

Key Clinical Message
We report a case of several autoimmune disorders eventually presenting as severe multi organ dysfunction syndrome caused by adult hemophagocytic lymphohistiocytosis (HLH). Clinical and laboratory tests might lead to fatal misinterpretation without awareness of its diagnostic evaluation, as HLH shares common features with sepsis and immune‐mediated systemic inflammatory response syndromes.
Keywords: Hemophagocytic lymphohistiocytosis, macrophage activation syndrome, multi organ dysfunction, MuSK‐Ab‐positive myasthenia gravis, systemic lupus erythematosus
Introduction
Hemophagocytic syndromes are commonly encountered and first described in children 1, but increasingly reported in adults 2. A recent review identified about 2200 adult cases worldwide, of which nearly 50% originated in Japan. The geographical variability with locally different predominant etiologies was attributed a specific genetic background or different trigger factors, such as infections. The mean age reported was about 50 years with a female‐to‐male ratio of 7:1 (in children, 1:1) 3. However, interpretation of epidemiologic data might be confounded by underdiagnosis 4; a retrospective case series from Sweden found that only 34% of diagnoses were made antemortem 3.
Several triggers and diseases, all of which either represent or induce an alteration of immune homeostasis 5, have been associated with the development of hemophagocytic lymphohistiocytosis (HLH), which is generally divided into primary (genetic) and secondary (acquired) forms. The latter can be subclassified into (1) infection‐associated, mostly by Epstein–Barr virus (EBV) and human immunodeficiency virus (HIV), (2) malignancy‐associated, (3) autoimmune‐associated, or (4) other syndromes 6.
The exact mechanism underlying HLH remains unclear, but all primary forms share a defect in the cytotoxic pathway of natural killer cells and/or cytotoxic lymphocytes required to eliminate activated macrophages 7. Studies of familial hemophagocytic lymphohistiocytoses (FHL) have identified mutations of genes encoding proteins for perforin synthesis (PRF1), cytolytic granule maturation (UNC13D), granule exocytosis (STX11) and granule release (STXBP2). Although FHL is considered an autosomal recessive inherited disorder, patients with presumed secondary HLH have been found to have heterocygosity for mutations in FHL‐associated genes 8. This impairment of cytotoxic function or other causes lead to a cytokine storm of interferon gamma (IFN‐γ), tumor necrosis factor alpha (TNF‐α), interleukins (IL–) 6, 10 and 12, and soluble IL‐2 receptor (sIL2R) which, along with less well‐characterized others, are thought to directly cause an ultimately fatal organ damage if the disease is undiagnosed and left untreated 7, 9. The associated clinical syndrome is characterized by acute or subacute nonspecific onsets with fever (>38.5°C), hepatosplenomegaly, bi‐ or pancytopenia, and frequently neurologic symptoms 2, 10. It typically progresses to multiple organ failure and disseminated intravascular coagulation (DIC), and resembles typical features of systemic inflammatory response syndrome (SIRS) requiring intensive care treatment 3, 6.
Case Report
A 52‐year‐old female presented to a nonaffiliated hospital with a myasthenic syndrome including exercise‐dependent muscle weakness, double‐vision and dysphagia. Her medical history was remarkable for a systemic lupus erythematosus (SLE) diagnosed at the age of 17 that led to an acute attack of lupus nephritis at the age of 36. Maintenance treatment with oral steroids was initiated, tapered to 4 mg prednisolone and continued up to the day of her current presentation.
Electrophysiological examinations revealed a decrement in repetitive nerve stimulation. CSF examination showed mild protein elevation (506 mg/L, normal range: 150–450 mg/L); white cell count, lactate, glucose and intrathecal immunoglobulin levels were normal. Antibody tests were positive for cardiolipin IgG/IgM and muscle‐specific kinase (MuSK; 0.3 nmol/L, normal range: <0.05 nmol/L). Importantly, tests were negative for acetylcholine receptor (AChR) and titin. The patient was eventually diagnosed with MuSK‐Ab‐positive generalized myasthenia gravis (MG). Accordingly, computed tomography (CT)‐imaging of the chest did not reveal evidence for a thymoma or thymus hyperplasia. About 3 weeks after admission, the patient developed progressive renal failure that was attributed to lupus nephritis and treated with 1000 mg intravenous methylprednisolone over 3 days. Her myasthenic symptoms gradually worsened and she developed respiratory failure. Despite subsequent treatment with oral pyridostigmine, steroids (70 mg oral methylprednisolone/day) and intravenous immunoglobulin (IVIg; 0.4 g/kg/day for 5 days) there was a persistent need for ventilatory support at the time the patient was referred to our intensive care unit.
On admission, neurologic examination revealed a symmetric proximal tetraparesis (2/5 on the MRC scale) and the need for mechanical ventilation. Plasmapheresis along with 250 mg intravenous methylprednisolone for 3 days with a subsequent maintenance dose of 70 mg oral prednisolone was initiated. CSF examination was repeated as the patient developed hyperactive delirium suggesting central nervous system (CNS) disease; it showed resolution of protein elevation but revealed an intrathecal synthesis of IgG, IgA, and IgM as well as oligoclonal bands. Autoimmune encephalitis was excluded by antibody testing (NMDA‐, AMPA‐, GABAb‐, and Glycin‐receptor) in CSF and serum. Subsequent magnetic resonance imaging (MRI) of the brain revealed contrast‐enhancing lesions compatible with CNS affection of SLE (Fig. 1).
Figure 1.
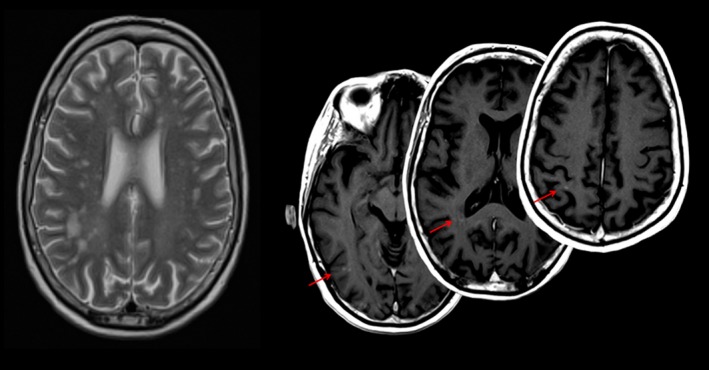
Axial T2‐weighted (left) and postcontrast T1‐weighted (right) MRI showing lesions consistent with the diagnosis of CNS involvement in systemic lupus erythematosus. Contrast‐enhancing lesions (right) indicate disease activity at the time of acquisition. MRI, magnetic resonance imaging; CNS, central nervous system.
Consistent with this finding, repeated antibody tests were positive for ANA (titer: 1:1280, normal range: <1:320), double‐stranded DNA (dsDNA) (39.8 U/mL, normal range: <20 U/mL), cardiolipin IgG/IgM (11.9 and 38.4 U/mL, normal range: <10 and <7 U/mL, respectively), beta2‐glycoprotein IgG/IgM (18.5 and 55.1 U/mL, normal range: both <8 U/mL), phosphatidylserine (referred to as APSA; 12 and 37 U/mL, normal range: both <10 U/mL), N‐type (ratio of 17.6, normal ratio: <10) and PQ‐type (35.8, normal range: <25 pmol/L) voltage gated‐calcium channels (VGCC), MuSK (0.3 mol/L, normal range: <0.05 nmol/L), thyroglobuline (1400 U/mL, normal range: 115 U/mL). Antibodies against AChR, titin and dsDNA (Crithidia luciliae immunofluorescence test, CLIFT) were negative. Results were interpreted as indicative of low lupus and antiphospholipid syndrome (APS) activity, as the CLIFT for dsDNA was negative and APS‐associated antibodies were predominantly of IgM‐subtype without clinical signs of venous thrombosis. The significance of the VGCC‐antibodies remained unclear but was ultimately regarded as insignificant as there were no electrophysiologic signs of Lambert–Eaton myasthenic syndrome 11. Furthermore, a whole‐body positron emission tomography of fluorodeoxyglucose distribution showed no evidence of malignant lesions except for pathological uptake in right axillary lymph nodes but histopathologic evaluation excluded malignancy.
Two months after admission, we observed moderate clinical improvement as a response to immunosuppressive treatment. Proximal weakness had improved by 1–2 points on the MRC scale, the delirium had resolved and a cerebral MRI showed resolution of contrast‐enhancing lesions. We considered a maintenance immunosuppressive regime with azathioprine, rituximab, or belimumab for an overlap syndrome of MuSK‐Ab‐positive MG and SLE. However, initiation of this maintenance treatment was hindered by a ventilator‐associated pneumonia (VAP) and progressive hepatic insufficiency. Seroconversion suggested an associated hepatitis E virus (HEV) infection, but PCR for HEV from different specimens (serum, feces, liver biopsy) was negative. The patient was treated with ribavirin as ultrasound and biopsy confirmed hepatitis could at that point not be attributed to any other autoimmune or infectious etiology. Although VAP was successfully treated with antibiotics, ribavirin caused agranulocytosis which was effectively reversed by granulocyte colony‐stimulating factor.
After clinical stabilization about 8 weeks later, the patient developed severe pneumonia with septic shock caused by a pneumothorax. Despite appropriate treatment for microbiologically confirmed multi‐drug resistant Klebsiella pneumoniae and Staphylococcus epidermidis there was only transient decline of inflammation markers and demand of circulatory support. Figure 2 illustrates the course of clinical and laboratory markers of inflammation and treatment. Due to the development of a DIC, we extended our search for infectious foci by repeated whole‐body CT‐imaging, white blood cell scan, extensive microbiological testing including repeated blood cultures and serology for viral and fungal infections. Results were negative except for high levels of EBV‐copies (15,000/mL) which were considered a reactivation during the course of sepsis, aggravated by SLE‐associated poor EBV‐control 12. Importantly, CT‐imaging showed resolution of prior lung infiltrate. We therefore considered noninfectious inflammatory disorders instead with features resembling sepsis. Initially, an eosinophilia of 75% gave rise to the suspicion of drug‐induced hypersensitivity syndrome (DIHS) 13. We changed the antibiotic regimen from vancomycin, which was the most likely drug to cause DIHS, to linezolid and initiated immunosuppressive treatment (0.4 g/kg/day IVIg for 5 days and 50 mg intravenous prednisolone followed by 20 mg daily over 7 days). Further differential diagnostic evaluation revealed high ferritin (11,599 μg/L) and soluble Il‐2 receptor (4586 μg/mL) levels. Splenomegaly, fever, anemia, thombo‐/leukocytopenia and hemophagocytosis in bone marrow biopsy suggested HLH, possibly associated with EBV infection 6. Typical histologic changes (Fig. 3) gave rise to the alternative term macrophage activation syndrome (MAS), which is preferentially used in the context of autoimmune disorders such as SLE 5. The patient was therefore treated with another course of IVIg (0.4 g/kg/day) and high‐dose intravenous steroids (500 mg methylprednisolone over 2 days with a consecutive taper to a daily maintenance dose of 7.5 mg). Additional treatment with cyclophosphamide (600/800 mg per induction dose) was administered for a combined syndrome of HLH, SLE, and MG. The patient's condition improved substantially (moderate tetraparesis of 4/5 on the MRC scale, no persistent need for mechanical ventilation) and she was eventually scheduled for rehabilitation.
Figure 2.
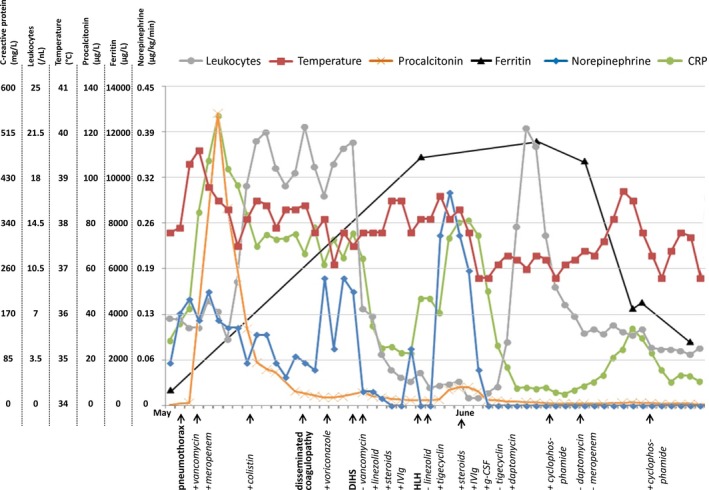
Diagram illustrating the course of clinical (temperature, norepinephrine) and laboratory inflammation markers (C‐reactive protein [CRP], leukocytes, procalcitonin) as well as markers of hemophagocytic lymphohistiocytosis (HLH) disease activity (ferritin) about 3 months after admission. The x‐axis labels in bold denote clinical diagnoses made that day; labels in regular print indicate drugs which were added (+) or withdrawn (−) that day. Note the initial sepsis‐associated increase in clinical and laboratory inflammation markers. Despite improvement of procalcitonin levels there was only minor improvement of CRP‐levels, leukocytosis and need for hemodynamic support and meanwhile a disseminated coagulopathy developed. Voriconazole was added for a possible fungal infection although blood cultures for fungi and serologic testing for candida and aspergillus were negative. Eosinophilia of about 75% in the presence of pronounced leukocytosis gave rise to the differential diagnosis of DIHS presumptively induced by vancomycin which was replaced with linezolid. Treatment was complemented by steroids and intravenous immunoglobulin (IVIg). Immunosuppressive treatment was discontinued after clinical stabilization whereas linezolid was replaced with tigecycline since leukopenia developed. At that time, the diagnosis of HLH was established which led to prompt reinitialization of immunosuppressive treatment (for details see text). About one and a half months after onset of clinical deterioration inflammation markers were normalized and ferritin levels had dropped substantially, indicating decrease in HLH activity.
Figure 3.
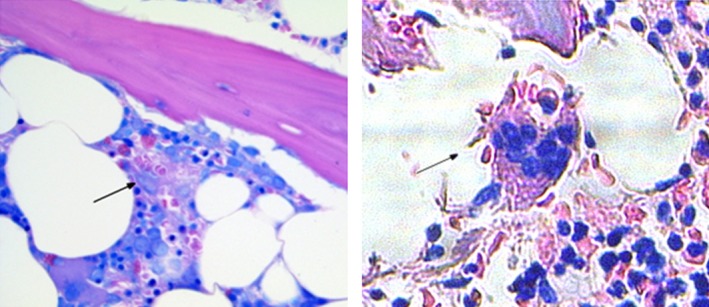
Giemsa‐stained bone marrow specimens obtained at diagnosis of hemophagocytic lymphohistiocytosis (HLH) (left) and at autopsy about 2 months later (right). Images show macrophages with erythro‐ (left and right) and leukophagocytic (right) activity representing key features of HLH disease activity.
Before discharge, however, she unexpectedly died of a fulminant subarachnoid hemorrhage caused by a ruptured 4 mm right middle cerebral artery aneurysm. Autopsy revealed diffuse lymphocytic infiltration of parenchymal organs, notably of liver, spleen, and bone marrow. Some macrophages revealed signs of erythrophagocytic activity (Fig. 3).
Discussion
In our patient, multiple possible HLH triggers were present; notably an infection with K. pneumoniae and S. epidermidis as well as EBV‐viremia, possibly related to poor EBV‐control in SLE aggravated by infection 12, and active SLE 2. Treating trigger factors such as infection or autoimmune disease activity in MAS, that is an autoimmune‐associated HLH variant, may suffice and allow deferring HLH‐specific treatment 5, 14. There are no randomized, controlled clinical trials investigating potential HLH treatment regimens in adults 2. In children, different protocols, that is HLH‐94 and HLH‐2004, have been established 6, 10; HLH‐2004 trial data, however, is not yet fully evaluated 6. Available results suggest efficacy of an induction treatment with dexamethasone and etoposide, and in case of nonresponding CNS involvement addition of intrathecal treatment with methotrexate and hydrocortisone. The role of ciclosporin A remains a matter of debate that might be resolved with the results of the HLH‐2004 trial. Noncontrolled and retrospective studies in adults have tested several combinations of corticosteroids, IVIg, etoposide, ciclosporin A, and alemtuzumab (for review see Ramos‐Casals et al., 2014) 2. The literature in both adults and children seems to justify an individual approach in all patients depending on trigger factors as well as organ manifestation 5. Thus, choosing an appropriate treatment can be challenging and requires due consideration. We eventually treated our patient with corticosteroids, IVIg and cyclophosphamide as (1) EBV‐associated HLH has been shown to respond to corticosteroids and IVIg 15, (2) cyclophosphamide is reported to improve SLE‐associated MAS 16, and (3) cyclophosphamide is a well‐established escalation and maintenance treatment for MG albeit rarely reported in MuSK‐positive variants 17; a combination of MuSK‐positive MG, SLE, and HLH in one patient has not yet been described in the literature.
Treatment response is expected to occur within 2–3 weeks after initiation. Salvage therapy with increasing or more frequent doses of immunosuppressive agents should be considered in those patients who do not display at least a partial response 5. Response to immunosuppressive treatment can, as in our case, be prompt; norepinephrine doses required for hemodynamic support, CRP‐levels and fever dropped substantially within only 2–3 days after treatment initiation (Fig. 2). Hence second line therapies, including hematopoietic stem cell transplant (HSCT) or alemtuzumab, were not considered 5, 18. In the HLH‐94 study, the overall mortality of patients suffering from HLH was reported to be around 45% 10. Refractory disease is considered to be associated with a poorer outcome 5.
It is unclear when the first HLH‐attack occurred in our patient as specific diagnostic tests were first conducted 3 months after admission. HLH might already have manifested during the preceding episode of noninfectious SIRS attributed to DIHS that responded to treatment with corticosteroids and IVIg. Notably, there was only scarce evidence for an HEV‐associated hepatitis about 1 month after admission and the biopsy confirmed lymphocytic hepatic infiltration arguing that hepatitis may also have been caused by HLH.
In summary, this case highlights that awareness of noninfectious causes of systemic inflammatory response syndromes and their specific diagnostic evaluation are paramount in the absence of conclusive evidence for septic foci. This is even more the case when patients suffer from autoimmune disorders given that such hyperinflammation (HLH) is treatable.
Consent
Written informed consent was obtained from the patient's next of kin (daughter) for publication of this case report and any accompanying images. A copy of the written consent is available for review.
Conflict of Interest
None declared.
Clinical Case Reports 2016; 4(2): 165–170
References
- 1. Scott, R. B. , and Robb‐Smith A. H.. 1939. Histiocytic medullary reticulocytosis. Lancet 2:194–198. [Google Scholar]
- 2. Ramos‐Casals, M. , Brito‐Zeron P., Lopez‐Guillermo A., Khamashta M. A., and Bosch X.. 2014. Adult haemophagocytic syndrome. Lancet 383:1503–1516. [DOI] [PubMed] [Google Scholar]
- 3. Henter, J. I. , and Elinder G.. 1991. Familial hemophagocytic lymphohistiocytosis. Clinical review based on the findings in seven children. Acta Paediatr Scand. 80:269–277. [DOI] [PubMed] [Google Scholar]
- 4. Raschke, R. A. , and Garcia‐Orr R.. 2011. Hemophagocytic lymphohistiocytosis: a potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults. Chest 140:933–938. [DOI] [PubMed] [Google Scholar]
- 5. Jordan, M. B. , Allen C. E., Weitzman S., Filipovich A. H., and McClain K. L.. 2011. How I treat hemophagocytic lymphohistiocytosis. Blood 118:4041–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henter, J. I. , Horne A., Arico M., Egeler R. M., Filipovich A. H., Imashuku S., et al. 2007. HLH‐2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 48:124–131. [DOI] [PubMed] [Google Scholar]
- 7. Filipovich, A. , McClain K., and Grom A.. 2010. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol. Blood Marrow Transplant. 16(1 Suppl):S82–S89. [DOI] [PubMed] [Google Scholar]
- 8. Zhang, K. , Jordan M. B., Marsh R. A., Johnson J. A., Kissell D., Meller J., et al. 2011. Hypomorphic mutations in PRF1, MUNC13‐4, and STXBP2 are associated with adult‐onset familial HLH. Blood 118:5794–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang, Y. , Xu X., Song H., Yang S., Shi S., Wei J., et al. 2008. Early diagnostic and prognostic significance of a specific Th1/Th2 cytokine pattern in children with haemophagocytic syndrome. Br. J. Haematol. 143:84–91. [DOI] [PubMed] [Google Scholar]
- 10. Trottestam, H. , Horne A., Arico M., Egeler R. M., Filipovich A. H., Gadner H., et al. 2011. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long‐term results of the HLH‐94 treatment protocol. Blood 118:4577–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oh, S. J. , Kurokawa K., Claussen G. C., and Ryan H. F. Jr. 2005. Electrophysiological diagnostic criteria of Lambert‐Eaton myasthenic syndrome. Muscle Nerve 32:515–520. [DOI] [PubMed] [Google Scholar]
- 12. Kang, I. , Quan T., Nolasco H., Park S. H., Hong M. S., Crouch J., et al. 2004. Defective control of latent Epstein‐Barr virus infection in systemic lupus erythematosus. J. Immunol. 172:1287–1294. [DOI] [PubMed] [Google Scholar]
- 13. Kardaun, S. H. , Sidoroff A., Valeyrie‐Allanore L., Halevy S., Davidovici B. B., Mockenhaupt M., et al. 2007. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br. J. Dermatol. 156:609–611. [DOI] [PubMed] [Google Scholar]
- 14. Brastianos, P. K. , Swanson J. W., Torbenson M., Sperati J., and Karakousis P. C.. 2006. Tuberculosis‐associated haemophagocytic syndrome. Lancet Infect Dis. 6:447–454. [DOI] [PubMed] [Google Scholar]
- 15. Ahn, J. S. , Rew S. Y., Shin M. G., Kim H. R., Yang D. H., Cho D., et al. 2010. Clinical significance of clonality and Epstein‐Barr virus infection in adult patients with hemophagocytic lymphohistiocytosis. Am. J. Hematol. 85:719–722. [DOI] [PubMed] [Google Scholar]
- 16. Carvalheiras, G. , Anjo D., Mendonca T., Vasconcelos C., and Farinha F.. 2010. Hemophagocytic syndrome as one of the main primary manifestations in acute systemic lupus erythematosus–case report and literature review. Lupus 19:756–761. [DOI] [PubMed] [Google Scholar]
- 17. De Feo, L. G. , Schottlender J., Martelli N. A., and Molfino N. A.. 2002. Use of intravenous pulsed cyclophosphamide in severe, generalized myasthenia gravis. Muscle Nerve 26:31–36. [DOI] [PubMed] [Google Scholar]
- 18. Marsh, R. A. , Allen C. E., McClain K. L., Weinstein J. L., Kanter J., Skiles J., et al. 2013. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr. Blood Cancer 60:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]