

Abstract
Incorporation of myristic acid onto the N terminus of a protein is a crucial modification that promotes membrane binding and correct localization of important components of signaling pathways. Recombinant expression of N‐myristoylated proteins in Escherichia coli can be achieved by co‐expressing yeast N‐myristoyltransferase and supplementing the growth medium with myristic acid. However, undesired incorporation of the 12‐carbon fatty acid lauric acid can also occur (leading to heterogeneous samples), especially when the available carbon sources are scarce, as it is the case in minimal medium for the expression of isotopically enriched samples. By applying this method to the brain acid soluble protein 1 and the 1–185 N‐terminal region of c‐Src, we show the significant, and protein‐specific, differences in the membrane binding properties of lauroylated and myristoylated forms. We also present a robust strategy for obtaining lauryl‐free samples of myristoylated proteins in both rich and minimal media.
Keywords: intrinsically disordered proteins, lauroylation, myristoylation, surface plasmon resonance, transferases
Introduction
Over the last twenty years, important progress in molecular biology and recombinant technologies has led to new applications and areas of research in the field of protein NMR. The ability to produce, by recombinant technologies, pure, highly concentrated, and isotopically labeled samples has allowed both solution‐ and solid‐state protein NMR to gain insight into the structural dynamics of many biomolecular systems with an atomic resolution. On the other hand, the very use of recombinant technology has led the field largely to neglect the importance of post‐translational modifications (PTMs), an indispensable step in the maturation of proteins towards their functional forms that regulate the activity, localization, stability, and physicochemical properties of proteins. PTMs have become especially relevant in the study of intrinsically disordered proteins (IDPs), which are attracting considerable interest among the biomolecular NMR community. Indeed, IDPs are more susceptible to undergoing PTMs than folded proteins.1 Because of the inherent flexibility of these proteins, PTMs will have an important impact on their activity, cellular localization, and interaction properties, by modulating their structural dynamics.2, 3, 4, 5, 6, 7, 8 For example, N‐terminal acetylation of α‐synuclein9 increases the helical propensity of the N‐terminal segment7 and enhances the affinity of α‐synuclein for calmodulin by a factor of 10.10 Thus, neglecting the impact of PTMs on the general properties of proteins will necessarily lead to an inaccurate description of their biochemical properties and ultimately of their physiological functions.
N‐Myristoylation is the covalent attachment, catalyzed by the enzyme N‐myristoyl transferase (NMT), of a 14‐carbon saturated fatty acid to the N‐terminal glycine residue through an amide bond; in eukaryotic systems it usually occurs co‐translationally. N‐Myristoylation is a very common PTM. By increasing the hydrophobicity of the modified protein, myristoylation is generally involved in membrane binding, targeting, and subcellular trafficking.11 As such, many myristoylated proteins are involved in important physiological processes such as signaling pathways, oncogenesis, or viral replication.12 Myristoylation can also be used to tune the activity of a protein through the effect known as the myristoyl‐electrostatic switch. This effect consists of a conformational change of the protein (usually triggered by the binding of a ligand) that will expose the myristoyl group, previously sequestered in a hydrophobic pocket.13 This mechanism is at the core of the function of proteins such as the ADP ribosylation factor or recoverin. Finally, myristoylation appears to play an important role in apoptosis, because many proteolitic products of caspase 3 are myristoylated and subsequently up‐ or down‐regulate apoptosis.14
Myristoylation does not naturally occur in Escherichia coli, the most commonly used organism for protein expression. Therefore, to obtain large amounts of isotopically labeled myristoylated proteins suitable for biomolecular NMR studies, two options are available. The first one is in vitro myristoylation, which requires, additionally to the protein of interest, purified NMT, myristoyl‐CoA (the co‐substrate of NMT), and an additional purification step to separate the myristoylated protein from the by‐products. An alternative approach is based on the co‐expression of the NMT and the substrate protein by use of a bicistronic vector, in order to enable in‐cell modification.15 However, Liu et al. reported that in minimal medium this procedure leads to mixtures of myristoylated and lauroylated forms of the recombinant protein.16 However, the factors leading to lauroylation remain unclear, and the degree of lauroylation seems to be highly variable and inconsistent. Indeed, other studies based on this myristoylation strategy (both in rich and in minimal media) did not report the formation of a lauroylated form.15, 17, 18
Here we confirm the observation of Liu et al. and demonstrate that myristoylated and lauroylated forms of the same protein exhibit significantly different lipid‐binding properties, emphasizing the need to ensure the homogeneity of the attached fatty acid chains. Consequently, we also report an optimized strategy for obtaining pure myristoylated proteins. This strategy has been applied to the production of two myristoylated proteins: brain acid soluble protein 1 (BASP1, also known as NAP‐22 and CAP‐23) and the first two domains of c‐Src: namely the Unique and SH3 domains (USH3). BASP1 is a 25 kDa intrinsically disordered protein highly abundant in the brain during development, involved in growth cone guidance and actin cytoskeleton organization19 and interacting with holo‐CaM specifically in its myristoylated form.20 c‐Src is the leading member of the Src family of non‐receptor tyrosine kinases (SFKs), which are involved in many signaling pathways. Deregulation of these kinases, and in particular of c‐Src itself, affects cell migration, proliferation, and survival, all of which contributes to its oncogenic potential. The N‐terminal (SH4) region of c‐Src is co‐translationally myristoylated at the N‐terminal glycine unit. SH4 is a basic peptide situated at the beginning of the Unique Domain of Src, an intrinsically disordered domain not conserved among the Src family. Myristoylated SH4 is responsible for c‐Src membrane anchoring, through concurrent hydrophobic and electrostatic interactions.
