

Abstract
Comprehensive genome-wide DNA methylation studies in neuroblastoma (NB), a childhood tumor that originates from precursor cells of the sympathetic nervous system, are scarce. Recently, we profiled the DNA methylome of 102 well-annotated primary NB tumors by methyl-CpG-binding domain (MBD) sequencing, in order to identify prognostic biomarker candidates. In this data descriptor, we give details on how this data set was generated and which bioinformatics analyses were applied during data processing. Through a series of technical validations, we illustrate that the data are of high quality and that the sequenced fragments represent methylated genomic regions. Furthermore, genes previously described to be methylated in NB are confirmed. As such, these MBD sequencing data are a valuable resource to further study the association of NB risk factors with the NB methylome, and offer the opportunity to integrate methylome data with other -omic data sets on the same tumor samples such as gene copy number and gene expression, also publically available.
Subject terms: DNA methylation, Paediatric cancer, Next-generation sequencing
Background & Summary
Neuroblastoma (NB), a neuro-ectodermal tumor that originates from precursor cells of the sympathetic nervous system, represents the most common extra-cranial solid tumor of early childhood and is considered a heterogeneous disease driven by genetic aberrations, as during the past decades mainly genetic factors have been described to influence the pathogenesis and disease course (including MYCN amplification, ALK amplification and mutation, hyperdiploidy, and gains and losses of specific chromosome arms (1p, 3p, 11q and 17q))1. Also, recent comprehensive whole-genome sequencing studies of primary NB tumors pinpointed chromothripsis and defects in neuritogenesis genes as important tumor-driving events in a subset of NB2, and indicated that MYCN, TERT and ATRX alterations define major subgroups of high-risk NB3,4. However, also epigenetic mechanisms, such as DNA methylation alterations, seem to contribute to the NB biology and clinical behaviour.
As reviewed in Decock et al.5, multiple DNA methylation alterations have been described in NB, but given the rare occurrence of the disease, the number of comprehensive genome-wide DNA methylation studies analyzing primary tumor samples is limited. Hence, most studies initially make use of NB cell lines and only validate the most obvious methylation alterations in primary NB tumors. For example, a frequently applied methodology to NB cell lines is assessment of gene expression reactivation upon 5’-aza-2’-deoxycytidine (DAC) treatment, a cytosine analogue that cannot be methylated, leading to progressive DNA demethylation upon cell division. However, major drawbacks of these studies are that their discovery phases fall short in covering the NB heterogeneity, as NB cell lines are considered models for aggressive high-risk tumors, and that DNA methylation detection is indirectly assessed, as the influence of the demethylating effect is measured at the transcriptional level6–8. To accommodate this, the Illumina 27 and 450 K methylation arrays, directly interrogating the status of approximately 27,000 and 485,000 methylation sites, respectively, recently were applied to primary NB tumors6,9–12. Yet, also this technology has important limitations: the design of the arrays is heavily biased to interrogation of CpG sites previously described in literature and covers less than 2% of all CpG sites in the human genome13.
Therefore, we generated a data set comprising of 102 primary NB tumors in which DNA methylation is assessed by massively parallel sequencing of methylation enriched DNA fragments. The applied method is based on the use of MeCP2, a member of the methyl-CpG-binding domain (MBD) protein family which specifically binds to methylated cytosines and enables precipitation of methylated DNA fragments. This data set is unique in the NB research field, as it is the first sample cohort in which the full tumor heterogeneity is being assessed by genome-wide methylation analysis using next-generation sequencing (NGS); it was originally collected for the identification of prognostic biomarker candidates. Selected candidates were validated in independent cohorts using methylation-specific PCR and we showed that MBD sequencing allowed selection of valuable markers which would not have been identified using the Illumina methylation arrays14.
Here, we provide a detailed description of the methodological approach and bioinformatics analyses, as well as easy access to the (analyzed) MBD sequencing data and analysis tools, allowing other researchers (inexperienced with MBD sequencing) to reuse it. Importantly, the analyzed samples are well annotated; besides overall and event-free survival data, also following NB characteristics are available: age of the patient at diagnosis, tumor stage according to the International Neuroblastoma Staging System (INSS)15 and MYCN amplification status. As such, these data offer the opportunity to further explore the association of these risk factors with the NB methylome. Furthermore, integration of methylome data with other -omic data sets should be examined in order to fully map the NB biology on a genome-wide level. The present MBD sequencing data greatly facilitate these integration analyses, considering that for part of the profiled samples matching expression and array comparative genomic hybridization (aCGH) data are available16–18 (see Methods for details).
In summary, this data descriptor outlines details on the generation and analysis of MBD sequencing data of 102 primary NB tumors (Fig. 1). As NB is a rare disease and comprehensive DNA methylation studies scarce, these MBD sequencing data are very valuable and permit further unravelling the role of DNA methylation in the NB biology.
Figure 1. The MBD sequencing data of 102 primary neuroblastoma tumors are processed using different analysis tools.

Depicted are the available MBD sequencing data sets and downstream data processing and technical validation steps. These steps are represented as arrows and circles, respectively. For each step, the applied tool or analysis is indicated. For the technical validation steps, also the corresponding data descriptor figures and tables are indicated. DMA, differential methylation analysis; IGV, Integrative Genomics Viewer; PE, paired-end; RPKM, reads per kilobase CpG island per million.
Methods
DNA sample collection
Two independent cohorts of 42 and 60 primary tumor DNA samples, respectively annotated as MBD cohort I and II, were sequenced. Samples of fresh frozen tumors were collected at the Ghent University Hospital (n=49; Ghent, Belgium), the Hospital Clínico Universitario (n=42; Valencia, Spain), the University Children’s Hospital Essen (n=8; Essen, Germany) and the Our Lady’s Children’s Hospital Dublin (n=3; Dublin, Ireland), according to previously published criteria7,14, and stage 4S tumors were also included. Detailed clinical characteristics of the patients are given in Table 1 (available online only). For samples 809 and 912, DNA was extracted from different parts of the same primary tumor. Informed consent was obtained from each patient’s guardian and the study was approved by the ethical committee of the Ghent University Hospital (approval number B67020109912). Matching expression data16,17 of 38 tumors are available through the NCBI Gene Expression Omnibus (GEO) database (GSE21713 and GSE32664; sample IDs in Table 1 (available online only)). Matching aCGH data18 of 38 tumors are available through ViVar19 (https://www.cmgg.be/vivar/; login: review, password: review, project: Kumps et al. 2013; sample IDs in Table 1 (available online only)).
