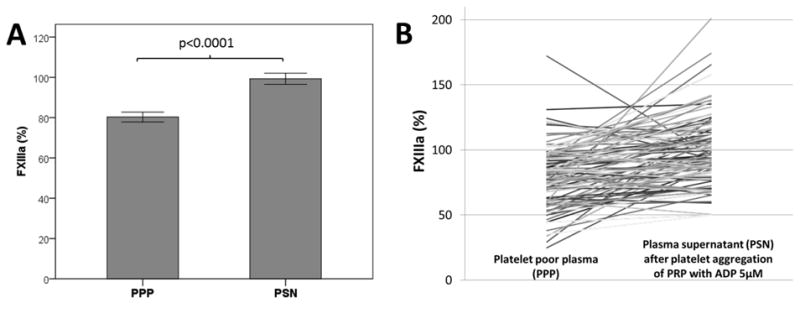
Comparison of FXIIIa concentration in non-stimulated platelet poor plasma (PPP) and plasma supernatant (PSN) after platelet aggregation with ADP 5μM. Mean ± SEM (Panel A) and individual paired data (Panel B).
Table 2.
Platelet aggregation measured by light transmittance aggregometry (LTA) and VerifyNow P2Y12. Thrombelastography (TEG) measurements: TEG-R (time to fibrin formation), TEG-K (time of initial clot formation), TEG-angle (angle of initial clot formation), TEG-MA and TEG-G (maximal clot strength). MPA: Maximal platelet aggregation. Values represent mean ± SD.
| Measure | Mean ± SD |
|---|---|
| MPA LTA ADP 5μM (%) | 32.2 ± 12 |
| VerifyNow P2Y12 (PRU) | 206 ± 77 |
| TEG – R (min) | 8.7 ± 2.3 |
| TEG – K (min) | 1.7 ± 1 |
| TEG – Angle | 66.3 ± 10 |
| TEG – MA (mm) | 36.8 ± 8 |
| TEG – G (dyn/cm2) | 3054 ± 1198 |
Pearson's correlation analysis of platelet aggregation, thrombelastography, and FXIIIa concentrations was performed and is shown in Table 3. There was a strong correlation between FXIIIa concentrations in PPP and PSN (Table 3, Fig 2.). Maximal platelet aggregation by LTA correlated strongly with VN P2Y12 PRU. Maximal plasma clot strength (TEG-G) significantly correlated with FXIIIa PSN and Δ FXIIIa, but not FXIII PPP (Table 3, Fig 2). There was no significant correlation between maximal platelet aggregation by LTA or VN P2Y12 and FXIIIa concentrations. When subjects were divided into clopidogrel responders and non-responders based on VN P2Y12 PRU, there was no significant difference in FXIIIa concentrations or ratio of Δ FXIII/platelet between responders and non-responders (Fig 3). Similarly, when grouped according to LTA (<42% MPA), there was no significant difference in Δ FXIIIa between responders and non-responders (20.7 ± 30 vs. 12.8 ± 24 %; p=0.27). Increasing quartiles of TEG-G were associated with increasing concentrations of FXIIIa in PPP and PSN (Fig 3). When analyzed in quartiles of platelet derived FXIIIa (Δ FXIIIa), TEG-G was highest in the highest quartile of Δ FXIIIa (Fig 3).
Table 3.
Pearson's correlation coefficient analysis of measures of platelet aggregation, FXIIIa, and thrombelastography (TEG). LTA: light transmittance aggregometry. VNP2Y12: VerifyNow P2Y12 assay. PPP: platelet poor plasma. PSN: plasma supernatant. PRP: platelet rich plasma. WB: whole blood. TEG-R: time to fibrin formation. TEG-K: time of initial clot formation. TEG-G: maximal clot strength. FXIII ADP PSN: FXIIIa concentration in plasma supernatant after platelet aggregation with ADP 5μM. FXIIIa Δ Platelet = FXIII ADP PSN – FXIIIa PPP. FXIIIa Δ/ Platelet = (FXIII ADP PSN – FXIIIa PPP)/WB platelet count.