Results and Discussion
Expression of recombinant N‐myristoylated protein in minimal medium leads to a mixture of N‐myristoylated and N‐lauroylated protein
We expressed recombinantly myristoylated BASP1 (MyrBASP1) by using the bicistrionic vector designed by Glück et al.15 In order to measure the extent of myristoylation by mass spectrometry, we first performed the expression in minimal medium supplemented with unlabeled (14N) ammonium chloride. The LC‐MS analysis revealed the presence of two species differing in mass by 27 Da (Figure 1), thus suggesting that both N‐myristoylated and N‐lauroylated forms of BASP1 (differing in mass by 28 Da) had been produced, as reported previously in the case of Arf1.16 NMR analysis of the putative (later confirmed) myristoylated BASP reveals that the N‐terminal acylation leads to a disappearance of the 1H,15N HSQC crosspeaks corresponding to the N terminus (Figures S1 and S2 in the Supporting Information), probably due to micelle formation.
Figure 1.
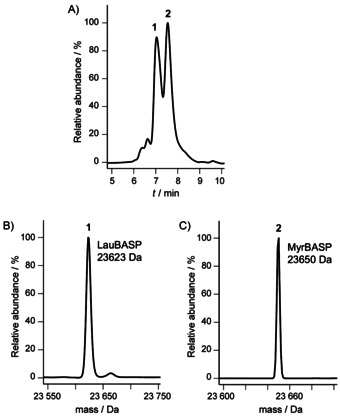
Expression of BASP1 in minimal medium leads to two different species. A) RP‐HPLC analysis of purified BASP1 co‐expressed with NMT in minimal medium. B) and C) MS analysis of products corresponding to peaks 1 and 2, respectively.
Further MS analysis of the two species by means of electron capture dissociation (ECD) and collisionally activated dissociation (CAD) confirmed that the difference in mass is 28.0302±0.0035 Da (CH2CH2, calcd mass 28.0313 Da) and is due to myristoylation and lauroylation at the N terminus of BASP1 (Supporting Information, data 1).
In order to explore the origin of the lauroylated form of BASP1 (LauBASP1), we measured the proportions of N‐myristoylated and N‐lauroylated forms of BASP1 in samples expressed under different conditions (Figure 2). When expressed in rich medium (lysogeny broth, LB), BASP1 is only found in its N‐myristoylated form (Figure 2 B). When expressed in minimal medium BASP1 is found in both its N‐myristoylated and N‐lauroylated forms, and the proportion of the N‐lauroylated form increases with the length of the expression time (Figure 2 A and C). This suggests that under scarce conditions (minimal medium and/or long expression times), myristoyl‐CoA is converted into lauroyl‐CoA through β‐oxidation and then used by the NMT (which is known to show weak discrimination between myristoyl‐CoA and shorter acyl‐CoA) to acylate BASP1. Expressing BASP1 in the presence of lauric acid only leads to the lauroylated form of BASP1 both in rich and in minimal medium (Figure 2 D and E). The same observations were made for USH3 (with a different expression strain of E. coli), regardless of whether it is expressed in rich or minimal medium (see below), thus suggesting that this problem is not specific to BASP1 but is instead a general problem that occurs especially in minimal medium but can also be observed in rich medium.
Figure 2.
