

Abstract
A general synthetic sequence involving simply prepared starting materials provides rapid access to diverse, novel tricyclic architectures inspired by pleuromutilin. SmII‐mediated radical cyclization cascades of dialdehydes, prepared using a new, one‐pot, copper‐catalyzed double organomagnesium addition to β‐chlorocyclohexenone, proceed with complete sequence selectivity and typically with high diastereocontrol to give analogues of the target core. Our expedient approach (ca. 7 steps) allows non‐traditional, de novo synthetic access to analogues of the important antibacterial that can′t be prepared from the natural product by semisynthesis.
Keywords: cyclization, domino reactions, electron‐transfer, radical reactions, samarium
Cascade processes have the potential to assemble complex molecular architectures in a single synthetic operation.1 If such processes can be mediated by simple, commercial reagents and can be harnessed to deliver structures possessing important biological or physical properties they become even more desirable. We have recently exploited carbonyl reduction by the well‐known electron‐transfer reductant SmI2 2 to trigger cascade processes that deliver natural and unnatural product structural motifs.3, 4 For example, we have prepared novel organic materials and bioactive targets using cascades involving phase‐tag removal and cyclization.3b–d In addition, we have developed radical cyclization cascades of dialdehydes4 and have used such a cascade4c,e in the first enantiospecific total synthesis of the antibacterial natural product pleuromutilin (Scheme 1 A).4e Electron‐transfer reduction of dialdehyde 2 using SmI2 gives 3 with complete diastereocontrol and excellent yield.4c, e A dialdehyde cascade has since been employed by Reisman in an elegant synthesis of (−)‐maoecrystal Z.5
Scheme 1.
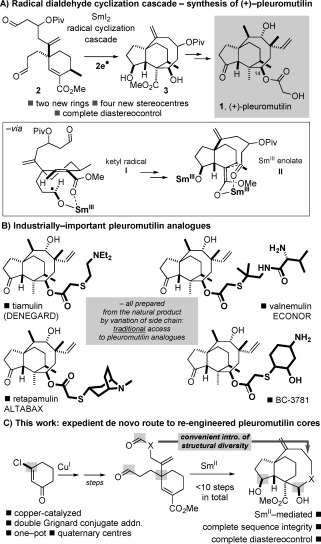
A) Radical cyclization cascade of a dialdehyde in the first enantiospecific synthesis of (+)‐pleuromutilin. B) Industrially‐important pleuromutilin analogues and the traditional method of analogue synthesis. C) This work: CuI and SmII in a short route to re‐engineered pleuromutilin cores.
The natural product pleuromutilin is an important lead for the development of new antibacterials6, 7 and much synthetic effort has been focused on the synthesis of analogues with increased activity and a more favourable pharmacokinetic profile. A handful of compounds are widely used in veterinary medicine, a single analogue has been licensed for topical use on humans, and others are currently undergoing clinical evaluation. Crucially, all of the analogues licensed or in clinical trials are prepared from the natural product and vary only in the C14 side chain (Scheme 1 B). As many of the undesirable properties of the pleuromutilins (e.g. poor solubility and poor pharmacokinetic properties) are associated with the core of the antibacterial natural product6, 7 re‐engineering of the pleuromutilin core is a crucial but long‐neglected approach to analogues that is orthogonal to traditional approaches involving variation of the C14 side chain only (Scheme 1 B). Importantly, our radical cascade approach to pleuromutilin allows us to consider the rapid de novo synthesis of simplified analogues that cannot be prepared from the natural product by semisynthesis—the approach universally adopted by industry in the pursuit of pleuromutilin analogues for use in humans.6, 7
Here we describe the development of a general synthetic sequence that rapidly converts simply prepared starting materials to diverse, novel tricyclic architectures inspired by pleuromutilin8 but that cannot be prepared from the natural product. Our studies have resulted in a new, one‐pot, copper‐catalyzed double organomagnesium addition to β‐chlorocyclohexenone and have also allowed us to evaluate the scope of the radical cyclization cascade of dialdehydes.
We began our studies by developing a route to simplified analogues of dialdehyde substrate 2. In our pursuit of an expedient route to re‐engineered pleuromutilin cores, we developed a one‐pot, sequential addition of two organomagnesiums to β‐chlorocyclohexenone 4 catalyzed by CuSPh⋅LiI (10 mol %). In situ trapping of the enolate intermediate with Comins’ reagent9 gave vinyl triflates 5 in good overall yield (Scheme 2). Thus, the framework of the cascade substrates 2 could be assembled in one‐pot. To the best of our knowledge, the one‐pot addition of two organometallic species to an enone bearing a β‐leaving group is unprecedented.10 Products of homo‐double addition were not isolated. After partial purification of 5, palladium‐catalyzed methoxycarbonylation gave unsaturated esters 6 a–g.