| Factors (R) |
LTA ADP5μM |
VNP2Y1 2 PRU |
TEG-R | TEG-K | TEG-G | FXIIIa PPP |
FXIIIa ADP PSN |
FXIIIa Δ Platelet |
FXIIIa Δ/Platelet | Platelets WB |
Platelets PRP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LTA ADP5μM |
1.0, NA | 0.66 P<0.0001 |
0.028 P=0.79 |
-0.03 P=0.75 |
-0.076 P=0.46 |
-0.005 P=0.96 |
-0.094 P=0.36 |
-0.084 P=0.42 |
-0.081 P=0.44 |
-0.16 P=0.31 |
-0.14 P=0.18 |
| VNP2Y1 2 PRU |
0.66 P<0.0001 |
1.0, NA | -0.078 P=0.45 |
-0.17 P=0.11 |
0.064 P=0.54 |
-0.18 P=0.086 |
-0.051 P=0.63 |
0.1 P=0.34 |
0.084 P=0.42 |
-0.062 P=0.55 |
-0.22 P=0.034 |
| TEG-R | 0.028 P=0.79 |
-0.078 P=0.46 |
1.0, NA | 0.38 P<0.001 |
0.26 P=0.012 |
0.017 P=0.87 |
0.11 P=0.28 |
0.09 P=0.38 |
-0.01 P=0.9 |
0.1 P=0.33 |
0.092 P=0.38 |
| TEG-K | -0.03 P=0.75 |
-0.17 P=0.11 |
0.38 P<0.001 |
1.0, NA | -0.21 P=0.039 |
-0.046 P=0.65 |
-0.14 P=0.19 |
-0.087 P=0.4 |
-0.13 P=0.22 |
-0.005 P=0.96 |
0.081 P=0.44 |
| TEG-G | -0.076 P=0.46 |
0.064 P=0.54 |
0.26 P=0.012 |
-0.21 P=0.039 |
1.0, NA | 0.15 P=0.14 |
0.48 P<0.0001 |
0.33 P=0.011 |
0.27 P=0.01 |
0.26 P=0.01 |
0.18 P=0.08 |
| FXIIIa PPP |
-0.005 P=0.96 |
-0.18 P=0.086 |
0.017 P=0.87 |
-0.046 P=0.65 |
0.15 P=0.14 |
1.0, NA | 0.37 P<0.001 |
-0.49 P<0.0001 |
-0.5 P<0.0001 |
0.15 P=0.15 |
0.17 P=0.11 |
| FXIIIa ADP PSN |
-0.094 P=0.36 |
-0.051 P=0.63 |
0.11 P=0.28 |
-0.14 P=0.19 |
0.48 P<0.0001 |
0.37 P<0.001 |
1.0, NA | 0.63 P<0.0001 |
0.58 P<0.0001 |
0.18 P=0.08 |
0.16 P=0.12 |
| FXIIIa Δ Platelet |
-0.084 P=0.42 |
0.1 P=0.34 |
0.09 P=0.38 |
-0.087 P=0.4 |
0.33 P=0.011 |
-0.49 P<0.0001 |
0.63 P<0.0001 |
1.0, NA | 0.58 P<0.0001 |
0.05 p=0.66 |
0.01 P=0.9 |
| FXIIIa Δ/Platelet | -0.081 P=0.44 |
0.084 P=0.42 |
-0.01 P=0.9 |
-0.13 P=0.22 |
0.27 P=0.01 |
-0.5 P<0.0001 |
0.58 P<0.0001 |
0.95 P<0.0001 |
1.0, NA | -0.16 P=0.11 |
-0.15 P=0.14 |
| Platelets WB |
-0.16 P=0.31 |
-0.062 P=0.55 |
0.1 P=0.33 |
-0.005 P=0.96 |
0.26 P=0.01 |
0.15 P=0.15 |
0.18 P=0.08 |
0.05 p=0.66 |
-0.16 P=0.11 |
1.0, NA | 0.86 P<0.0001 |
| Platelets PRP |
-0.1 P=0.32 |
-0.17 P=0.1 |
0.092 P=0.38 |
0.081 P=0.44 |
0.18 P=0.08 |
0.17 P=0.11 |
0.16 P=0.12 |
0.01 P=0.9 |
-0.15 P=0.14 |
0.86 P<0.0001 |
1.0, NA |
Figure 2.