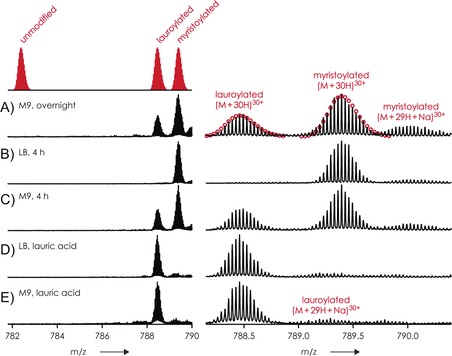
Proportion of N‐myristoylated and N‐lauroylated forms of BASP under different expression conditions. ESI spectra of human BASP co‐expressed with hNMT in A) M9 overnight, supplemented with myristic acid, B) LB for 4 h, supplemented with myristic acid, C) M9 for 4 h, supplemented with myristic acid, D) and E) LB and M9, respectively, supplemented with lauric acid; red circles in (A) show calculated isotopic profiles for [M+30 H]30+ ions of lauroylated and myristoylated protein.
N‐Myristoylated and N‐lauroylated forms of the same protein have different biochemical properties
To test whether the biochemical properties of BASP1 and USH3 are affected by the length of the N‐acyl chain, we performed surface plasmon resonance (SPR) experiments with immobilized liposomes. Because this method does not require isotopic labeling, we obtained fully myristoylated or lauroylated samples by expression in rich medium supplemented with the corresponding fatty acid (lauric or myristic acid). In the case of BASP1 (Figure 3 A), the binding to 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine/l‐α‐phosphatidylserine (DMPC/PS) liposomes of LauBASP1 (K D=86 nm) shows an affinity reduced by a factor of 10 relative to MyrBASP1 (K D=7.9 nm). Additionally, the length of the acyl chain also seems to affect the dissociation behavior because MyrBASP1 dissociates from the liposome more slowly than LauBASP1 (Figure 3 A).
Figure 3.
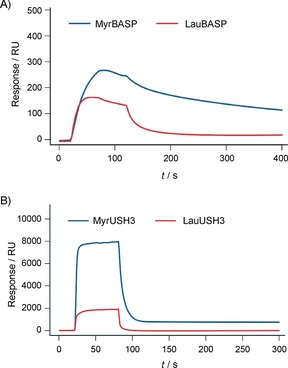
SPR analysis of the binding properties of different acylated forms of BASP and USH3 towards liposomes. A) Real‐time SPR response showing binding of MyrBASP and LauBASP to surface‐immobilized DMPC/PS (3:1) liposomes. The protein concentration was 0.2 μm. B) Real‐time SPR response showing binding of MyrUSH3 and LauUSH3 to surface‐immobilized DMPC/DMPG (2:1) liposomes; the protein concentration was 20 μm.
In the case of myristoylated USH3 (MyrUSH3), the difference is even more pronounced. Lauroylated USH3 (LauUSH3) binding to DMPC/DMPG (2:1; DMPG=1,2‐dimyristoyl‐sn‐glycero‐3‐phospho‐rac‐(1‐glycerol)) liposomes is four times weaker than for MyrUSH3 (Figure 3 B). Dissociation of the lauroylated form from the liposomes is also faster than for the myristoylated form.
Our SPR results clearly show that the length of the N‐acyl chain influences biochemical properties. Therefore the two species should be separated in order to obtain biochemically pure samples.
Production of pure N‐myristoylated USH3
Our SPR results show that the lengths of the N‐acyl chains influence the properties of N‐acylated proteins. Therefore, it is necessary to obtain a pure N‐myristoylated sample for the pursuit of subsequent biochemical and NMR studies. As described above, the lauroylated and myristoylated forms of BASP1 can be easily separated by RP‐HPLC. However, the use of organic solvent is not generally applicable to proteins containing folded domains, as in the case of USH3.
After protein expression, the soluble fraction of USH3 elutes from the size‐exclusion column in the form of three different peaks (Figure 4 A). SDS‐PAGE (Figure 4 B) reveals that peak 1 contain both USH3 and NMT, whereas peak 2 only contain USH3 (peak 3 contains a degraded form of the protein). Further MS analysis (Figure 4 C and D) showed that in peak 1 (which contains both USH3 and NMT) USH3 is exclusively myristoylated whereas in peak 2 USH3 (Figure 4 E) is only found in its unmodified and lauroylated forms. Hence, it appears that MyrUSH3 co‐purifies with NMT whereas LauUSH3 remains free of NMT due to the lower affinity of the enzyme for the shorter acyl chain forms.
Figure 4.
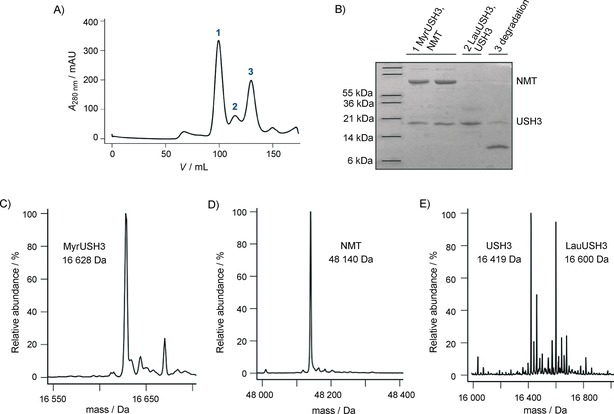
A) Size‐exclusion chromatography profiles of acylated USH3 purified from the supernatant. B) SDS‐PAGE analysis of pooled fractions of the peaks observed in (A). C) and D) MS analysis of peaks 1. E) MS analysis of the peaks 2.