Scheme 2.
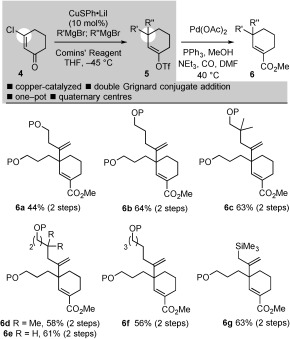
One‐pot, copper‐catalyzed, double conjugate addition of organomagnesiums with enolate trapping in the expedient synthesis of unsaturated esters 6. Comins’ reagent=N‐(5‐chloro‐2‐pyridyl)bis(trifluoromethanesulfonimide); P=TBS, tert‐butyl dimethylsilyl.
Unsaturated esters 6 a–f were then converted to dialdehydes 7 a–f in two straightforward steps (bis‐desilylation and ‐oxidation—see Scheme 3 for illustrative conditions).11 Thus, cascade cyclization substrates 7 a–f were accessible in only four steps from 4. A cascade substrate bearing an oxygenated tether was constructed from 6 g by Sakurai coupling12 with 3‐[(tert‐butyldimethylsilyl)oxy]propanal, protection of the resulting secondary alcohol as the pivaloate, followed by bis‐desilylation and ‐oxidation to give dialdehyde 9 in good overall yield (6 steps from 4) (Scheme 3).
Scheme 3.
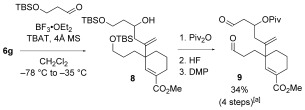
Sakurai coupling in the synthesis of a cascade substrate bearing an oxygenated tether. DMP=Dess–Martin periodinane; TBAT=tetrabutylammonium difluorotriphenylsilicate. [a] 1:1 mixture of diastereoisomers.
Pleasingly, in all cases, complete sequence integrity was observed in the cascade cyclizations of 7 a–f, 9 and synthetic pleuromutilin scaffolds containing 6, 7, 8, and even 9‐membered right‐hand rings (10 a–g) were obtained in moderate to good yield (Scheme 4). In all but one case, the cascades proceeded with complete diastereocontrol at the four newly‐formed stereocentres. It is important to note that general approaches to such scaffolds from the natural product pleuromutilin would be impossible.6, 7
Scheme 4.
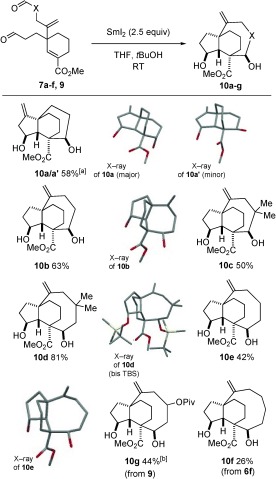
Selective cascade cyclizations to six‐, seven‐, eight‐, and nine‐membered ring‐containing analogues of the pleuromutilin scaffold. [a] 2:1 mixture of diastereoisomers. [b] 1:1 mixture of diastereoisomers at the centre bearing the OPiv group.
Remarkably, in the cascade cyclization of 7 a, a substrate in which both aldehydes are attached to three‐carbon tethers, complete sequence integrity was observed in the dialdehyde cyclization cascade: the aldehyde on the tether containing the sp2‐hybridized alkene carbon atom undergoes initial coupling (Scheme 5).4 This most likely results from the restricted conformation (‘Thorpe–Ingold Effect′) of the alkene‐containing tether resulting in more facile cyclization. Thus reversible formation of radical anion 11 13 leads selectively to a SmIII‐enolate14 that undergoes aldol cyclization through closed transition structure 12 to give major product 10 a. The minor diastereoisomer 10 a′ may arise from open aldol transition‐structure 12′. The low diastereoselectivity observed in the second stage of this particular cascade may result from unfavorable 1,3‐trans‐diaxial interactions in the closed transition‐structure 12 leading to the relative stereochemistry typically formed in the cascades (vide infra). The relative stereochemistry of 10 a and 10 a′ was determined by X‐ray crystallographic analysis (Scheme 4).15
Scheme 5.