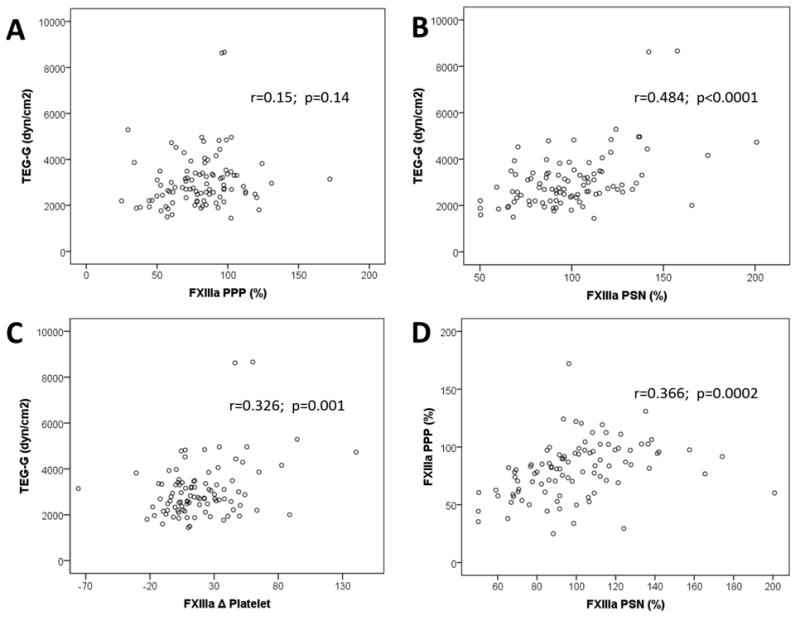
Scatterplots of measures of thrombelastography and FXIIIa concentrations. PPP: platelet poor plasma. PSN: plasma supernatant. TEG-G: maximal clot strength. FXIII PSN: FXIIIa concentration in plasma supernatant after platelet aggregation with ADP 5μM. FXIIIa Δ Platelet = FXIII ADP PSN – FXIIIa PPP.
Figure 3.
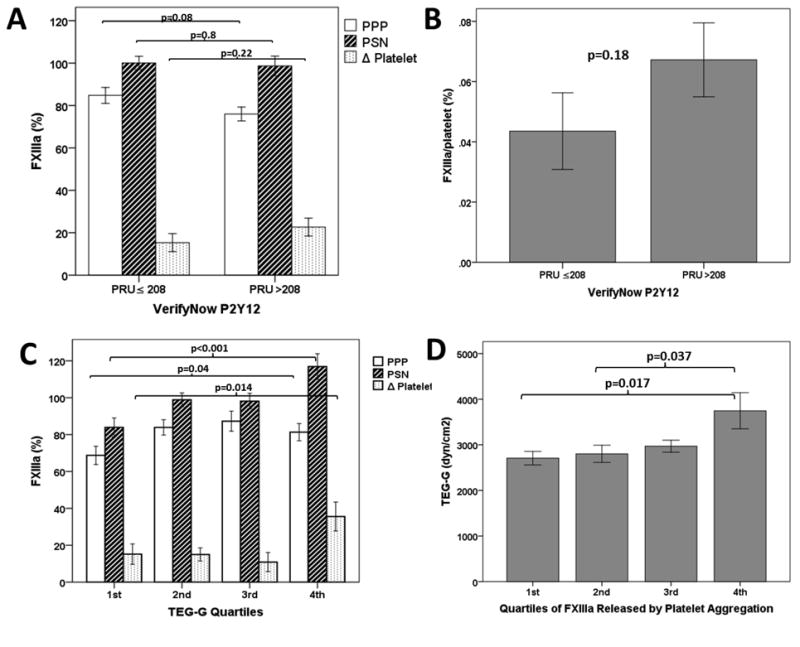
Comparison of FXIIIa concentrations (Panel A) and ratio of platelet derived FXIIIa to platelet count (FXIIIa/platelet) (Panel B) between clopidogrel responders (PRU≤208) and clopidogrel non-responders (PRU>208). Panel C) FXIIIa concentrations according to quartiles of maximal plasma clot strength (TEG-G). Panel D) Quartiles of FXIIIa derived from platelets (FXIIIa Δ Platelet) and TEG-G. PRU: platelet reactivity units. PPP: Platelet poor plasma. PSN: FXIIIa concentration in plasma supernatant after platelet aggregation with ADP 5μM. Δ Platelet = FXIII ADP PSN – FXIIIa PPP.