Table 1. In total, 102 annotated primary neuroblastoma DNA samples were profiled by MBD sequencing.
| Source Name | Characteristics[organism] | Characteristics[organism part] | Method | Sample Name | Factor Value[cohort] | Factor Value[age at diagnosis (months)] | Factor Value[INSS stage] | Factor Value[MYCN amplification status] | Factor Value[overall survival time (days)] | Factor Value[event-free survival time (days)] | Factor Value[overall survival status] | Factor Value[event-free survival status] | Factor Value[expression data_GEO ID] | Factor Value[aCGH data_ViVar ID] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Each sample is characterized by a unique Sample Name and is assigned to a specific cohort (MBD cohort I or MBD cohort II). Clinical characteristics given are the age at diagnosis in months, International Neuroblastoma Staging System (INSS) stage, MYCN amplification status (non-amplified or amplified), and overall survival (OS) and event-free survival (EFS) status and time in days after diagnosis. The OS status indicates whether the patient was alive at the last known follow-up or died of disease. Similarly, the EFS status indicates events such as relapse, progression or death. For each sample, the GEO accession IDs are provided. For samples for which expression and/or aCGH data are available, also the corresponding accession IDs to these data are indicated. NAs represent missing data. | ||||||||||||||
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 811 | MBD cohort I | 66.70684932 | 4 | non-amplified | 944 | 587 | died of disease | event | GSE21713—GSM541724 | id 4883 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1429 | MBD cohort I | 64.63561644 | 4 | non-amplified | 1188 | 867 | died of disease | event | GSE21713—GSM541703 | id 4758 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1467 | MBD cohort I | 0 | 4 | amplified | 1 | 1 | died of disease | event | GSE21713—GSM541689 | id 4817 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1473 | MBD cohort I | 7.528767123 | 4 | amplified | 239 | 116 | died of disease | event | GSE21713—GSM541691 | id 4882 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1477 | MBD cohort I | 78.70684932 | 4 | amplified | 1246 | 898 | died of disease | event | NA | id 4600 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1517 | MBD cohort I | 34.88219178 | 4 | non-amplified | 547 | 433 | died of disease | event | GSE21714—GSM541705 | id 11565 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1520 | MBD cohort I | 39.74794521 | 4 | amplified | 1279 | 552 | died of disease | event | GSE21713—GSM541694 | id 11771 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1522 | MBD cohort I | 22.61917808 | 3 | amplified | 728 | 594 | died of disease | event | GSE21713—GSM541696 | id 5098 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1527 | MBD cohort I | 107.9342466 | 4 | amplified | 319 | NA | died of disease | event | GSE21713—GSM541698 | id 4762 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1648 | MBD cohort I | 53.16164384 | 4 | non-amplified | 1221 | 341 | died of disease | event | GSE21713—GSM541701 | id 4603 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E061 | MBD cohort I | 30.70684932 | 4 | non-amplified | 1445 | 497 | died of disease | event | GSE32664—GSM810696 | id 5143 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E069 | MBD cohort I | 59.44109589 | 4 | non-amplified | 2836 | 2016 | died of disease | event | GSE32664—GSM810694 | id 5095 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E282 | MBD cohort I | 16.99726027 | 4 | amplified | 711 | 433 | died of disease | event | GSE32664—GSM810699 | id 5142 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E290 | MBD cohort I | 6.443835616 | 4 | amplified | 539 | 351 | died of disease | event | GSE32664—GSM810682 | id 5085 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 526 | MBD cohort I | 26.43287671 | 4 | amplified | 2009 | 2009 | alive | no event | GSE21713—GSM541725 | id 4866 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1017 | MBD cohort I | 22.32328767 | 3 | amplified | 1758 | 1758 | alive | no event | GSE21713—GSM541728 | id 4910 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1431 | MBD cohort I | 23.04657534 | 4 | amplified | 953 | 953 | alive | no event | GSE21713—GSM541704 | id 4550 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1521 | MBD cohort I | 38.03835616 | 4 | amplified | 2163 | 2163 | alive | no event | GSE21713—GSM541695 | id 4602 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1524 | MBD cohort I | 23.30958904 | 3 | amplified | 2387 | 2387 | alive | no event | GSE21713—GSM541697 | id 4766 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1616 | MBD cohort I | 18.34520548 | 4 | amplified | 2137 | 2137 | alive | no event | GSE21713—GSM541690 | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 2857 | MBD cohort I | 28.83287671 | 3 | amplified | 1295 | 1295 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 2863 | MBD cohort I | 10.81643836 | 4 | amplified | 1237 | 1237 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 2868 | MBD cohort I | 157.1835616 | 4 | non-amplified | 1159 | 1159 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E579 | MBD cohort I | 13.15068493 | 4 | non-amplified | 3534 | 3534 | alive | no event | GSE32664—GSM810692 | id 4913 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E598 | MBD cohort I | 48.85479452 | 4 | non-amplified | 3219 | 3219 | alive | no event | GSE32664—GSM810689 | id 5137 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E685 | MBD cohort I | 14.16986301 | 4 | non-amplified | 3011 | 3011 | alive | no event | GSE32664—GSM810685 | id 5043 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | E700 | MBD cohort I | 20.35068493 | 4 | non-amplified | 1536 | 1536 | alive | no event | GSE32664—GSM810680 | id 5041 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 278 | MBD cohort I | 14.53150685 | 1 | non-amplified | 3404 | 3404 | alive | no event | GSE21713—GSM541713 | id 4884 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 397 | MBD cohort I | 18.47671233 | 2 | non-amplified | 3555 | 3555 | alive | no event | GSE21713—GSM541720 | id 10138 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 410 | MBD cohort I | 1.676712329 | 1 | non-amplified | 2910 | 2910 | alive | no event | GSE21713—GSM541714 | id 4878 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 529 | MBD cohort I | 1.249315068 | 3 | non-amplified | 2264 | 2264 | alive | no event | GSE21713—GSM541707 | id 4863 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 530 | MBD cohort I | 1.545205479 | 1 | non-amplified | 2216 | 2216 | alive | no event | GSE21713—GSM541717 | id 4785 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 566 | MBD cohort I | 0.098630137 | 2 | non-amplified | 1615 | 1615 | alive | no event | GSE21713—GSM541718 | id 4826 