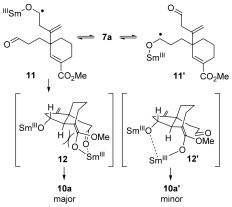
Challenging the sequence integrity of the dialdehyde cascade: Cyclization of 7 a using SmI2.
Pleasingly, cascade substrate 7 b, designed to deliver a 5,6,7‐tricyclic architecture, underwent highly selective reaction with SmI2 to give 10 b as a single diastereoisomer in 63 % yield (Scheme 4). Remarkably, efficient cascade cyclization was also observed for related substrate 7 c despite significant steric crowding adjacent to the second aldehyde group, and 10 c was isolated in 50 % yield as a single diastereoisomer. gem‐Disubstitution in the longer tether of cascade substrates resulted in more efficient cascade cyclization. For example, gem‐dimethyl‐substituted 5,6,8‐tricyclic cascade product 10 d was formed in 81 % yield while products possessing less‐substituted eight‐membered rings 10 e and 10 g were isolated as single diastereoisomers in 42 and 44 % yields, respectively. In a bid to identify the limits of the dialdehyde cascade process, we attempted to construct a nine‐membered ring in the final stage of the cascade. The construction of nine‐membered carbocycles is challenging and, to our knowledge, the formation of nine‐membered carbacycles by using a SmI2‐mediated aldol reaction is unprecedented.16 Impressively, treatment of substrate 7 f with SmI2 gave the 5,6,9‐tricyclic architecture and 10 f was isolated as a single diastereoisomer in 26 % yield. The relative stereochemistry in the cascade products was determined by X‐ray crystallographic analysis of 10 a,a′, 10 b, and 10 e and a derivative of 10 d 15 and inferred for the remainder.
To illustrate the potential of our concise route to pleuromutilin analogues, diol 10 d, possessing a 5,6,8‐tricyclic scaffold, was treated with one equivalent of Dess–Martin periodinane17 to give C3 ketone 11. Installation of the C14 glycolate ester8b gave 12 and completed a seven‐step synthesis of a model pleuromutilin analogue (Scheme 6).
Scheme 6.
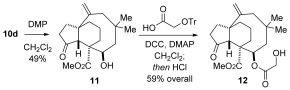
Concluding a seven‐step synthesis of pleuromutilin analogue 12. DCC=N,N‐dicyclohexylcarbodiimide; DMAP=4‐dimethylaminopyridine.
In summary, a general synthetic sequence involving simply prepared starting materials provides rapid access to diverse, novel tricyclic architectures inspired by pleuromutilin. At the heart of the short route, SmII‐mediated radical cyclization cascades of dialdehydes, prepared using a new one‐pot, copper‐catalyzed double organomagnesium addition to β‐chlorocyclohexenone, proceed with complete sequence selectivity and typically with high diastereocontrol. Our expedient approach (ca. 7 steps) allows non‐traditional, de novo synthetic access to analogues of the important antibacterial natural product that can′t be prepared from the natural product by semisynthesis.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
We thank the BBSRC (DTP Studentship to R.E.R.), EPSRC (Established Career Felllowship to D.J.P. and DTA Studentship to N.J.F.), and the University of Manchester (President’s Scholarship to H.H.). We also thank Miles Aukland for his help optimizing the route to 10 a/a′.