Discussion
FXIIIa is essential in cross linking of soluble fibrin strands and the generation of stable fibrin clot. It has been estimated that up to half of circulating FXIIIa is stored in platelets, with a smaller pool found in monocytes, while the remainder is present in plasma and bound to FXIIIB [18]. It has not been previously established whether FXIIIa found in platelets is released upon platelet activation and whether it is influenced by the degree of platelet aggregation in patients treated with dual antiplatelet therapy. We demonstrate a mean 24% increase in FXIIIa plasma concentration after aggregation and activation of platelets with ADP. Variability of platelet inhibition was not significantly associated with differences in FXIIIa release from platelets. In contrast, maximal clot strength by TEG correlated strongly with FXIIIa levels after platelet aggregation. TEG is a sensitive method to detect functional deficiency in FXIIIa activity [19]. Since FXIIIa is necessary for fibrin cross linking, it is possible that platelet derived FXIIIa contributes to the higher clot strength observed in individuals with higher levels of FXIIIa release. Maximal whole blood clot strength has been associated with increased risk for recurrent thrombotic events after coronary stenting and after coronary artery bypass grafting [11,20]. Thus, variability in platelet derived FXIIIa may contribute to differences in prothrombotic phenotypes and risk of recurrent ischemic events. We have previously shown that thrombin induced whole blood TEG-G is predominantly influenced by platelet count, whereas plasma fibrin TEG-G is higher in clopidogrel responders as compared to non-responders [21]. Plasma TEG-G in our study correlated weakly with platelet count, similar to the extent previously published [21]. We have previously demonstrated that increased c-reactive protein in patients with coronary artery disease is associated with increased plasma TEG-G [17]. Chronic inflammation is often associated with mild thrombocytosis, and could thus be a possible explanation for this correlation.
Arguably, FXIIIa deposited locally by aggregating platelets at sites of developing platelet-fibrin thrombus, could be pivotal in accelerating thrombus stabilization under high shear conditions. FXIIIa has been demonstrated on the surface of activated platelets and has been shown to enhance platelet adhesion under flow conditions [13,22]. Alternatively, subjects with increased levels of platelet derived FXIIIa may concomitantly deposit other coagulation factors that could in part influence maximal clot formation phenotype, such as Factor V and Factor VII which are also found in platelet granules [9]. While the exact mechanisms of FXIIIa release from cells have not been elucidated, data from Cordell at al. suggest a novel subcellular tracking mechanism of FXIIIa in monocytes and macrophages [23]. Platelet derived microparticles have been shown to contain cFXIIIa and this may be a mechanism of release of platelet FXIIIa into plasma upon platelet activation [24].
We could not find an association between clopidogrel response status and FXIIIa release in our study population. While maximal platelet aggregation is blunted in clopidogrel responders, platelet activation and granule secretion nevertheless occurs, and may be sufficient to overcome the inhibitory effects of clopidogrel [25]. ADP was chosen as agonist, since ADP induced on treatment aggregation is a strong predictor of recurrent coronary thrombosis after coronary stenting, and correlates strongly with P2Y12 receptor inhibition. While P2Y12 receptor activation was in part inhibited by clopidogrel, P2Y1 receptor activation also occurred when PRP was stimulated by ADP in our study. P2Y1 activation triggers mobilization of calcium from internal platelet stores, which results in platelet shape change and weak, transient aggregation in response to ADP [26]. It may therefore be possible that the release of FXIIIa from platelets in response to ADP could in part be mediated by P2Y1 receptor. Alternatively, steps involved in sequential centrifugation of PRP could have led to additional release of FXIIIa from platelets despite treatment with aspirin and clopidogrel.
Since FXIIIa has a role in facilitating platelet recruitment in flowing blood, future studies utilizing platelet deposition assays in contrast to aggregation based assays, could help further characterize the functional implications of FXIIIa release from platelets.
In conclusion, we demonstrate a substantial release of FXIIIa by platelets after stimulation with ADP in patients with coronary artery disease treated with clopidogrel and aspirin. Platelet derived FXIIIa may contribute to the generation of shear resistant, non-soluble plasma fibrin thrombus and in part explain variability in maximal clot strength phenotype observed in patients with coronary artery disease.
Acknowledgments
This study was supported in part, by the Indiana Clinical and Translational Sciences Institute funded, in part by Grant Number (RR025761) from the National Institutes of Health, National Center for Research Resources, Clinical and Translational Sciences Award, the Indiana University Health Value Grant, the Department of Medicine, Indiana University School of Medicine, and the Indiana University School of Medicine – Indiana University Health Strategic Research Initiative.