This co‐purification might be exploitable, because the purification of the complex could lead to a fully myristoylated sample. However, we were not able to dissociate the complex fully, either by chromatographic methods (such as ion exchange or hydrophobic separation) or by the use of myristoylated coenzyme A as a competitor as suggested elsewhere.21 Consequently, we applied and refined an alternative method originally proposed by Ha et al.22 This exploits the strong affinity of the myristoylated protein towards membrane: a significant amount of myristoylated protein remains in the cell pellet upon centrifugation, whereas most of the NMT remains in the supernatant. This procedure relies on the resuspension and resolubilization of the membrane‐bound protein, with use of Triton, before further purification. This procedure leads to a mixture of lauroylated and myristoylated USH3 that can be separated by size‐exclusion chromatography. As can been seen from the size‐exclusion profile (Figure 5 A, red curve), the first peak corresponds to LauUSH3 (Figure 5 B) whereas the second corresponds to MyrUSH3 (Figure 5 C). Intriguingly, when large amounts of LauUSH3 are produced (Figure 5 A, green curve), a third peak corresponding to a mixture of Myr and LauUSH3 is observed (Figure 5 D). This last peak merges with the MyrUSH3 peak, compromising its purity. When the expression conditions are optimized to minimize the production of LauUSH3 (see below for a detailed explanation), pure MyrUSH3 can easily be obtained (Figure 5 A, blue curve). Thus, in order to be able to prepare pure myristoylated protein efficiently, the expression protocols have to be optimized to minimize the presence of lauroylated forms before the purification. In addition, size separation of the two acylated forms is a property of our particular system and might not occur for other proteins, thus emphasizing the necessity to use conditions that ensure the presence of pure myristoylated proteins.
Figure 5.
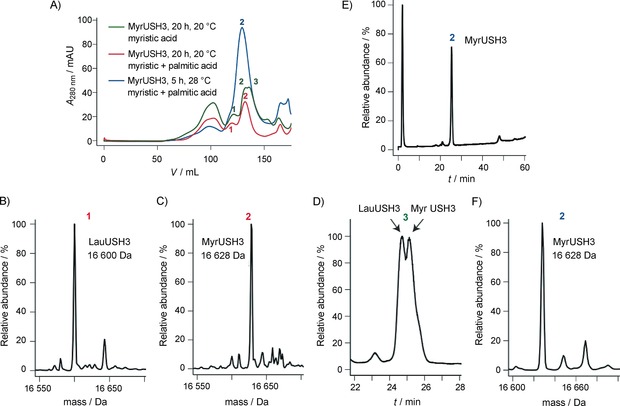
A) Size‐exclusion chromatography profiles of acylated USH3 expressed under different conditions. Red curve: in Rosetta cells, in the presence of both myristic and palmitic acid (20 h, 20 °C), two elution peaks are observed. Green curve: in T7 cells, in the presence only of myristic acid (20 h, 20 °C), three elution peaks are observed. Blue curve: in T7 cells, in the presence of both myristic and palmitic acid (5 h, 28 °C), one elution peak is observed. B) and C) Mass spectrometric analysis after separate pooling of the fractions corresponding to peaks 1 and 2, respectively. D) RP‐HPLC analysis of peak 3 of the red curve in (A) after pooling of its fractions. E) and F) RP‐HPLC and MS analysis of peak 2 of the blue curve after pooling of its fractions.
We hypothesized that lauroylCoA originates from β‐oxidation of added myristic acid. This hypothesis would explain the observation that lauroylation increases under conditions of limited availability of carbon sources (minimal medium) or after extended expression (presumably associated with nutrient depletion). Thus, one of the strategies that we tested was the addition of palmitic acid. NMT does not incorporate palmitoyl groups, but β‐oxidation of palmitic acid would contribute to replenish the myristoylCoA pool.