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 711 | MBD cohort I | 16.99726027 | 2 | non-amplified | 1885 | 1885 | alive | no event | GSE21713—GSM541710 | id 11562 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 744 | MBD cohort I | 16.0109589 | 2 | non-amplified | 1850 | 1850 | alive | no event | GSE21713—GSM541716 | id 4870 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 747 | MBD cohort I | 7.989041096 | 1 | non-amplified | 2302 | 2302 | alive | no event | GSE21713—GSM541712 | id 11563 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 809 | MBD cohort I | 0.164383562 | 1 | non-amplified | 2904 | 2904 | alive | no event | GSE21713—GSM541723 | id 4868 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 914 | MBD cohort I | 1.249315068 | 1 | non-amplified | 2425 | 2425 | alive | no event | GSE21713—GSM541711 | id 11564 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 916 | MBD cohort I | 9.994520548 | 1 | non-amplified | 3830 | 3830 | alive | no event | GSE21713—GSM541726 | id 5372 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 926 | MBD cohort I | 0.920547945 | 1 | non-amplified | 1861 | 1861 | alive | no event | GSE21713—GSM541715 | id 4921 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1650 | MBD cohort I | 0.854794521 | 2 | non-amplified | 1264 | 187 | alive | event | GSE21713—GSM541702 | id 4813 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1699 | MBD cohort I | 7.463013699 | 3 | non-amplified | 2882 | 999 | alive | event | GSE21713—GSM541706 | id 10132 |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 41 | MBD cohort II | 49.90684932 | 4 | non-amplified | 569 | NA | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 610 | MBD cohort II | 34.02739726 | 4 | amplified | 850 | NA | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 928 | MBD cohort II | 11.7369863 | 4 | amplified | 412 | NA | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1430 | MBD cohort II | 35.4739726 | 4 | non-amplified | 520 | 391 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1507 | MBD cohort II | 14.33424658 | 4 | amplified | 285 | NA | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1713 | MBD cohort II | 40.99726027 | 4 | non-amplified | 581 | 449 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1780 | MBD cohort II | 30.90410959 | 3 | amplified | 569 | NA | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1782 | MBD cohort II | 41.49041096 | 4 | amplified | 707 | 441 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1783 | MBD cohort II | 13.6109589 | 4 | amplified | 316 | 288 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1784 | MBD cohort II | 76.5369863 | 4 | amplified | 1819 | 972 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1786 | MBD cohort II | 15.02465753 | 4 | amplified | 950 | 607 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1790 | MBD cohort II | 101.2931507 | 4 | non-amplified | 989 | 306 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1791 | MBD cohort II | 51.32054795 | 4 | non-amplified | 377 | 357 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1795 | MBD cohort II | 134.0383562 | 4 | non-amplified | 671 | 214 | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1796 | MBD cohort II | 24.2630137 | 4 | non-amplified | 414 | NA | died of disease | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 11 | MBD cohort II | 15.22191781 | 4 | non-amplified | 4396 | 4396 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1030 | MBD cohort II | 54.64109589 | 4 | non-amplified | 2653 | 2653 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1381 | MBD cohort II | 21.46849315 | 4 | non-amplified | 1981 | 1981 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1382 | MBD cohort II | 70.09315068 | 4 | non-amplified | 1891 | 1891 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1384 | MBD cohort II | 16.37260274 | 3 | amplified | 1620 | 1620 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1501 | MBD cohort II | 131.3753425 | 4 | non-amplified | 1558 | 1558 | alive | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1515 | MBD cohort II | 15.97808219 | 4 | non-amplified | 1687 | 1687 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1647 | MBD cohort II | 10.06027397 | 2 | amplified | 2136 | 2136 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1649 | MBD cohort II | 8.745205479 | 4 | amplified | 2174 | 2174 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1789 | MBD cohort II | 25.18356164 | 3 | amplified | 5616 | 5616 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1793 | MBD cohort II | 32.54794521 | 3 | amplified | 2328 | 2328 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1794 | MBD cohort II | 33.23835616 | 3 | amplified | 5096 | 5096 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1800 | MBD cohort II | 71.07945205 | 4 | amplified | 1862 | 1862 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1803 | MBD cohort II | 19.36438356 | 4 | amplified | 2410 | 2410 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1863 | MBD cohort II | 168.6246575 | 4 | non-amplified | 1053 | 560 | alive | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 820 | MBD cohort II | 10.88219178 | 2 | non-amplified | 4794 | 4794 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 822 | MBD cohort II | 12.82191781 | 1 | non-amplified | 1153 | 1153 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 823 | MBD cohort II | 4.24109589 | 1 | non-amplified | 1090 | 1090 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 912 | MBD cohort II | 0.164383562 | 1 | non-amplified | 2904 | 2904 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1028 | MBD cohort II | 3.484931507 | 2 | non-amplified | 2932 | 2932 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1038 | MBD cohort II | 11.4739726 | 2 | non-amplified | 1071 | 1071 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1039 | MBD cohort II | 7.956164384 | 3 | non-amplified | 1394 | 1394 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1469 | MBD cohort II | 1.446575342 | 1 | non-amplified | 3275 | 3275 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1476 | MBD cohort II | 22.22465753 | 2 | non-amplified | 2075 | 2075 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1483 | MBD cohort II | 0.690410959 | 1 | non-amplified | 2554 | 2554 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1484 | MBD cohort II | 1.347945205 | 1 | non-amplified | 2566 | 2566 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1486 | MBD cohort II | 11.50684932 | 1 | non-amplified | 2328 | 2328 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1488 | MBD cohort II | 2.432876712 | 2 | non-amplified | 1827 | 1827 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1509 | MBD cohort II | 1.019178082 | 3 | non-amplified | 2597 | 2597 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1646 | MBD cohort II | 4.109589041 | 2 | non-amplified | 2096 | 2096 