Footnotes
Disclosure: Recruitment of subjects occurred during employment of Y. Jin at Indiana University. Y. Jin is currently an employee of Eli Lilly and Co, USA. The other authors have no conflicts of interest to declare.
References
- 1.Bagoly Z, Koncz Z, Hársfalvi J, Muszbek L. Factor XIII, clot structure, thrombosis. Thromb Res. 2012;129:382–7. doi: 10.1016/j.thromres.2011.11.040. [DOI] [PubMed] [Google Scholar]
- 2.Schroeder V, Chatterjee T, Kohler HP. Influence of blood coagulation factor XIII and FXIII Val34Leu on plasma clot formation measured by thrombelastography. Thromb Res. 2001;104:467–74. doi: 10.1016/s0049-3848(01)00395-4. [DOI] [PubMed] [Google Scholar]
- 3.Levy JH, Greenberg C. Biology of Factor XIII and clinical manifestations of Factor XIII deficiency. Transfusion. 2013;53(5):1120–31. doi: 10.1111/j.1537-2995.2012.03865.x. [DOI] [PubMed] [Google Scholar]
- 4.Inbal A, Lubetsky A, Krapp T, Castel D, Shaish A, Dickneitte G, Modis L, Muszbek L, Inbal A. Impaired wound healing in factorXIII deficient mice. Thromb Haemost. 2005;94:432–7. doi: 10.1160/TH05-04-0291. [DOI] [PubMed] [Google Scholar]
- 5.Dardik R, Loscalzo J, Inbal A. Factor XIII (FXIII) and angiogenesis. J Thromb Haemost. 2006;4:19–25. doi: 10.1111/j.1538-7836.2005.01473.x. [DOI] [PubMed] [Google Scholar]
- 6.Adány R, Bárdos H. Factor XIII subunit A as an intracellular transglutaminase. Cell Mol Life Sci. 2003;60(6):1049–60. doi: 10.1007/s00018-003-2178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ádány R. Intracellular factor XIII: cellular distribution of factor XIII subunit a in humans. Semin Thrombos Haemostas. 1996;22:399–408. [PubMed] [Google Scholar]
- 8.Sixma JJ, van den Berg A, Schiphorst M, Gezue JH, McDonagh J. Immunocytochemical localization of albumin and factor XIII in thin cryo sections of human blood platelets. Throm Haemost. 1984;51:388–391. [PubMed] [Google Scholar]
- 9.Dashty M, Akbarkhanzadeh V, Zeebregts CJ, Spek CA, Sijbrands EJ, Peppelenbosch MP, Rezaee F. Characterization of coagulation factor synthesis in nine human primary cell types. Sci Rep. 2012;2:787. doi: 10.1038/srep00787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasahara K, Kaneda M, Miki T, Iida K, Sekino-Suzuki N, Kawashima I, Suzuki H, Shimonaka M, Arai M, Ohno-Iwashita Y, Kojima S, Abe M, Kobayashi T, Okazaki T, Souri M, Ichinose A, Yamamoto N. Clot retraction is mediated by factor XIII-dependent fibrin-αIIbβ3-myosin axis in platelet sphingomyelin-rich membrane rafts. Blood. 2013;122(19):3340–8. doi: 10.1182/blood-2013-04-491290. [DOI] [PubMed] [Google Scholar]
- 11.Gurbel PA, Bliden KP, Guyer K, Cho PW, Zaman KA, Kreutz RP, Bassi AK, Tantry US. Platelet Reactivity in Patients and Recurrent Events Post-Stenting: Results of the PREPARE POST-STENTING Study. J Am Coll Cardiol. 2005;46:1820–1826. doi: 10.1016/j.jacc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 12.Bitar A, Kreutz RP. Role of Thrombelastography (TEG) in Risk Assessment and Guidance of Antithrombotic Therapy in Patients with Coronary Artery Disease. Drug Development Research. 2013;74:533–540. [Google Scholar]
- 13.Magwenzi SG, Ajjan RA, Standeven KF, Parapia LA, Naseem KM. Factor XIII supports platelet activation and enhances thrombus formation by matrix proteins under flow conditions. J Thromb Haemost. 2011;9:820–33. doi: 10.1111/j.1538-7836.2011.04234.x. [DOI] [PubMed] [Google Scholar]
- 14.Kreutz RP, Tantry US, Bliden KP, Gurbel PA. Inflammatory changes during the ‘common cold’ are associated with platelet activation and increased reactivity of platelets to agonists. Blood Coagul Fibrinolysis. 2007;18:713–8. doi: 10.1097/MBC.0b013e328201c77e. [DOI] [PubMed] [Google Scholar]
- 15.Kreutz RP, Alloosh M, Mansour K, Neeb Z, Kreutz Y, Flockhart DA, Sturek M. Morbid obesity and metabolic syndrome in Ossabaw miniature swine are associated with increased platelet reactivity. Diabetes Metab Syndr Obes. 2011;4:99–105. doi: 10.2147/DMSO.S17105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stone GW, Witzenbichler B, Weisz G, Rinaldi MJ, Neumann FJ, Metzger DC, Henry TD, Cox DA, Duffy PL, Mazzaferri E, Gurbel PA, Xu K, Parise H, Kirtane AJ, Brodie BR, Mehran R, Stuckey TD, ADAPT-DES Investigators Platelet reactivity and clinical outcomes after coronary artery implantation of drug-eluting stents (ADAPT-DES): a prospective multicentre registry study. Lancet. 2013;382(9892):614–23. doi: 10.1016/S0140-6736(13)61170-8. [DOI] [PubMed] [Google Scholar]
- 17.Kreutz RP, Owens J, Breall JA, Lu D, von der Lohe E, Bolad I, Sinha A, Flockhart DA. C-reactive protein and fibrin clot strength measured by thrombelastography after coronary stenting. Blood Coagul Fibrinolysis. 2013;24:321–6. doi: 10.1097/MBC.0b013e32835cc193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonagh J, McDonagh RP, Jr, Delâge JM, Wagner RH. Factor XIII in human plasma and platelets. J Clin Invest. 1969;48(5):940–6. doi: 10.1172/JCI106053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen VG, Gurley WQ, Jr, Burch TM. The impact of factor XIII on coagulation kinetics and clot strength determined by thrombelastography. Anesth Analg. 2004;99(1):120–3. doi: 10.1213/01.ANE.0000123012.24871.62. [DOI] [PubMed] [Google Scholar]
- 20.Zacho M, Rafiq S, Kelbæk H, Johansson PI, Nielsen MB, Steinbrüchel DA, Kofoed KF. Hypercoagulability in relation to coronary artery bypass graft patency and clinical outcome. Scand Cardiovasc J. 2013;47(2):104–8. doi: 10.3109/14017431.2012.754934. [DOI] [PubMed] [Google Scholar]
- 21.Lu D, Owens J, Kreutz RP. Plasma and whole blood clot strength measured by thrombelastography in patients treated with clopidogrel during acute coronary syndromes. Thromb Res. 2013;132(2):e94–8. doi: 10.1016/j.thromres.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagy B, Jr, Simon Z, Bagoly Z, Muszbek L, Kappelmayer J. Binding of plasma factor XIII to thrombin-receptor activated human platelets. Thromb Haemost. 2009;102(1):83–9. doi: 10.1160/TH09-01-0054. [DOI] [PubMed] [Google Scholar]
- 23.Cordell PA, Kile BT, Standeven KF, Josefsson EC, Pease RJ, Grant PJ. Association of coagulation factor XIII-A with Golgi proteins within monocyte-macrophages: implications for subcellular trafficking and secretion. Blood. 2010;115(13):2674–81. doi: 10.1182/blood-2009-08-231316. [DOI] [PubMed] [Google Scholar]
- 24.Holme PA, Brosstad F, Solum NO. The difference between platelet and plasma FXIII used to study the mechanism of platelet microvesicle formation. Thromb Haemost. 1993;70(4):681–6. [PubMed] [Google Scholar]
- 25.Mueller T, Dieplinger B, Poelz W, Calatzis A, Haltmayer M. Utility of whole blood impedance aggregometry for the assessment of clopidogrel action using the novel Multiplate analyzer--comparison with two flow cytometric methods. Thromb Res. 2007;121(2):249–58. doi: 10.1016/j.thromres.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 26.Jin J, Daniel JL, Kunapuli SP. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J Biol Chem. 1998;273(4):2030–2034. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]