Table 1 summarizes the effects of different expression factors on the relative amounts of MyrUSH3 and LauUSH3 obtained after the Triton wash procedure, nickel column purification, and size‐exclusion chromatography. The induction time appears to be the most important factor, because longer expression times lead to greater amounts of lauroylated form. Nevertheless, some factors such as the expression strain or the addition of palmitic acid to the growth medium also have a significant impact on the myristoylation level. More specifically, expression in T7 cells yields lower lauroylation levels than expression in Rosetta cells. In rich medium, USH3 is found as a mixture of myristoylated and lauroylated forms even after short induction times (whereas under the same conditions BASP1 was fully myristoylated). However, adding palmitic acid to the growth medium leads to almost pure myristoylated USH3. To sum up, the set of best conditions consists of a short induction time in the presence of both palmitic and myristic acid in the expression medium. Use of these conditions together with the Triton wash purification procedure leads to pure and homogeneous natural‐abundance MyrUSH3 samples (Figure 5 A, blue curve, and E/F). However, expression in minimal media, required in order to produce isotopically labeled proteins, did not provide pure MyrUSH3, presumably due to limited availability of carbon sources. This limitation was overcome by using the Marley method, which consists of generating cell mass in unlabeled rich media and subsequent transfer into labeled media just before induction.23 In that case we were able to obtain sufficient amounts of labeled proteins (95 %) to enable NMR measurement. Finally, this protocol was also successfully used for the expression and purification of MyrBASP1, thus demonstrating the robustness of the method.
Table 1.
Effects of expression conditions in LB medium on the relative amounts of myristoylated and lauroylated USH3.
| Cell | Acyl chain addition | t [h] after | Lauroylated | Myristoylated |
|---|---|---|---|---|
| type | induction | USH3 [%] | USH3 [%] | |
| Rosetta | myristic acid | 5[a] | 20 | 80 |
| T7 | myristic acid | 5[a] | 20 | 80 |
| Rosetta | myristic+palmitic acid | 5[a] | 5 | 95 |
| T7 | myristic+palmitic acid | 5[a] | 2 | 98 |
| Rosetta | myristic acid | 20[b] | 80 | 20 |
| T7 | myristic acid | 20[b] | 35[c] | 65[c] |
| Rosetta | myristic+palmitic acid | 20[b] | 18 | 82 |
| T7 | myristic+palmitic acid | 20[b] | 3 | 97 |
The relative amounts of myristoylated and lauroylated USH3 were determined by integrating the elution peaks from size‐exclusion chromatography performed on the mixtures obtained after the Triton wash purification procedure. [a] At 28 °C. [b] At 20 °C. [c] Myristoylated and lauroylated species are poorly separated.
Conclusion
Myristoylation is an important post‐translation modification generally involved in subcellular trafficking and membrane association. Correct structural characterization of myristoylated proteins requires the production of their isotopically labeled forms. Co‐expression of yeast NMT with the protein of interest in E. coli provides an efficient method but is complicated by the simultaneous formation of lauroylated and myristoylated proteins. Although they are chemically similar, SPR experiments showed that the variation in chain length greatly alters the lipid‐binding properties. Therefore, a suitable method to obtain purely myristoylated proteins in E. coli needs to be developed. We suggest here that the addition of the shorter acyl chain appears to be due to the conjunction of two factors: 1) the availability of a shorter acyl‐CoA generated through β‐oxidation of the myristic acid that needs to be supplemented to the growth medium, and 2) the poor discrimination of the human NMT for the shorter acyl‐CoA.24 On the basis of this hypothesis, we have shown that addition of palmitic acid, which is not incorporated into the protein by NMT but can resupply the MyrCoA pool, contributes, under some conditions, to minimize the formation of the lauroylated form.
The capacity to isolate lauroylated and myristoylated proteins from mixtures of the two is dependent upon the physiochemical properties of the proteins of interest. In the case of a fully disordered protein (BASP1), the two forms can be separated by RP‐HPLC thanks to their different hydrophobicities. In the case of a partially or fully folded protein (USH3), we had to develop a more sophisticated procedure based on 1) optimization of the expression conditions to minimize the formation of LauUSH3, 2) use of the Triton wash purification protocol to obtain NMT‐free myristoylated protein with a low proportion of the lauroylated form, and 3) final purification by affinity and size‐exclusion chromatography. Interestingly, although both proteins have acyl moieties attached to a disordered segment, their biochemical properties seem to be affected in different ways by the length of the acyl chain. It is known that myristoylation by itself is not enough for stable membrane attachment. A second binding event is usually required.11 Examples include electrostatic interactions, a second aliphatic anchor, or additional contacts with amphipathic regions of the peptide backbone. Clearly, the myristoyl chain plays a stronger role in lipid binding by USH3 than by BASP1. This might be due to the proximity of the charged residues and the acyl chain in USH3, coupling the strength of the electrostatic interaction with the insertion of the acyl chain in the lipid bilayer, or might be the result of an indirect effect caused by modulation of the SH4 and SH3 interactions by the presence of distinct fatty acid chains attached to the SH4 domain.25
Thus, if pure lauroylated and myristoylated forms of the same protein can be obtained independently or separated from mixtures, the comparison of their lipid‐binding properties provides additional insight into the processes involved in fatty‐acid‐mediated lipid interactions.
