

Abstract
The synthesis of an all‐halogen‐bonding rotaxane for anion recognition is achieved by using active‐metal templation. A flexible bis‐iodotriazole‐containing macrocycle is exploited for the metal‐directed rotaxane synthesis. Endotopic binding of a CuI template facilitates an active‐metal CuAAC iodotriazole axle formation reaction that captures the interlocked rotaxane product. Following copper‐template removal, exotopic coordination of a more sterically demanding rhenium(I) complex induces an inversion in the conformation of the macrocycle component, directing the iodotriazole halogen‐bond donors into the rotaxane’s interlocked binding cavity to facilitate anion recognition.
Keywords: halogen bonding, host–guest systems, rotaxanes, supramolecular chemistry, template synthesis
The template‐directed synthesis of mechanically interlocked molecular architectures, such as rotaxanes and catenanes, is a rapidly expanding area of supramolecular chemistry, facilitating the preparation of a wide range of mechanically bonded systems with utility in molecular machines and prototypical nano‐technological applications.1–4 Such species also offer considerable advantages in the field of molecular recognition.5–9 In particular, we have developed a discrete anion‐templation methodology10 as a means of constructing interlocked structures, which are able to bind anions with high affinity within the cavity formed between the interlocked components through convergent hydrogen‐6, 7 or halogen‐bonding (XB)11–14 interactions. An impressive feature of this method of rotaxane and catenane assembly is that the resultant interlocked molecular structure retains a degree of functionality based on the anion template itself.7, 8
In 2006, Leigh and co‐workers described an active‐metal template synthesis of a [2]rotaxane by using a CuI‐catalysed azide–alkyne cycloaddition (CuAAC) click reaction.15 The CuI metal cation plays a dual role in the synthesis: orientating the respective azide‐ and alkyne‐functionalised half‐axle components and catalysing the covalent bond formation between the two species. This approach relies on the endotopic binding of the metal cation within the cavity of the macrocycle, such that the formation of the axle component occurs through the annulus of the cyclic component. Subsequently, a range of transition‐metal‐catalysed reactions have been exploited for active‐metal interlocked molecule synthesis16 to construct a plethora of rotaxanes,17–19 catenanes,20, 21 higher‐order interlocked structures22, 23 and molecular knots.24
The template‐directed synthesis of interlocked hosts for cation or anion recognition typically requires the respective‐charged guest as the templating species: that is, for example, employing an anion template to direct the formation of an anion‐binding host. Indeed, the use of cations to template the formation of an anion‐binding interlocked host is rare25 and has not been achieved by using an active‐metal template approach, despite the wide range of mechanical bond‐forming reactions and sophisticated interlocked molecular architectures accessible through metal‐cation templation strategies.26
Herein, we report as a proof of concept the first use of active‐metal templation to synthesise an all‐halogen‐bonding rotaxane host system for anion recognition by exploiting a bis‐iodotriazole ligand incorporated within a flexible macrocyclic framework.
The utilisation of metal cations to template the synthesis of anion‐binding rotaxanes is hindered by the fundamentally antagonistic intermolecular interactions required: namely, Lewis basic sites for metal‐cation coordination and Lewis acidic sites for anion binding. To overcome this limitation, we envisaged a strategy for active‐metal template rotaxane synthesis in which a functional motif that contains both Lewis basic and Lewis acidic (halogen‐bond donor) sites, integrated into a flexible macrocycle component, can undergo inversion of conformation of the donor sites under metal‐cation binding control (Scheme 1). Endo‐complexation of the catalytic metal cation M1, favoured by the exo‐topic preference of the bulky iodine atoms, facilitates the active‐metal template formation of a [2]rotaxane that contains a halogen‐bond‐donor axle component. Removal of M1 and subsequent complexation of a more sterically demanding metal M2, which requires exo‐coordination to the macrocycle component, affords a rotaxane‐host cavity with convergent polarised halogen‐bond‐donor groups for anion recognition.
Scheme 1.
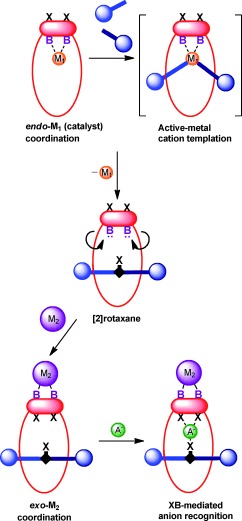
Schematic representation of an active‐metal templation route to a halogen‐bonding rotaxane for anion recognition by inversion of the macrocycle geometry. B=Lewis base, X=Lewis acid (halogen‐bond‐donor group), M1=catalytic metal cation, M2=more sterically demanding metal complex.
The bis‐triazole motif has been previously reported to act as a bidentate planar N‐donor ligand for transition‐metal complexation.27–30 We anticipated that the analogous bidentate bis‐iodotriazole motif could also act as a N‐donor ligand whilst simultaneously binding anions through XB interactions with the XB‐donor iodine atoms, which are themselves polarised by complexation to the transition metal. Endotopic‐binding of the CuI cation facilitates the active‐metal template synthesis of the rotaxane by directing and catalysing the CuAAC click reaction through the annulus of the macrocycle.
Whilst the analogous proto‐click reaction using a terminal alkyne has previously been exploited for active‐metal rotaxane synthesis, the use of an iodo‐alkyne31 for such a reaction is unprecedented. Removal of the CuI template and subsequent metalation with a more sterically demanding rhenium(I) metal complex inverts the geometry of the macrocycle, forcing the transition metal to adopt an exotopic orientation and directing the XB‐donor iodine atoms into the macrocycle’s cavity.
Macrocycle 2 was prepared by using a CuI‐mediated CuAAC click reaction between di‐iodobutadiyne and bis‐azide 1 under high‐dilution conditions (Scheme 2). The active‐metal templation procedure for rotaxane synthesis was achieved by stirring an equimolar mixture of macrocycle 2 and Cu(CH3CN)4PF6 with four equivalents of both azide‐terminated terphenyl stopper 3 and iodo‐alkyne terphenyl stopper 4 in CH2Cl2 (Scheme 3). After 48 hours, the alkyne and azide starting materials were completely consumed, and the reaction mixture was washed with an aqueous basic solution of ethylenediaminetetraacetate (EDTA) to remove the copper template. 1H NMR analysis of the crude reaction mixture revealed that the [2]rotaxane 5 was formed in 30 % yield (for comparison, in the presence of one equivalent of azide and iodo‐alkyne starting materials the crude yield was 15 %). The remaining 70 % was accounted for by non‐interlocked macrocycle 2 and axle by‐product. Purification by preparative thin‐layer silica‐gel chromatography afforded the tris‐iodotriazole rotaxane 5 in 25 % isolated yield, which was fully characterised by 1H and 13C NMR techniques and high‐resolution mass spectrometry. The partial 1H NMR spectra of rotaxane 5, macrocycle 2 and the corresponding non‐interlocked axle13 (isolated from the reaction mixture) are compared in Figure 1.
Scheme 2.
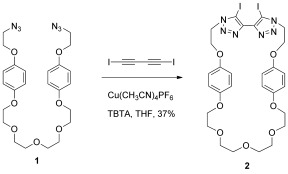
Synthesis of bis‐iodotriazole macrocycle 2
Scheme 3.
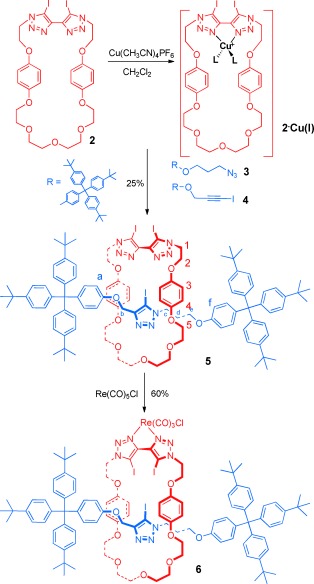
Active‐metal template synthesis of halogen‐bonding rotaxane 6.
Figure 1.
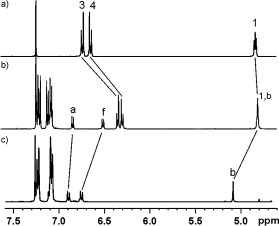
1H NMR spectra of a) macrocycle 2, b) rotaxane 5 and c) the axle (CDCl3, 500 MHz). For atom labels see Scheme 3.
Of particular note is the significant upfield perturbation of the macrocycle hydroquinone protons 3 and 4, due to donor–acceptor interactions between the relatively electron‐deficient iodotriazole of the axle component and the electron‐rich hydroquinone motifs, and is diagnostic of rotaxane formation. Furthermore, axle protons b and f are perturbed upfield in the rotaxane spectrum. Additional evidence for the interlocked nature of the rotaxane was obtained in the 1H–1H ROESY NMR spectrum in which multiple through‐space interactions between macrocycle and axle protons are observed (Figure 2).
Figure 2.
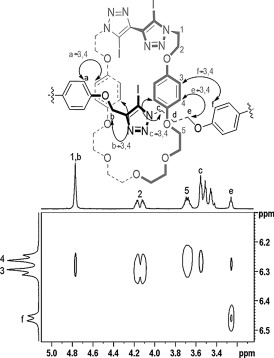
Partial 1H–1H ROESY NMR spectrum of rotaxane 5 showing selected through‐space interactions between the interlocked macrocycle and axle components (CDCl3, 500 MHz). For further details see the Supporting Information.
Exotopic metalation of rotaxane 5 with rhenium(I) was achieved by stirring a solution of the rotaxane in CH2Cl2 with Re(CO)3(CH3CN)2Cl (prepared by stirring Re(CO)5Cl in CH3CN at reflux), affording ReI‐rotaxane 6 in 60 % yield following purification by preparative thin‐layer silica‐gel chromatography. The rotaxane was fully characterised by 1H and 13C NMR spectroscopy and high‐resolution mass spectrometry (see the Supporting Information). The 1H NMR spectra of ReI‐rotaxane 6 and the metal‐free rotaxane 5 are compared in Figure 3. Importantly, the hydroquinone protons 3 and 4 remain shifted upfield relative to the free macrocycle, which confirms that the macrocycle and axle components remain interlocked following metalation.
Figure 3.
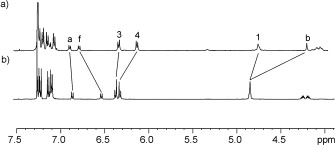
1H NMR spectra of a) Re‐rotaxane 6 and b) rotaxane 5 (CDCl3, 500 MHz). For atom labels see Scheme 2.
To demonstrate that rotaxane 6, having been prepared through metal‐cation templation, is capable of XB‐mediated anion binding, a preliminary 1H NMR chloride binding titration was conducted in CDCl3. Rotaxane 6 is notable in that the binding domain is composed of three iodotriazole halogen‐bond‐donor groups and that no hydrogen‐bond‐donor protons are available for anion binding. It was therefore not surprising that upon addition of chloride only small perturbations of protons 1, 4, b and f—which are located in the vicinity of the interlocked cavity and indicative of halide binding—were observed in the 1H NMR spectrum (see the Supporting Information).
As observed with previously reported ReI[27] and RuII[28–30] bis‐prototriazole complexes, ReI‐rotaxane 6 is not luminescent. Nevertheless, quantitative anion‐binding data was determined by monitoring the UV/Vis absorption spectrum of the rotaxane in CHCl3 upon addition of anions (see the Supporting Information). In a typical titration experiment, a decrease in the broad metal‐to‐ligand charge transfer (MLCT)27 absorption centered around 340 nm was observed, with an isosbestic point at 320 nm suggesting the formation of a single rotaxane–anion complex in solution. The 1:1 stoichiometric anion association constants were determined by a global fit procedure using SPECFIT32 and are shown in Table 1 together with quantitative anion‐binding data obtained with an acyclic bis‐iodotriazole ReI receptor 7 (Figure 4) with pendent octyl chains.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Figure 4.
Acyclic bis‐iodotriazole ReI receptor 7.
Importantly, the neutral ReI‐rotaxane host exhibits a selectivity preference for chloride over the larger halides and acetate, suggesting that the rotaxane’s XB‐donor cavity is of complementary size and shape for the smaller halide guest species. By contrast, the acyclic XB‐donor receptor 7 displays no selectivity, binding the halides and acetate with near equal strength. Furthermore, analogous titration experiments with the metal‐free rotaxane 5 and the metal‐free acyclic receptor 7 gave no evidence of anion binding, which highlights the requirement of the rhenium metal to polarize and pre‐organise the iodotriazole XB donors. These observations demonstrate that the XB‐ReI‐rotaxane 6, prepared initially by cation templation with one metal and subsequent complexation with a different metal, is capable of enhanced XB‐mediated chloride recognition compared to an XB ReI‐acyclic analogue. This represents a rare example of an interlocked host that binds anions through solely halogen‐bonding interactions.33, 34
In conclusion, an active‐metal template strategy has been utilised for the first time to construct a halogen‐bonding rotaxane for anion recognition. Exploiting the endo‐/exo‐conformational flexibility of a bis‐iodotriazole‐containing macrocycle expedites a CuI‐catalysed CuAAC active‐metal iodotriazole axle component formation within the macrocycle’s cavity to form an all‐XB [2]rotaxane. Subsequent removal of the copper template and re‐metallation with a more sterically demanding rhenium(I) complex directs the iodotriazole XB donors into the rotaxane’s binding cavity to facilitate XB anion recognition. The preparation of halogen‐bonding interlocked anion hosts by using novel template‐directed synthesis strategies is continuing in our laboratories.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
M.J.L. thanks the EPSRC for a Doctoral Award and the European Research Council for funding under the European Union’s Framework Program (FP7/2007‐2013) ERC Advanced Grant Agreement no. 267426. Y.X. is supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) and by a University of Oxford Clarendon Fund Scholarship.
References
- 1. Balzani V., Credi A., Silvi S., Venturi M., Chem. Soc. Rev. 2006, 35, 1135–1149. [DOI] [PubMed] [Google Scholar]
- 2. Kay E. R., Leigh D. A., Zerbetto F., Angew. Chem. Int. Ed. 2007, 46, 72–191; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 72–196. [Google Scholar]
- 3. Kay E. R., Leigh D. A., Pure Appl. Chem. 2008, 80, 17–29. [Google Scholar]
- 4. Fang L., Olson M. A., Benítez D., Tkatchouk E., W. A. Goddard III , Stoddart J. F., Chem. Soc. Rev. 2010, 39, 17–29. [DOI] [PubMed] [Google Scholar]
- 5. Langton M. J., Beer P. D., Acc. Chem. Res. 2014, 47, 1935–1949. [DOI] [PubMed] [Google Scholar]
- 6. Wisner J. A., Beer P. D., Drew M. G. B., Sambrook M. R., J. Am. Chem. Soc. 2002, 124, 12469–12476. [DOI] [PubMed] [Google Scholar]
- 7. Hancock L. M., Beer P. D., Chem. Eur. J. 2009, 15, 42–44. [DOI] [PubMed] [Google Scholar]
- 8. Langton M. J., Duckworth L. C., Beer P. D., Chem. Commun. 2013, 49, 8608–8610. [DOI] [PubMed] [Google Scholar]
- 9. Collins C. G., Peck E. M., Kramer P. J., Smith B. D., Chem. Sci. 2013, 4, 2557–2563. [Google Scholar]
- 10. Spence G. T., Beer P. D., Acc. Chem. Res. 2013, 46, 571–586. [DOI] [PubMed] [Google Scholar]
- 11. Beale T. M., Chudzinski M. G., Sarwar M. G., Taylor M. S., Chem. Soc. Rev. 2013, 42, 1667–1680. [DOI] [PubMed] [Google Scholar]
- 12. Gilday L. C., Robinson S. W., Barendt T. A., Langton M. J., Mullaney B. R., Beer P. D., Chem. Rev. 2015, 115, 7118–7195. [DOI] [PubMed] [Google Scholar]
- 13. Kilah N. L., Wise M. D., Serpell C. J., Thompson A. L., White N. G., Christensen K. E., Beer P. D., J. Am. Chem. Soc. 2010, 132, 11893–11895. [DOI] [PubMed] [Google Scholar]
- 14. Langton M. J., Robinson S. W., Marques I., Félix V., Beer P. D., Nat. Chem. 2014, 6, 1039–1043. [DOI] [PubMed] [Google Scholar]
- 15. Aucagne V., Hänni K. D., Leigh D. A., Lusby P. J., Walker D. B., J. Am. Chem. Soc. 2006, 128, 2186–2187. [DOI] [PubMed] [Google Scholar]
- 16. Crowley J. D., Goldup S. M., Lee A.‐L., Leigh D. A., McBurney R. T., Chem. Soc. Rev. 2009, 38, 1530–1541. [DOI] [PubMed] [Google Scholar]
- 17. Saito S., Takahashi E., Nakazono K., Org. Lett. 2006, 8, 5133–5136. [DOI] [PubMed] [Google Scholar]
- 18. Aucagne V., Berná J., Crowley J. D., Goldup S. M., Hänni K. D., Leigh D. A., Lusby P. J., Ronaldson V. E., Slawin A. M. Z., Viterisi A., J. Am. Chem. Soc. 2007, 129, 11950–11963. [DOI] [PubMed] [Google Scholar]
- 19. Hoekman S., Kitching M. O., Leigh D. A., Papmeyer M., Roke D., J. Am. Chem. Soc. 2015, 137, 7656–7659. [DOI] [PubMed] [Google Scholar]
- 20. Sato Y., Yamasaki R., Saito S., Angew. Chem. Int. Ed. 2009, 48, 504–507; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 512–515. [Google Scholar]
- 21. Goldup S. M., Leigh D. A., Long T., McGonigal P. R., Symes M. D., Wu J., J. Am. Chem. Soc. 2009, 131, 15924–15929. [DOI] [PubMed] [Google Scholar]
- 22. Langton M. J., Matichak J. D., Thompson A. L., Anderson H. L., Chem. Sci. 2011, 2, 1897. [Google Scholar]
- 23. Neal E. A., Goldup S. M., Chem. Sci. 2015, 6, 2398–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barran P. E., Cole H. L., Goldup S. M., Leigh D. A., McGonigal P. R., Symes M. D., Wu J., Zengerle M., Angew. Chem. Int. Ed. 2011, 50, 12280–12284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12488–12492. [Google Scholar]
- 25. Knighton R. C., Beer P. D., Chem. Commun. 2014, 50, 1540–1542. [DOI] [PubMed] [Google Scholar]
- 26. Beves J. E., Blight B. A., Campbell C. J., Leigh D. A., McBurney R. T., Angew. Chem. Int. Ed. 2011, 50, 9260–9327; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9428–9499. [Google Scholar]
- 27. Monkowius U., Ritter S., König B., Zabel M., Yersin H., Eur. J. Inorg. Chem. 2007, 4597–4606. [Google Scholar]
- 28. Fletcher J. T., Bumgarner B. J., Engels N. D., Skoglund D. A., Organometallics 2008, 27, 5430–5433. [Google Scholar]
- 29. Welby C. E., Grkinic S., Zahid A., Uppal B. S., Gibson E. A., Rice C. R., Elliott P. I. P., Dalton Trans. 2012, 41, 7637–7646. [DOI] [PubMed] [Google Scholar]
- 30. Hohloch S., Suntrup L., Sarkar B., Organometallics 2013, 32, 7376–7385. [Google Scholar]
- 31. Hein J. E., Tripp J. C., Krasnova L. B., Sharpless K. B., Fokin V. V., Angew. Chem. Int. Ed. 2009, 48, 8018–8021; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8162–8165. [Google Scholar]
- 32. SPECFIT v.2.02, Spectrum Software Associates, Chapel Hill, NC, v.2.02.
- 33. Caballero A., Zapata F., White N. G., Costa P. J., Félix V., Beer P. D., Angew. Chem. Int. Ed. 2012, 51, 1876–1880; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1912–1916. [Google Scholar]
- 34. Mullaney B. R., Thompson A. L., Beer P. D., Angew. Chem. Int. Ed. 2014, 53, 11458–11462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11642–11646. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information