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1530 | MBD cohort II | 0.098630137 | 4S | non-amplified | 2039 | 2039 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1494 | MBD cohort II | 1.084931507 | 4S | non-amplified | 1590 | 1590 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1615 | MBD cohort II | 0.295890411 | 4S | non-amplified | 1562 | 1562 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1613 | MBD cohort II | 0.460273973 | 4S | non-amplified | 1503 | 1503 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1191 | MBD cohort II | 1.709589041 | 4S | non-amplified | 2105 | 2105 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1750 | MBD cohort II | 0.723287671 | 4S | non-amplified | 1322 | 1322 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 277 | MBD cohort II | 2.367123288 | 4S | non-amplified | 2099 | 101 | alive | event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1392 | MBD cohort II | 3.484931507 | 4S | non-amplified | 3190 | 3190 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 821 | MBD cohort II | 2.038356164 | 4S | non-amplified | 2652 | 2652 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 750 | MBD cohort II | 8.482191781 | 4S | non-amplified | 1567 | 1567 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1383 | MBD cohort II | 0.493150685 | 4S | non-amplified | 2670 | 2670 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1013 | MBD cohort II | 2.432876712 | 4S | non-amplified | 2252 | 2252 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 511 | MBD cohort II | 3.682191781 | 4S | non-amplified | 3837 | 3837 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 520 | MBD cohort II | 0.328767123 | 4S | non-amplified | 3703 | 3703 | alive | no event | NA | NA |
| Neuroblastoma patient | Homo sapiens | Primary tumor | DNA sample collection | 1537 | MBD cohort II | 0.55890411 | 4S | amplified | 2178 | 2178 | alive | no event | NA | NA |
Methyl-CpG-binding domain (MBD) sequencing
DNA fragmentation
For each sample, between 400 to 1000 ng DNA was sheared to obtain DNA fragments with an average length of 200 bp. The DNA was loaded in 120 μl TE buffer (1:5), transferred to a Snap Cap microTUBE (Covaris) and exposed to Covaris S2 Adaptive Focused Acoustics. Fragment distribution and concentration was determined on a High Sensitivity DNA chip (Agilent Technologies).
Methylated DNA capturing
Subsequently, capturing of methylated DNA fragments was done according to the MethylCap kit protocol of Diagenode using 200–500 ng DNA. Elution of the captured fraction was performed in 150 μl High Elution Buffer and DNA was purified using the MinElute PCR purification kit (Qiagen). For MBD cohort II, also input samples (10%) were prepared.
Library preparation
As MBD cohort I and II were profiled in a different time frame and NGS methodologies evolve at rapid pace, a different library preparation protocol and sequencing technology was applied to each of them. For MBD cohort I, DNA library preparation was performed using the NEBNext DNA Library Prep Master Mix Set for Illumina (New England Biolabs) in combination with the Multiplexing Sample Preparation Oligonucleotide Kit (Illumina) for paired-end adapter ligation. Size selection of the library is done on a 2% agarose gel (Bio-Rad). Fragments between 250 and 350 bp were excised and purified using a Qiagen Gel Extraction Kit. For MBD cohort II, library preparation was automated on an Apollo 324 Next Generation Sequencing Library Preparation System (IntegenX), making use of the PrepX ILM DNA Library Kit (IntegenX). For paired-end adapter ligation the Multiplexing Sample Preparation Oligonucleotide Kit was used. Size selection was done with 1X AMPure XP beads (Agencourt) and PEG-Bead Solution.
Library amplification
PCR library amplification with appropriate Index Primers for each sample was performed using the Multiplexing Sample Preparation Oligonucleotide Kit and following PCR conditions: 30 s at 98 °C, 21 amplification cycles (10 s at 98 °C, 30 s at 65 °C and 30 s at 72 °C), 5 min at 72 °C, and held at 4 °C. PCR product purification was done using the High Pure PCR Purification Kit (Roche). QC was performed on a DNA 1000 chip (Agilent) and concentration was determined by qPCR according to the qPCR Quantification Protocol Guide of Illumina. Samples were pooled and profiled on an Illumina GAIIx (PE 2×45 bp) for MBD cohort I and on an Illumina HiSeq2000 (PE 2×51 bp) for MBD cohort II.
Data processing and analysis
Sequencing data
All crucial steps in the processing and analysis of the MBD sequencing data are summarized in Fig. 1. Raw sequencing data were demultiplexed and converted to FASTQ files (with sequencing reads and quality scores). Quality control on the raw data was performed by FASTQC (version 0.9.2; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Read mapping
Next, the sequencing reads were mapped/aligned to the human reference genome (hg19), using the Bowtie2 (ref. 20) mapper (version 2.0.0 beta7) and FASTQ files as input. For each sample, two paired FASTQ files are available (as we performed paired-end sequencing), in which the data lines correspond to each other. To improve the mapping quality, reads were only taken into account if the sequences in both files could be mapped to the reference genome (maximum 500 bp between both paired ends). Also sequencing quality scores were used in the mapping process. The BAM format was used as output file type. PCR duplicates were marked with Picard (version 1.79; http://broadinstitute.github.io/picard/) and the BAM files were sorted and indexed using SAMtools21 (version 0.1.18) and index commands. These files have been deposited as raw data files in the NCBI Gene Expression Omnibus (GEO) database (Data Citation 1 for MBD cohort I; Data Citation 2 and Data Citation 3 for MBD cohort II). FASTQ records can be extracted from the sequence alignments in the BAM files using the BEDTools bamtofastq conversion utility22. Starting from the SRA files, the NCBI SRA Toolkit (fastq-dump) can be used to generate the FASTQ files. Mapping quality was evaluated using SAMStat23 (version 1.08) and BamUtil (version 1.0.2; http://genome.sph.umich.edu/wiki/BamUtil). Technical validation of MBD enrichment is performed by fragment CpG plot analysis24 and by plotting the densities of the median numbers of mapped reads per kilobase per million (RPKM25) in all CpG islands (n=28,691) across the different subcohorts.
Peak calling
The process of converting mapped sequencing reads to coverage vectors and the detection of enriched regions (peaks) is referred to as peak detection or peak calling. Here, peak calling was done using the MACS26 software tool (version 1.4.0 beta) and BAM files as input. BED files were generated (Data Citation 1 for MBD cohort I; Data Citation 2 and Data Citation 3 for MBD cohort II), indicating the location and score (linked to the P-value) of the identified peaks.