To summarize, we provide evidence as to why the production of myristoylated proteins in E. coli by co‐expressing NMT results in a heterogeneously N‐acylated sample. We have demonstrated that the different N‐acylated forms have different biochemical properties and we have developed an expression/purification protocol to generate a homogeneously N‐myristoylated sample. We expect these observations to be especially relevant for the conduct of NMR studies in which the homogeneity of the sample is essential.
Experimental Section
Protein expression and purification of myristoylated BASP1: Expression was achieved with E. coli strain T7 Express (New England BioLabs) and the bicistronic vector pETDuet‐1Δ6His_hNMT_hBASP1_6His as already described.15 Expression of BASP1 for the analysis of the protein by mass spectrometry was done either in rich medium (LB medium) or in M9 minimal medium with unlabeled (14N) ammonium chloride (1 g L−1) and (12C) glucose (4 g L−1). In order to produce myristoylated protein, myristic acid was added to the growth medium at a final concentration of 50 μm 10 min before induction. Fresh myristic acid stock solution was prepared as described elsewhere.15 The cells were grown at 37 °C to an OD600 of 0.8 and induced by adding isopropyl β‐d‐thiogalactopyranoside (IPTG, 0.8 mm). The expression temperature was 28 °C for 4 h or overnight. The cells were pelleted by centrifugation at 2862 g at 4 °C for 20 min. The pellet was resuspended in lysis buffer (PBS) with protease inhibitors (cOmplete, Mini Protease Inhibitor Tablets, EDTA‐free, Roche) and sonicated on ice with a Branson W‐450 D sonifier with a microtip (3 min, 50 % amplitude) before centrifugation at 36 223 g for 20 min at 4 °C. The supernatant was applied to a Ni2+‐loaded HisTrap HP column (5 mL, GE Healthcare) that was pre‐equilibrated with PBS. The column was washed with PBS and high‐salt PBS [NaCl (1.5 m), imidazole (20 mm)] and five column volumes of PBS before elution with a linear gradient of PBS/PBS with imidazole (0.5 m).
FT‐ICR mass spectrometry: Protein samples were desalted with Vivaspin 500 centrifugal concentrators (Sartorius, Germany, PES membrane, MWCO 5000) as described previously26 Briefly, a protein solution (≈10 μm, 500 μL) was concentrated to 100 μL, and an aqueous ammonium acetate solution (100 mm, 400 μL) was added. The process was repeated five times, followed by six cycles of concentration and dilution with H2O. For ESI (flow rate 1.5 μL min−1), desalted protein was diluted to ≈2 μm in H2O/CH3OH 1:1 with acetic acid (1 % vol) as additive. H2O was purified to 18 MΩ⋅cm at room temperature with a Milli‐Q system (Millipore, Austria); CH3OH (Acros, Austria) was HPLC‐grade. Experiments were performed with a 7 Tesla Fourier transform ion cyclotron resonance (FT‐ICR) mass spectrometer equipped with an electrospray ionization source, a collision cell for CAD, and a hollow dispenser cathode for ECD.
Myristoylated USH3 expression: USH3 expression was performed in E. coli Rosetta (DE3)pLysS cells (Novagen) or T7 Express (New England BioLabs) cells and using the bicistronic vector pETDuet‐1Δ6His_hNMT_USH3_6His. The cells were grown at 37 °C to an OD600 of 0.8, and 10 min before induction with IPTG (final concentration 1 mm, Melford), freshly prepared myristic and/or palmitic acid (Sigma) were added to the cell culture, at a final concentration of 200 μm. For the expression of isotopically labeled samples the protocol developed by Marley et al. was used:23 cells were first grown in LB to an OD600 of 0.4 and were then centrifuged at 1000 g and 4 °C for 20 min. The pellet was resuspended in half the volume of minimal medium containing ammonium chloride and glucose. After 20 min at 37 °C, myristic and palmitic acid were added as described above. The expression temperature was either 28 °C (for 5 h) or 20 °C (for 20 h). The cells were pelleted by centrifugation at 3993 g at 4 °C for 30 min. The pellet was resuspended in lysis buffer [Tris⋅HCl (20 mm), NaCl (300 mm), imidazole (5 mm), pH 8], to which protease inhibitors (protein inhibitor cocktail and phenylmethanesulfonyl fluoride (PMSF); 1 mm), both from Sigma) were added.