References
- 1.For selected reviews on cascade reactions, see:
- 1a. Anderson E. A., Org. Biomol. Chem. 2011, 9, 3997; [DOI] [PubMed] [Google Scholar]
- 1b. Nicolaou K. C., Edmonds D. J., Bulger P. G., Angew. Chem. Int. Ed. 2006, 45, 7134; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7292; [Google Scholar]
- 1c. Nicolaou K. C., Chen J. S., Chem. Soc. Rev. 2009, 38, 2993; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Tietze L. F., Chem. Rev. 1996, 96, 115. [DOI] [PubMed] [Google Scholar]
- 2.For recent reviews of SmI2, see:
- 2a. Just‐Baringo X., Procter D. J., Acc. Chem. Res. 2015, 48, 1263; [DOI] [PubMed] [Google Scholar]
- 2b. Szostak M., Fazakerley N. J., Parmar D., Procter D. J., Chem. Rev. 2014, 114, 5959; [DOI] [PubMed] [Google Scholar]
- 2c. Szostak M., Spain M., Procter D. J., Chem. Soc. Rev. 2013, 42, 9155; [DOI] [PubMed] [Google Scholar]
- 2d. Szostak M., Spain M., Parmar D., Procter D. J., Chem. Commun. 2012, 48, 330; [DOI] [PubMed] [Google Scholar]
- 2e. Szostak M., Procter D. J., Angew. Chem. Int. Ed. 2012, 51, 9238; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9372; [Google Scholar]
- 2f. Beemelmanns C., Reißig H.‐U., Chem. Soc. Rev. 2011, 40, 2199; [DOI] [PubMed] [Google Scholar]
- 2g. Beemelmanns C., Reißig H.‐U., Pure Appl. Chem. 2011, 83, 507; [Google Scholar]
- 2h. Nicolaou K. C., Ellery S. P., Chen J. S., Angew. Chem. Int. Ed. 2009, 48, 7140; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7276; [Google Scholar]
- 2i. Procter D. J., Flowers II R. A., Skrydstrup T., Organic Synthesis using Samarium Diiodide: A Practical Guide, RSC Publishing, Cambridge, 2009; [Google Scholar]
- 2j. Edmonds D. J., Johnston D., Procter D. J., Chem. Rev. 2004, 104, 3371; [DOI] [PubMed] [Google Scholar]
- 2k. Kagan H. B., Tetrahedron 2003, 59, 10351; [Google Scholar]
- 2l. Krief A., Laval A.‐M., Chem. Rev. 1999, 99, 745; [DOI] [PubMed] [Google Scholar]
- 2m. Molander G. A., Harris C. R., Chem. Rev. 1996, 96, 307. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Yalavac I., Lyons S. E., Webb M. R., Procter D. J., Chem. Commun. 2014, 50, 12863; [DOI] [PubMed] [Google Scholar]
- 3b. Levick M. T., Grace I., Dai S.‐Y., Kasch N., Muryn C., Lambert C., Turner M. L., Procter D. J., Org. Lett. 2014, 16, 2292; [DOI] [PubMed] [Google Scholar]
- 3c. Levick M. T., Coote S. C., Grace I., Lambert C., Turner M. L., Procter D. J., Org. Lett. 2012, 14, 5744; [DOI] [PubMed] [Google Scholar]
- 3d. Coote S. C., Quenum S., Procter D. J., Org. Biomol. Chem. 2011, 9, 5104; [DOI] [PubMed] [Google Scholar]
- 3e. Parmar D., Matsubara H., Price K., Spain M., Procter D. J., J. Am. Chem. Soc. 2012, 134, 12751; [DOI] [PubMed] [Google Scholar]
- 3f. Sautier B., Lyons S. E., Webb M. R., Procter D. J., Org. Lett. 2012, 14, 146; [DOI] [PubMed] [Google Scholar]
- 3g. Parmar D., Price K., Spain M., Matsubara H., Bradley P. A., Procter D. J., J. Am. Chem. Soc. 2011, 133, 2418. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Helm M. D., Sucunza D., Da Silva M., Helliwell M., Procter D. J., Tetrahedron Lett. 2009, 50, 3224; [Google Scholar]
- 4b. Helm M. D., Da Silva M., Sucunza D., Helliwell M., Procter D. J., Tetrahedron 2009, 65, 10816; [Google Scholar]
- 4c. Helm M. D., Da Silva M., Sucunza D., Findley T. J. K., Procter D. J., Angew. Chem. Int. Ed. 2009, 48, 9315; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9479; [Google Scholar]
- 4d. Findley T. J. K., Sucunza D., Miller L. C., Helm M. D., Helliwell M., Davies D. T., Procter D. J., Org. Biomol. Chem. 2011, 9, 2433; [DOI] [PubMed] [Google Scholar]
- 4e. Fazakerley N. J., Helm M. D., Procter D. J., Chem. Eur. J. 2013, 19, 6718. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Cha J. Y., Yeoman J. T. S., Reisman S. E., J. Am. Chem. Soc. 2011, 133, 14964; [DOI] [PubMed] [Google Scholar]
- 5b. Yeoman J. T. S., Cha J. Y., Mak V. W., Reisman S. E., Tetrahedron 2014, 70, 4070. [Google Scholar]
- 6.For selected, recent reviews on pleuromutilin analogues, see:
- 6a. Shang R., Wang J., Guo W., Liang J., Current Topics Med. Chem. 2013, 13, 3013; [DOI] [PubMed] [Google Scholar]
- 6b. Tang Y.‐Z., Liu Y.‐H., Chen J.‐X., Mini‐Rev. Med. Chem. 2012, 12, 53; [DOI] [PubMed] [Google Scholar]
- 6c. Novak R. in Drug Discovery from Natural Products (Eds.: O. Genilloud, F. Vicente) RSC, 2012, pp. 326; [Google Scholar]
- 6d. Novak R., Annals New York Acad. Sci. 2011, 1241, 71; [DOI] [PubMed] [Google Scholar]
- 6e. Jarvest R. in Functional Molecules from Natural Sources (Eds.: S. K. Wrigley, R. Thomas, N. Nicholson, C. Bedford), RSC, 2010, pp. 106; [Google Scholar]
- 6f. Novak R., Shlaes D. M., Curr. Opin. Investig. Drugs 2010, 11, 182; [PubMed] [Google Scholar]
- 6g. Hu C., Zou Y., Mini‐Rev. Med. Chem. 2009, 9, 1397; [DOI] [PubMed] [Google Scholar]
- 6h. Butler M. S., Buss A. D., Biochem. Pharmacol. 2006, 71, 919; [DOI] [PubMed] [Google Scholar]
- 6i. Hunt E., Drugs Future 2000, 25, 1163; for studies on pleuromutilin binding to the ribosome; [Google Scholar]
- 6j. Schlünzen F., Pyetan E., Fucini P., Yonath A., Harms J. M., Mol. Microbiol. 2004, 54, 1287; [DOI] [PubMed] [Google Scholar]
- 6k. Davidovich C., Bashan A., Auerbach‐Nevo T., Yaggie R. D., Gontarek R. R., Yonath A., Proc. Natl. Acad. Sci. USA 2007, 104, 4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For a recent review of the synthesis of pleuromutilin analogues from the natural product, see: Fazakerley N. J., Procter D. J., Tetrahedron 2014, 70, 6911. [Google Scholar]
- 8.For an alternative approach to pleuromutilin analogues, see:
- 8a. Liu J., Lotesta S. D., Sorensen E. J., Chem. Commun. 2011, 47, 1500; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Lotesta S. D., Liu J., Yates E. V., Krieger I., Sacchettini J. C., Freundlich J. S., Sorensen E. J., Chem. Sci. 2011, 2, 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Comins D. L., Dehghani A., Tetrahedron Lett. 1992, 33, 6299. [Google Scholar]
- 10.For the addition of bis‐organocuprates to β‐halocyclohexenones to give spirocycles, see:
- 10a. Wender P. A., White A. M., J. Am. Chem. Soc. 1988, 110, 2218; [Google Scholar]
- 10b. Wender P. A., White A. W., McDonald F. E., Org. Synth. 1992, 70, 204. [Google Scholar]
- 11.See the Supporting Information.
- 12.
- 12a. Hosomi A., Endo M., Sakurai H., Chem. Lett. 1976, 5, 941; [Google Scholar]
- 12b. Hosomi A., Sakurai H., Tetrahedron Lett. 1976, 17, 1295; for a recent example in target synthesis using BF3 ⋅OEt2, see: [Google Scholar]
- 12c. Wanner M. J., Willard N. P., Koomen G.‐J., Pandit U. K., J. Chem. Soc. Chem. Commun. 1986, 396. [Google Scholar]
- 13.For a discussion of reversible radical formation from aldehydes/ketones using SmI2, see: Chopade P. R., Prasad E., Flowers R. A., J. Am. Chem. Soc. 2004, 126, 44.14709051 [Google Scholar]
- 14. Rudkin I. M., Miller L. C., Procter D. J., Organomet. Chem. 2008, 34, 19. [Google Scholar]
- 15.See the Supporting Information for CCDC numbers.
- 16.For alternative methods for nine‐membered carbocycle formation using SmI2, see:
- 16a. Saadi J., Lentz D., Reissig H.‐U., Org. Lett. 2009, 11, 3334; [DOI] [PubMed] [Google Scholar]
- 16b. Nandanan E., Dinesh C. U., Reissig H.‐U., Tetrahedron 2000, 56, 4267; [Google Scholar]
- 16c. Tamiya H., Goto K., Matsuda F., Org. Lett. 2004, 6, 545; [DOI] [PubMed] [Google Scholar]
- 16d. Molander G. A., Brown G. A., de Garcia I. S., J. Org. Chem. 2002, 67, 3459; [DOI] [PubMed] [Google Scholar]
- 16e. Molander G. A., Le Huérou Y., Brown G. A., J. Org. Chem. 2001, 66, 4511. [DOI] [PubMed] [Google Scholar]
- 17. Dess D. B., Martin J. C., J. Org. Chem. 1983, 48, 4155. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information