Visualization
MACS is also used to output WIG files (Data Citation 1 for MBD cohort I; Data Citation 2 and Data Citation 3 for MBD cohort II), which are transformed to a binary format (TDF file; Data Citation 1 for MBD cohort I; Data Citation 2 and Data Citation 3 for MBD cohort II) by igvtools (https://www.broadinstitute.org/igv/igvtools) for visualization in the Integrative Genomics Viewer (IGV)27. An example IGV XML-session file for MBD cohort II and instructions on how to make use of this file are included in the GitHub repository (see Code availability).
Differential methylation analyses
Differential methylation analyses between sample groups are described in detail in Decock et al.14. Briefly, for each subcohort, two count data sets were constructed, in which for each sample the numbers of mapped reads in the promoter region of the different Ensembl Transcripts or 5 kb genomic windows are indicated. Here, we provide access to these count data sets (Supplementary Tables 3,4,5,6,7,8), which can directly be used for differential methylation analyses in DESeq14,28.
Code availability
All tools and code that are necessary to generate the described file types are provided in a Docker container (Docker Hub; https://hub.docker.com/r/mateongenaert/mbdtoolbox/). More advanced analysis scripts can be found in the GitHub repository (https://github.com/mateongenaert/MBDToolBox).
Data Records
An overview of the sample annotation and data outputs is given in Table 1 (available online only). The outputs of each step in the data processing (read mapping: BAM files, peak calling: BED files, and visualization: WIG and TDF files) have been deposited in the GEO database. For MBD cohort I, the accession number is GSE69224 (Data Citation 1), for MBD cohort II, GSE69243 (Data Citation 2) and GSE69268 (Data Citation 3). In GEO, these data sets were submitted as SubSeries of the SuperSeries GSE69279 (Data Citation 4). We also provide a Docker container, made available through Docker Hub, that embeds all necessary tools to generate the data files and illustrates the analysis pipeline. More advanced analysis scripts are given in the GitHub repository (see Code availability).
Technical Validation
Validation of raw and mapped sequencing data
The total read number and percentage of duplicate and properly paired reads in each sample are given in Supplementary Table 1, and a summary of these sequencing statistics across the different sample cohorts can be found in Table 2.
Table 2. Using BamUtil, basic sequencing statistics of MBD cohort I and II are computed.
| Statistic |
MBD cohort I—enriched samples |
MBD cohort II—enriched samples |
MBD cohort II—input samples |
||||||
|---|---|---|---|---|---|---|---|---|---|
| range | mean | median | range | mean | median | range | mean | median | |
| Total read number: the total number of reads in the two paired FASTQ files of a sample; duplicate reads as a percentage of the total read number; properly paired reads as a percentage of the total read number. | |||||||||
| total read number (e6) | 4.65–18.20 | 13.38 | 14.17 | 29.74–66.59 | 45.09 | 44.41 | 20.86–59.51 | 36.00 | 33.19 |
| duplicate reads (%) | 0.70–72.00 | 6.46 | 3.39 | 2.55–79.69 | 31.04 | 19.89 | 2.24–10.47 | 4.17 | 3.68 |
| properly paired reads (%) | 48.29–94.51 | 85.64 | 89.29 | 86.86–97.57 | 95.33 | 95.72 | 94.78–97.55 | 96.50 | 96.59 |
To ensure raw data quality, FASTQC analyses were performed to determine the per base sequence quality which reflects the probability that a base has been called incorrectly29. Quality scores between 41 and 28, 28 and 20, and below 20 are considered base calls of very good quality, calls of reasonable quality and calls of poor quality, respectively. In order to obtain a general overview of the range of quality values across all bases at each position, the median quality score for each position in each FASTQ file was determined. Fig. 2 shows the distribution of these median per base quality scores across the different sample cohorts. In general, the quality scores of both MBD cohort I and II are of reasonable to very good quality. Given the different sequencing technologies that were used for MBD cohort I (Illumina GAIIx) and II (Illumina HiSeq2000), it is expected that the read quality of MBD cohort II is higher than that of MBD cohort I. The steadily increase and subsequent decrease in quality along the read is also expected for Illumina-based experiments29,30.
Figure 2. The per base sequence quality scores indicate that the raw sequencing data are of good quality.

Shown are the distributions of the median per base quality score (determined by FASTQC) of the enriched samples of MBD cohort I (a), and of the enriched (b) and input (c) samples of MBD cohort II. In the boxplots, the lower and upper hinge of the boxes represents the 25th and 75th percentile, respectively. The whiskers extend to the lowest and highest value that is within 1.5 times the interquartile range. Data beyond the end of the whiskers are outliers and plotted as dots.
Mapping quality is ensured by analyzing the mapping quality scores of the alignments in each sample (Supplementary Table 2). In Fig. 3, the distributions of the percentages of mapped reads across the different mapping quality ranges are shown. For all subcohorts, the reads are clearly mapped with high accuracy, as almost for every sample, more than half of the mapped reads has a MAPQ≥30 (ref. 23).
Figure 3. The mapping quality scores illustrate high mapping accuracy.
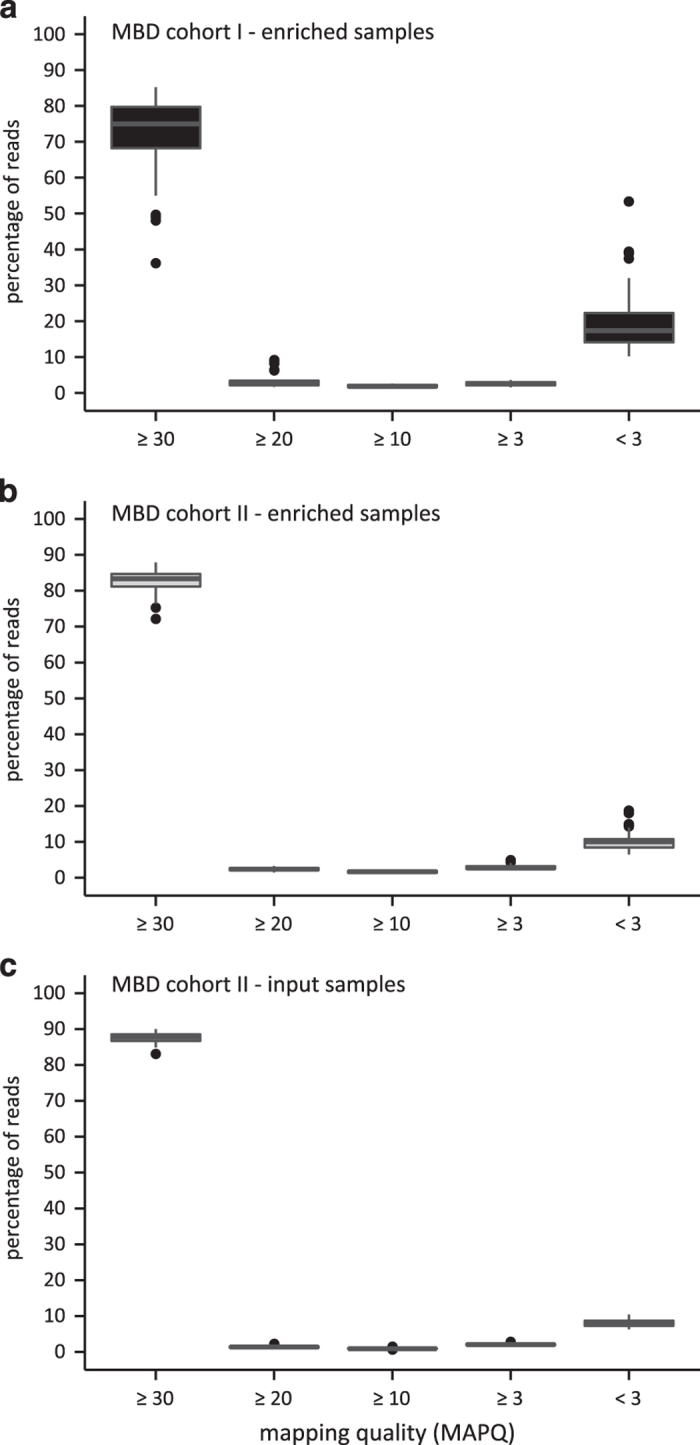
Shown are the distributions of the percentages of mapped reads across the different mapping quality ranges, as determined by SAMStat ((a) enriched samples of MBD cohort I, (b) enriched samples of MBD cohort II and (c) input samples of MBD cohort II). In the boxplots, the lower and upper hinge of the boxes represents the 25th and 75th percentile, respectively. The whiskers extend to the lowest and highest value that is within 1.5 times the interquartile range. Data beyond the end of the whiskers are outliers and plotted as dots.
Validation of MBD-based enrichment
Over the past years several companies developed commercial kits for MBD-based capturing of methylated fragments. Although all of them claim to be of high quality, differences in performance exist. Careful kit selection is thus of utmost importance24. Here, sheared tumor DNA was enriched towards methylated fragments using the MethylCap kit of Diagenode, that makes use of the methylCap protein, consisting of the MBD of human MeCP2 fused with gluthatione-S-transferase (GST) containing an N-terminal His6-tag. A previous evaluation assessed the quality of this kit for combination with NGS by comparison with four other commercially available kits24. This study also compared the MBD sequencing data with reduced representation bisulfite sequencing (RRBS) and Illumina 27 K methylation array data of the same samples. Together, these analyses showed that the MethylCap kit outperforms the others, due to a consistent combination of high yield, sensitivity and specificity24. In order to demonstrate that the samples of MBD cohort I and II were enriched for methylated DNA fragments after MBD-based capturing, we made use of the fragment CpG plot24. As this plot depicts the CpG content of the mapped fragments and the MethylCap kit theoretically only captures methylated cytosines in a CpG dinucleotide context, the fragment CpG plot can be used to evaluate the MBD-based enrichment. An overview of the CpG content of the mapped fragments per sample cohort is depicted in Fig. 4. This fragment CpG plot clearly illustrates that the MBD-enriched samples of MBD cohort I and II have a high fraction of CpG dense fragments, while the input (non-MBD-enriched) samples of MBD cohort II are not enriched in CpG content. Additionally, using the number of mapped reads per kilobase CpG island per million (RPKM) values25, the methylation level of each CpG island across the different subcohorts was determined. The density plot in Fig. 5 indicates that the MBD-enriched samples have a higher fraction of CpG islands with an RPKM>1 compared to the input samples of MBD cohort II. Based on these analyses, it can be concluded that the MBD-based capture successfully led to the enrichment of methylated DNA fragments.
Figure 4. Fragment CpG plots demonstrate that the MBD-enriched samples have a high fraction of CpG dense sequencing fragments.
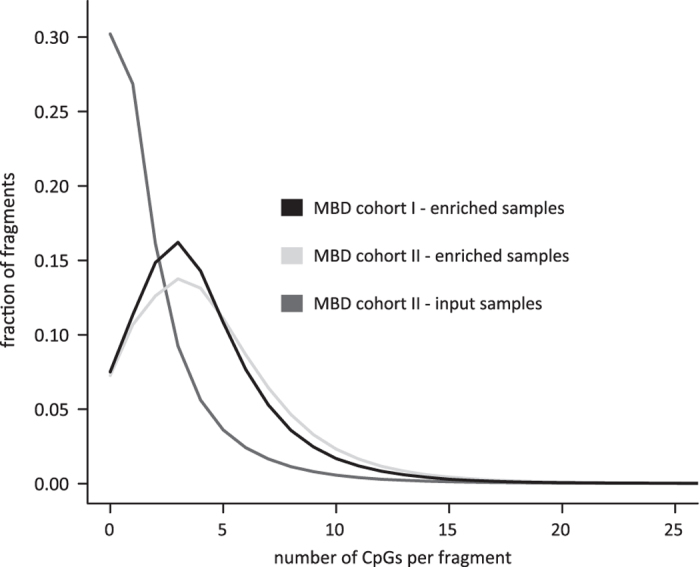
Shown are the fractions of mapped MBD sequencing fragments with different CpG counts. Per cohort, 100,000 randomly selected fragments of each sample were used to construct the plots.
Figure 5. CpG island RPKM values confirm enrichment towards methylated DNA fragments upon MBD capture.
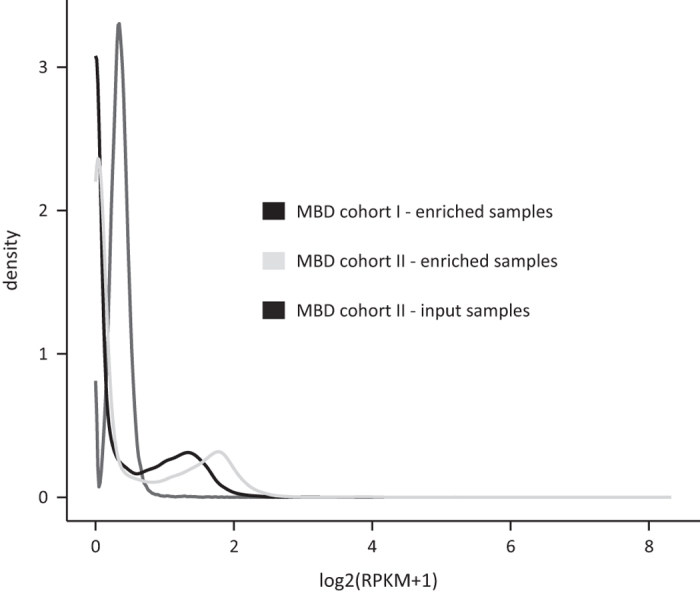
Shown are the densities of the median RPKM values per subcohort. RPKM: reads per kilobase CpG island per million.
Validation of methylated genes in neuroblastoma
Finally, TDF and BED files, containing sequence coverage and peak locations respectively, were loaded into IGV to visually inspect genes previously described to be methylated in NB. As an example, the MBD sequencing data of the PCDHB gene cluster is shown in Fig. 6. This gene cluster is frequently methylated in NB5,31, which is confirmed by the MBD sequencing data of both MBD cohort I and II. Additionally, 78 regions identified in the MBD sequencing data as being methylated, were validated in two independent patient cohorts using methylation-specific PCR (MSP)14. These data confirm the validity of MBD sequencing in identifying methylated regions in NB.
Figure 6. Visualization of the MBD sequencing data in IGV confirms methylation of the PCDHB gene cluster.

In (a) the data of MBD cohort I is shown, in (b) the data of MBD cohort II. The upper panels show the genes in the cluster, the location of CpG islands and the GC percentage. In the lower panels, sequence coverage of 6 high-risk patient samples is shown (peak pattern), as well as the location of identified peaks (horizontal bars).
Usage Notes
The MBD sequencing data can be downloaded from the GEO database via accession numbers GSE69224 (for MBD cohort I; Data Citation 1), GSE69243 and GSE69268 (for MBD cohort II; Data Citation 2 and Data Citation 3; SuperSeries GSE69279 (Data Citation 4)). The unique GEO sample accession IDs and clinical annotation can be found in Table 1 (available online only). This table also contains the accession IDs of the matching expression and aCGH data, which allows easy data access and facilitates integration analyses.
All output files from the different steps in the MBD sequencing data processing are provided through GEO. Analysis tools and scripts have been embedded in a Docker container, to deliver an environment that runs on any supported host platform (Windows, MAC, Linux). This Docker container, and all instructions on how it is made and how analyses can be run on the data, are made available through Docker Hub and GitHub (see Code availability). This allows researchers to try out the analysis pipeline that was used to generate the publically available data, without the need of additional infrastructure or software versions. The Docker container guarantees that the provided commands work and allows researchers to start exploring the data at the level they are experienced with.
Alternative processing tools can be tested for read mapping (e.g., BWA32) or identification of enriched regions (e.g., PeakRanger33 or BALM34), or absolute methylation scores can be calculated (MEDIPS35; see Code availability). Researchers inexperienced with MBD sequencing can easily visualize their genes of interest by downloading the BED and TDF files (see Code availability). Downstream differential methylation analyses can be done with DESeq28 (as described in Decock et al.14) using count data sets provided in Supplementary Tables 3,4,5,6,7,8, or other software can be used, such as DiffBind36 and edgeR37. Differences in absolute methylation scores can be used for RankProd38 analyses.
Additional information
Table 1 is only available in the online version of this paper.
How to cite this article: Decock, A. et al. DNA methylation profiling of primary neuroblastoma tumors using methyl-CpG-binding domain sequencing. Sci. Data 3:160004 doi: 10.1038/sdata.2016.4 (2016).
Supplementary Material
Using BamUtil, basic sequencing statistics of each sample of MBD cohort I and II are computed. Given are the total read numbers, and the number and percentage of properly paired and duplicate reads of each sample of MBD cohort I (a) and II (enriched samples in (b); input samples in (c)).
Using SAMStat, the mapping quality scores of each sample of MBD cohort I and II are analyzed. Given are the numbers and percentages of mapped reads across the different mapping quality ranges, as determined by SAMStat ((a) enriched samples of MBD cohort I, (b) enriched samples of MBD cohort II and (c) input samples of MBD cohort II).
Promoter count data for the MBD-enriched samples of MBD cohort I. For each MBD-enriched sample of MBD cohort I, the number of mapped reads in each Ensembl Transcript promoter region (-1500 bp to +500 bp around TSS) is given.
Promoter count data for the MBD-enriched samples of MBD cohort II. For each MBD-enriched sample of MBD cohort II, the number of mapped reads in each Ensembl Transcript promoter region (-1500 bp to +500 bp around TSS) is given.
Promoter count data for the input samples of MBD cohort II. For each input sample of MBD cohort II, the number of mapped reads in each Ensembl Transcript promoter region (-1500 bp to +500 bp around TSS) is given.
Window count data for the MBD-enriched samples of MBD cohort I. For each MBD-enriched sample of MBD cohort I, the number of mapped reads in each 5 kb genomic window (2.5 kb overlapping moving windows) is given.
Window count data for the MBD-enriched samples of MBD cohort II. For each MBD-enriched sample of MBD cohort II, the number of mapped reads in each 5 kb genomic window (2.5 kb overlapping moving windows) is given.
Window count data for the input samples of MBD cohort II. For each input sample of MBD cohort II, the number of mapped reads in each 5 kb genomic window (2.5 kb overlapping moving windows) is given.
Acknowledgments
The authors thank Rosa Noguera, Johannes H. Schulte and Raymond L. Stallings for sharing primary tumor samples, and NXTGNT for the sequencing services. The work is further supported by the Emmanuel Van der Schueren Foundation (scientific partner of the Flemish League Against Cancer (VLK)), the Fournier-Majoie Foundation (FFM) and the Belgian National Lottery. A.D. was a recipient of a grant of the Research Foundation Flanders (FWO) and an Emmanuel Van der Schueren research grant (VLK).
Footnotes
The authors declare no competing financial interests.
Data Citations
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69224
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69243
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69268
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69279
References
- Brodeur G. M. Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer 3, 203–216 (2003). [DOI] [PubMed] [Google Scholar]
- Molenaar J. J. et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593 (2012). [DOI] [PubMed] [Google Scholar]
- Valentijn L. J. et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 47, 1411–1414 (2015). [DOI] [PubMed] [Google Scholar]
- Peifer M. et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700–704 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock A., Ongenaert M., Vandesompele J. & Speleman F. Neuroblastoma epigenetics: from candidate gene approaches to genome-wide screenings. Epigenetics 6, 962–970 (2011). [DOI] [PubMed] [Google Scholar]
- Carén H. et al. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 11, 66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock A. et al. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 13, R95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duijkers F. A. et al. Epigenetic drug combination induces genome-wide demethylation and altered gene expression in neuro-ectodermal tumor-derived cell lines. Cell. Oncol. 36, 351–362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayol G. et al. DNA hypomethylation affects cancer-related biological functions and genes relevant in neuroblastoma pathogenesis. PLoS ONE 7, e48401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez Y. et al. Two independent epigenetic biomarkers predict survival in neuroblastoma. Clin. Epigenetics 7, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez S. et al. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics 7, 1137–1153 (2015). [DOI] [PubMed] [Google Scholar]
- Schramm A. et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 47, 872–877 (2015). [DOI] [PubMed] [Google Scholar]
- Stirzaker C., Taberlay P. C., Statham A. L. & Clark S. J. Mining cancer methylomes: prospects and challenges. Trends Genet. 30, 75–84 (2014). [DOI] [PubMed] [Google Scholar]
- Decock A. et al. Methyl-CpG-binding domain sequencing reveals a prognostic methylation signature in neuroblastoma. Oncotarget 7, 1960–1972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur G. M. et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 11, 1466–1477 (1993). [DOI] [PubMed] [Google Scholar]
- Mestdagh P. et al. The miR-17-92 microRNA cluster regulates multiple components of the TGF-β pathway in neuroblastoma. Mol. Cell 40, 762–773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenburg G., Eggert A., Schramm A., Lode H. N. & Hundsdoerfer P. Smac mimetic LBW242 sensitizes XIAP-overexpressing neuroblastoma cells for TNF-α-independent apoptosis. Cancer Res. 72, 2645–2656 (2012). [DOI] [PubMed] [Google Scholar]
- Kumps C. et al. Focal DNA copy number changes in neuroblastoma target MYCN regulated genes. PLoS ONE 8, e52321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sante T. et al. ViVar: a comprehensive platform for the analysis and visualization of structural genomic variation. PLoS ONE 9, e113800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. & Hall I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T., Hayashizaki Y. & Daub C. O. SAMStat: monitoring biases in next generation sequencing data. Bioinformatics 27, 130–131 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer T. et al. Quality evaluation of methyl binding domain based kits for enrichment DNA-methylation sequencing. PLoS ONE 8, e59068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L. & Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H., Robinson J. T. & Mesirov J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S. & Huber W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoche A. E., Dohm J. C. & Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 12, R112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. et al. Comprehensive RNA-Seq transcriptomic profiling across 11 organs, 4 ages, and 2 sexes of Fischer 344 rats. Sci. Data 1, 140013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banelli B. et al. A pyrosequencing assay for the quantitative methylation analysis of the PCDHB gene cluster, the major factor in neuroblastoma methylator phenotype. Lab. Invest. 92, 458–465 (2012). [DOI] [PubMed] [Google Scholar]
- Li H. & Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X., Grossman R. & Stein L. PeakRanger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics 12, 139 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan X. et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-seq technology. PLoS ONE 6, e22226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard M., Grimm C., Morkel M., Herwig R. & Chavez L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 30, 284–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R. & Brown G. D. DiffBind: differential binding analysis of ChIP-seq peak data. http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf (2011).
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F. et al. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22, 2825–2827 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69224
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69243
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69268
- Decock A., Ongenaert M., Speleman F., Vandesompele J. 2015. Gene Expression Omnibus . GSE69279
Supplementary Materials
Using BamUtil, basic sequencing statistics of each sample of MBD cohort I and II are computed. Given are the total read numbers, and the number and percentage of properly paired and duplicate reads of each sample of MBD cohort I (a) and II (enriched samples in (b); input samples in (c)).
Using SAMStat, the mapping quality scores of each sample of MBD cohort I and II are analyzed. Given are the numbers and percentages of mapped reads across the different mapping quality ranges, as determined by SAMStat ((a) enriched samples of MBD cohort I, (b) enriched samples of MBD cohort II and (c) input samples of MBD cohort II).
Promoter count data for the MBD-enriched samples of MBD cohort I. For each MBD-enriched sample of MBD cohort I, the number of mapped reads in each Ensembl Transcript promoter region (-1500 bp to +500 bp around TSS) is given.
Promoter count data for the MBD-enriched samples of MBD cohort II. For each MBD-enriched sample of MBD cohort II, the number of mapped reads in each Ensembl Transcript promoter region (-1500 bp to +500 bp around TSS) is given.
Promoter count data for the input samples of MBD cohort II. For each input sample of MBD cohort II, the number of mapped reads in each Ensembl Transcript promoter region (-1500 bp to +500 bp around TSS) is given.
Window count data for the MBD-enriched samples of MBD cohort I. For each MBD-enriched sample of MBD cohort I, the number of mapped reads in each 5 kb genomic window (2.5 kb overlapping moving windows) is given.
Window count data for the MBD-enriched samples of MBD cohort II. For each MBD-enriched sample of MBD cohort II, the number of mapped reads in each 5 kb genomic window (2.5 kb overlapping moving windows) is given.
Window count data for the input samples of MBD cohort II. For each input sample of MBD cohort II, the number of mapped reads in each 5 kb genomic window (2.5 kb overlapping moving windows) is given.