MyrUSH3 purification: The resuspended pellet was sonicated on ice before centrifugation at 25 000 rpm for 45 min at 4 °C. The protein appeared to be distributed between the supernatant and the pellet, so two different purification methods were used. For the soluble fraction, the supernatant was applied to a Ni‐NTA column (Qiagen) followed by size‐exclusion chromatography in a Superdex 75 column in sodium phosphate buffer (Na3PO4) [NaP (50 mm), NaCl (150 mm), EDTA (0.2 mm), pH 7.5]. For the insoluble fraction, the pellet was resuspended in lysis buffer containing Triton X‐100 (1 %). The resuspended pellet was centrifuged again for 30 min at 75 600 g, and the procedure was repeated twice or three times. The supernatant from the Triton washes was purified by immobilized metal affinity chromatography as described above. If lauroylated species were present, they eluted from the size‐exclusion chromatographic column at an apparent higher molecular weight than the myristoylated ones. This method enabled the two different acylated species to be separated and their respective amounts quantified. A comparison of various expression and purification protocols is presented in the Results section.
LC‐MS: The purities and identities of the products were established by UPLC coupled to MS [Acquity chromatograph with a BioSuite pPhenyl column (1000RPC 2.0×75 mm)] coupled to a LCT‐Premier spectrometer (Waters corporation).
Liposome preparation: DMPC, DMPG, and PS were purchased from Avanti Polar Lipids, Inc. The lipids were dissolved in chloroform or, in the case of DMPG, in chloroform/methanol/H2O (65:35:8). Liposomes were prepared by mixing the appropriate amount of lipids in the solvent. The solvent was removed in a rotary evaporator, followed by rehydration and vortexing at 40 °C with the buffer used for SPR analysis, with a final lipid concentration of 1 mm. The different liposomes were prepared with a DMPC/DMPG ratio of 2:1 or a DMPC/PS ratio of 4:1. Large unilamellar vesicles were mechanically extruded at 40 °C by use of a Thermobarrel extruder (10 mL Thermobarrel extruder; Lipex Northerns Lipids Inc. Burnaby, Canada) with at least ten cycles of extrusion and use of a polycarbonate filter (100 nm). To verify the appropriate size of the liposomes, the mean diameter was checked by dynamic light scattering (Zetasizer Nanoseries S, Malvern Instruments).
Surface plasmon resonance (SPR): SPR experiments with BASP1 were carried out with a Biacore 2000 instrument (Biacore, GE Healthcare) and SPR sensor chip (L1, Biacore, GE Healthcare). Liposomes were injected for 500 s at a flow rate of 5 μL min−1. The reference channel was coated with BSA by use of a 200 s injection of BSA (1 mg mL−1, Sigma, fatty acid free) at a flow rate of 10 μL min−1. Protein binding experiments were performed at 50 μL min−1. The interaction of MyrBASP1 or LauBASP1 with liposomes was followed by observing the SPR response when a solution of protein was injected for 100 s (association phase), followed by a 300 s washing period (dissociation phase). MyrBASP1 binding to DMPC/PS liposomes was monitored by injections at concentrations ranging from 10 nm to 2.5 μm. LauBASP1 binding to DMPC/PS liposomes was monitored by injections at concentrations ranging from 20 nm to 7.5 μm. All experiments were performed in the running buffer, which consisted of Na3PO4 (20 mm), NaCl (50 mm), pH 7.4. The surface was regenerated with a series of CHAPS (20 mm)/HCl (10 mm)/CHAPS (20 mm) pulses, each pulse for 30 s at 100 μL min−1. Each experiment was started with freshly captured liposomes. Liposome coating was reproducible, with a variation smaller than 4 % between subsequent coatings, ensuring very reproducible protein binding curves.
SPR experiments with MyrUSH3 were performed in a very similar fashion, with slight modifications. The SPR chip (a 2D carboxymethyldextran surface) was purchased from Xantec and modified by covalent attachment of phytosphingosine (TebuBio) to allow the capture of DMPC:DMPG (2:1) liposomes. Liposomes were injected at 10 μL min−1 for 200 s before the protein binding experiment: MyrUSH3 or LauUSH3 was injected at 50 μL min−1 for 60 s, and dissociation was allowed for 300 s. The running buffer was composed of Na3PO4 (50 mm), NaCl (150 mm), EDTA (0.2 mm, pH 7.5). Liposome coating was reproducible, with a variation of about 1 % between the subsequent coatings, ensuring very reproducible protein binding curves. Data analysis was performed with the Biacore Biaevalution software.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The bicistronic vector encoding NMT was a kind gift from Dr. Dieter Willbold (Forschungszentrum Juelich Institute of Complex Systems). This work was supported by grants from the Austrian Science Foundation (FWF P26317 and Y372, to K.B.), as well as by funds from MINECO (BIO2013–45793‐R) and la Fundació Marató TV3. A.L.L.R. holds an IRB/La Caixa fellowship. We thank Markus Hartl and the Mass Spectrometry Facility (Campus Science support Facilities) of the F. Max Perutz Laboratories in Vienna, as well as Christian Becker and Aleksandr Kravchuk from the Institute of Biological Chemistry in Vienna. We gratefully acknowledge the technical help of Marta Taules (CCiTUB) in the SPR experiments and the support of Maria Antonia Busquets (Physical Chemistry Department, Faculty of Pharmacy, UB) in the preparation of liposomes, as well as the Mass Spectrometry Core Facility, in particular Marta Vilaseca (Institute for Research in Biomedicine). We also thank Mariano Maffei for his input.
A. G. Flamm, A.-L. Le Roux, B. Mateos, M. Díaz-Lobo, B. Storch, K. Breuker, R. Konrat, M. Pons, N. Coudevylle, ChemBioChem 2016, 17, 82.
References
- 1. Khoury G. A., Baliban R. C., Floudas C. A., Sci. Rep. 2011, 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Errington N., Doig A. J., Biochemistry 2005, 44, 7553–7558. [DOI] [PubMed] [Google Scholar]
- 3. Meyer B., Möller H., Top. Curr. Chem. 2006, 267, 187–251. [Google Scholar]
- 4. Liu J., Faeder J. R., Camacho C. J., Proc. Natl. Acad. Sci. USA 2009, 106, 19819–19823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deribe Y. L., Pawson T., Dikic I., Nat. Struct. Mol. Biol. 2010, 17, 666–672. [DOI] [PubMed] [Google Scholar]
- 6. Mao A. H., Crick S. L., Vitalis A., Chicoine C. L., Pappu R. V., Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maltsev A. S., Ying J., Bax A., Biochemistry 2012, 51, 5004–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Theillet F. X., Binolfi A., Frembgen-Kesner T., Hingorani K., Sarkar M., Kyne C., Li C., Crowley P. B., Gierasch L., Pielak G. J., Elcock A. H., Gershenson A., Selenko P., Chem. Rev. 2014, 114, 6661–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartels T., Choi J. G., Selkoe D. J., Nature 2011, 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gruschus J. M., Yap T. L., Pistolesi S., Maltsev A. S., Lee J. C., Biochemistry 2013, 52, 3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Resh M. D., Biochim. Biophys. Acta Mol. Cell Res. 1999, 1451, 1–16. [DOI] [PubMed] [Google Scholar]
- 12. Resh M. D., Nat. Chem. Biol. 2006, 2, 584–590. [DOI] [PubMed] [Google Scholar]
- 13. McLaughlin S., Aderem A., Trends Biochem. Sci. 1995, 20, 272–276. [DOI] [PubMed] [Google Scholar]
- 14. Martin D. D., Beauchamp E., Berthiaume L. G., Biochimie 2011, 93, 18–31. [DOI] [PubMed] [Google Scholar]
- 15. Glück J. M., Hoffmann S., Koenig B. W., Willbold D., PLoS One 2010, 5, e10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y., Kahn R. A., Prestegard J. H., Structure 2009, 17, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Breuer S., Gerlach H., Kolaric B., Urbanke C., Opitz N., Geyer M., Biochemistry 2006, 45, 2339–2349. [DOI] [PubMed] [Google Scholar]
- 18. Lim S., Peshenko I., Dizhoor A., Ames J. B., Biochemistry 2009, 48, 850–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frey D., Laux T., Xu L., Schneider C., Caroni P., J. Cell Biol. 2000, 149, 1443–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsubara M., Nakatsu T., Kato H., Taniguchi H., EMBO J. 2004, 23, 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morgan C. R., Miglionico B. V., Engen J. R., Biochemistry 2011, 50, 3394–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ha V. L., Thomas G. M., Stauffer S., Randazzo P. A., Methods Enzymol. 2005, 404, 164–174. [DOI] [PubMed] [Google Scholar]
- 23. Marley J., Lu M., Bracken C., J. Biomol. NMR 2001, 20, 71–75. [DOI] [PubMed] [Google Scholar]
- 24. Neubert T. A., Johnson R. S., Hurley J. B., Walsh K. A., J. Biol. Chem. 1992, 267, 18274–18277. [PubMed] [Google Scholar]
- 25. Maffei M., Arbesu M., Le Roux A. L., Amata I., Roche S., Pons M., Structure 2015, 23, 893–902. [DOI] [PubMed] [Google Scholar]
- 26. Hartl M., Mitterstiller A. M., Valovka T., Breuker K., Hobmayer B., Bister K., Proc. Natl. Acad. Sci. USA 2010, 107, 4051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary