

Abstract
We report the preparation of thermally tunable hydrogels displaying angle‐independent structural colors. The porous structures were formed with short‐range order using colloidal amorphous array templates and a small amount of carbon black (CB). The resultant porous hydrogels prepared using colloidal amorphous arrays without CB appeared white, whereas the hydrogels with CB revealed bright structural colors. The brightly colored hydrogels rapidly changed hues in a reversible manner, and the hues varied widely depending on the water temperature. Moreover, the structural colors were angle‐independent under diffusive lighting because of the isotropic nanostructure generated from the colloidal amorphous arrays.
Keywords: amorphous arrays, colloids, hydrogels, polymers, structural colors
Stimuli‐sensitive soft materials composed of three‐dimensional polymer networks or block copolymers with fine periodic dielectric structures of dimensions comparable to the wavelength of visible light can display bright structural colors.1–5 The hues of the structural colors can reversibly change by varying the periodic dielectric structures with different stimuli. Extensive research has been performed in recent decades to develop such stimuli‐sensitive structurally colored soft materials for application to displays2, 4, 6 and sensor systems.5, 7 In contrast to any other display and sensor technologies, these stimuli‐sensitive structurally colored soft materials can exhibit a wide range of colors on their own, without requiring color filters and complicated control devices.2 However, the colors produced by the periodic dielectric structures show distinct variations depending on the viewing and light illumination angles.8 The iridescence originates from the Bragg reflection, resulting from the long‐range order of the periodic structures. For potential applications to reflective full‐color displays and sensor systems with wide viewing angles, the angular dependence of the structural color is a major issue. In this study, we report the development of thermally tunable hydrogels displaying angle‐independent structural colors. We demonstrate that the application of colloidal amorphous arrays as templates and the introduction of a small amount of black particles into the thermosensitive hydrogels are effective for obtaining tunable and angle‐independent brightly colored systems.
In the past, structurally colored materials most likely possessed periodic dielectric structures with long‐range order, such as opals9 and multi‐layer films.3 Recent research has shown, however, that if the materials have short‐range order in the dielectric structures, then these materials also can display structural color.8, 10, 11–13 Moreover, materials with only short‐range order in the dielectric structures display angle‐independent structural colors originating from wavelength‐specific constructive interference in the visible region. We demonstrated that colloidal amorphous arrays composed of fine submicron‐sized particles with a short‐range order could reveal bright, angle‐independent structural colors.13, 14 If we can introduce short‐range order in the dielectric structure of stimuli‐sensitive soft materials, then the materials should be able to display angle‐independent structural colors and should be able to change these colors in response to external stimuli.14
In the initial step, porous thermosensitive hydrogels, the volumes of which change reversibly with the water temperature, were prepared using the colloidal amorphous arrays as templates (Scheme 1). A pre‐gel solution in which an N‐isopropylacrylamide monomer (NIPA), a cross‐linker, and an initiator were dissolved in dimethyl sulfoxide, was impregnated into the colloidal amorphous arrays and was converted into hydrogels. The hydrogels and colloidal amorphous arrays were immersed in a 10 wt % NaOH aqueous solution to remove the silica component, leaving behind porous poly(N‐isopropylacrylamide) (PNIPA) hydrogels. Thus, a porous structure with short‐range order in the dielectric structure was constructed in the thermosensitive PNIPA hydrogels using colloidal amorphous arrays as templates.
Scheme 1.

Preparation of a porous thermosensitive hydrogel using a colloidal amorphous array.
In Figure 1 a, the temperature dependence of swelling is shown for the disk‐shaped porous PNIPA hydrogel in water, which was measured with reference to the diameter of the hydrogel at 15 °C in water (d 0). The reference size of the disk‐shaped porous PNIPA hydrogel was 1 cm in diameter and 3 mm in thickness at 15 °C in water. As expected, the porous PNIPA hydrogel underwent a continuous but large volume change at approximately 33 °C.15
Figure 1.
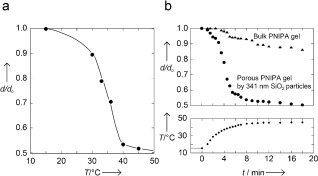
Swelling behaviors of the porous PNIPA hydrogel. a) The temperature dependence of the swelling ratio (d/d 0) of the porous PNIPA hydrogel, prepared by a colloidal amorphous array composed of fine silica particles (341 nm diameter) in water. d is the observed diameter of the porous PNIPA hydrogel. b) The progression of swelling in the bulk and porous disk‐shaped PNIPA hydrogels in water in response to rapid temperature change. The lower curve shows the change in temperature.
In a previous study, we reported that the interconnecting porous PNIPA hydrogels, prepared using a colloidal crystal composed of fine silica particles as a template, shrunk rapidly when the water temperature suddenly changed from 25 to 40 °C.16 The rate was more than 1000 times faster than that of homogeneous bulk PNIPA hydrogels of the same shape and size. Figure 1 b shows the shrinkage of the disk‐shaped hydrogels, prepared using the colloidal amorphous array as a template and a bulk PNIPA hydrogel with the same shape and size, after a sudden change in the water temperature from 15 to 45 °C. The required time for the hydrogels to reach 45 °C from 15 °C was approximately 10 minutes in our experimental system (lower plot in Figure 1 b). The bulk PNIPA hydrogel contracted in response to the rise in temperature, and immediately, the surface became opaque with the formation of a surface skin layer. Following this event, the shrinking process appeared to cease for a certain period, after which the hydrogel continued to gradually shrink. More than 50 h were required to reach an equilibrated collapsed state of the bulk hydrogel. The kinetics of gel shrinking is governed by the collective diffusion of the polymer network relative to the solvent. The relaxation time τ to reach the equilibrium swelling state of the gel is proportional to the square of a characteristic gel size and can be expressed as follows: τ∝a 2/D, where a is the thickness of the disk, and D is the collective diffusion coefficient of the polymer network.17 The value of D is typically considered to be constant and on the order of 10−7 cm2 s−1 if the initial and the final states of the swelling are fixed. Considering the size of the disk gel and the formation of a skin layer, it was expected that a long time would be required to reach the fully collapsed state after the rapid temperature increase. However, an extremely fast response could be observed in the porous PNIPA hydrogel. The deswelling of the porous PNIPA hydrogel was accomplished in approximately 10 minutes, and the change in volume was followed by a change in the water temperature. The fast response is attributed to the interconnecting pore structure generated from the colloidal amorphous array.16, 18
The porous PNIPA hydrogels remained opaque and whitish, regardless of the equilibrated volume. The opacity and the whiteness of the porous hydrogels were caused by the porous structure prepared from the colloidal amorphous arrays and were not caused by phase separation of the polymer networks. The appearances of the relatively thick colloidal amorphous arrays are also white, regardless of the size of the submicron‐sized silica particles. Because of the incoherent multiple scattering of light throughout the entire visible region by the inner microstructure of the colloidal amorphous arrays, the arrays appear white to the naked eye.12, 19 The porous PNIPA hydrogels also appeared white because of the same optical effect as in the colloidal amorphous arrays.
Materials with a short‐range order and refractive index variations comparable to the wavelength of visible light can exhibit constructive interference of light in this wavelength region. For example, as shown in Figure 2 a, the colloidal amorphous array, composed of fine silica particles 341 nm in diameter, coherently scatters light at approximately 640 nm. Although the colloidal amorphous array can reveal the wavelength‐specific constructive interference of light, the observation of structural color from the relatively thick array is difficult because of the large contribution from the incoherent multiple scattering of light.11 By adding black components, such as a carbon black (CB), to the amorphous arrays (Figure 2 b), the overall magnitude of the scattering greatly decreases with the amount of CB, while the position and intensity of the peak components appear to remain constant (Figure 2 a).11, 12, 20–22 As a result, we can observe structural colors from colloidal amorphous arrays due to the enhancement of the structural color saturation by reducing the incoherent wavelength‐independent scattering of light across the entire visible region (Figure 2 c). The hue of the structural colors, which is only slightly influenced by the addition of CB, is determined by the characteristic length of the short‐range order and by the average refractive index of the colloidal amorphous arrays. As shown in Figure 2 c, the hue of the array can change when using different silica particle sizes.20 Figure 2 d shows the scattering spectra of a colloidal amorphous array, composed of silica particles 341 nm in diameter and containing 0.05 wt % of CB, measured by changing only the detection angle, while the incidence angle remained fixed. The incidence angle relative to the normal angle of the surface of the array was 0°. The detection angle θ was varied from 0 to 45°. In the case of a colloidal crystal film, the Bragg reflection peak in the scattering spectrum disappears when the detection angle deviates from the specular direction.22 However, the scattering peak in the spectrum from the colloidal amorphous array could be observed from every angle due to its isotropic nanoscale structure.23 As a result, we could observe a distinct peak from coherent scattering in the scattering spectrum as the detection angle changed from 0 to 45°, whereas the intensity of the peak became smaller as the angle increased. The position of the peak shifted slightly towards shorter wavelengths with increasing detection angle (inset in Figure 2 d). This result suggests that the scattering peak was not caused by the Mie resonance of single particle scattering, but rather, is caused by the constructive interference of light scattered only once in the colloidal amorphous array with a short‐range order.23 Under diffuse lighting, such as sunlight, however, the colors from the colloidal amorphous arrays are invariant with the viewing angle or array orientation because of the isotropic nanostructure of the arrays (Figure 2 e). This is because the scattering spectrum under diffuse lightning is the sum of the spectra under directional illumination over many angles.
Figure 2.
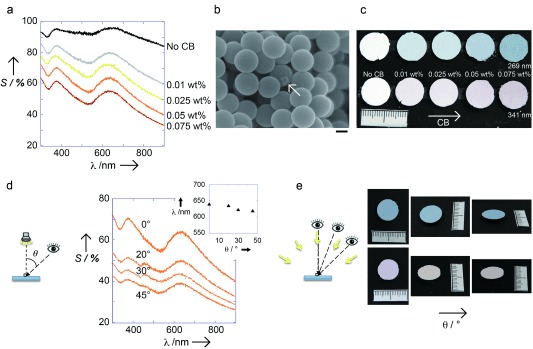
Physical properties of the colloidal amorphous arrays. a) Scattering spectra of colloidal amorphous arrays composed 341 nm silica particles with different amounts of CB. The incident angle relative to the normal angle of the planar membrane surface was 0°. The measurement angle was also approximately 0° relative to the normal angle of the planar membrane surface. b) SEM image of the colloidal amorphous arrays composed of 341 nm silica particles of with 0.05 wt % CB. The CB is indicated by an arrow. Scale bar=100 nm. c) Optical photographs showing the color change with varying quantities of CB, and with the size of silica particles. d) The scattering spectra of the array of 341 nm silica particles with 0.05 wt % CB as a function of the measurement angle. The incident angle relative to the normal membrane surface was 0°. The measurement angle θ varied from 0 to 20, 30 and 45° relative to the normal surface. Inset: Plots of the position of the peak wavelength λ max versus the measurement angle θ. e) Optical photographs of the colloidal amorphous arrays composed of 269 and 341 nm particles with 0.075 wt % CB at different viewing angles under diffusive lighting conditions.
Considering the results with the addition of CB, we can expect that the porous PNIPA hydrogels prepared using colloidal amorphous arrays as templates will also display structural color with the introduction of CB. Therefore, we studied the effect of CB addition on the structural coloration from the porous PNIPA hydrogels prepared using the colloidal amorphous arrays including a small amount of CB as the template. Prior to the preparation of the hydrogels, six versions of the colloidal amorphous arrays with different amounts of CB were prepared. The silica particles are soluble in a NaOH aqueous solution, while the PNIPA polymer network and CB are chemically stable in this solution. Thus, the resultant hydrogels contained CB even after immersion in the solution (Scheme 2). Figure 3 a shows the change in appearance of the six porous PNIPA hydrogels prepared with the colloidal amorphous arrays, composed of the 269 nm diameter silica particles with different amounts of CB. The white hydrogel situated at the centre of the five colored hydrogels did not contain CB. The surrounding numbered hydrogels included

0.01,

0.025,

0.05,

0.075, and
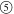
0.1 wt % CB. The porous PNIPA hydrogel without CB always appeared white, regardless of the observed water temperature. In contrast, the porous PNIPA hydrogels with CB displayed bright structural colors with different hues, depending on the water temperature. For example, the porous PNIPA hydrogels in Figure 3 a exhibited a dusty red color at 15 °C, which changed to green and then blue with increasing temperature (Figure 3 a). By varying the amount of CB, the saturation changed, and the brightness of the structural color was altered because the resultant hydrogels changed. For the hydrogels containing 0.1 wt % CB, the colors were nearly black, regardless of the used particle size and temperature. The hydrogels with a CB content less than 0.1 wt % could display brilliant structural colors, which changed with their volume for different water temperatures. The structural colors from the hydrogels prepared using the colloidal amorphous array composed of 269 nm silica particles were barely visible when the hydrogels were completely collapsed at approximately 45 °C. This was because the scattering peaks caused by constructive interference were situated in the ultraviolet region (Figure 3 b,c). By using larger silica particle sizes for the templates, structural colors from the porous hydrogels could be observed, even at a high temperature. The hydrogel obtained using 341 nm particles displayed a bright blue color, even in the collapsed state at 45 °C (Supporting information Figure S5, and Movie S2) because the characteristic size of the short‐range order structured by the templates was larger. Figure 3 c shows the position of the scattering peak as a function of water temperature observed in the porous hydrogel using 269 nm silica particles and with 0.075 wt % CB. This plot demonstrates the same trend as that shown in Figure 1 a. As observed in Figure 3 b, the intensity of the scattering peak, which is proportional to the magnitude of the refractive index contrast, increased at higher temperatures. The increase in the refractive index contrast as the temperature increases was caused by an increase in the refractive index of the gel portion and a slight decrease in that of water. Therefore, the structural colors observed in the porous hydrogels at the higher temperature were brighter (Figure 3 a). Similar to the case of the colloidal amorphous arrays with a small amount of CB, the colors from the porous hydrogels with CB did not vary with the viewing angle under diffuse lightning conditions (Figure 3 d).
Scheme 2.

Preparation of a structurally colored porous thermosensitive hydrogel using a colloidal amorphous array including CB.
Figure 3.

Optical properties of the porous PNIPA hydrogels. a) Optical photographs of the porous PNIPA hydrogels prepared using an array of 269 nm silica particles and different amounts of CB in water at several temperatures. b) The scattering spectra of the porous PNIPA hydrogels prepared using 269 nm silica particles and 0.075 wt % CB in water at several temperatures. The incident angle relative to the normal planar membrane surface was 0°. The measurement angle was also approximately 0° relative to the normal surface of the membrane. c) Plots of the position of the peak wavelength λ max versus the water temperature for Figure 3 b. d) Optical photographs of the porous PNIPA hydrogels prepared using 269 nm silica particles and 0.075 wt % CB in water at several temperatures, under different viewing angles and diffusive lighting conditions.
Many examples of stimuli‐sensitive structurally colored soft materials with rapid response and hue changes have been reported; however, such systems without angle‐independent structural colors have yet to be reported. The porous PNIPA hydrogels prepared using colloidal amorphous array templates can exhibit rapid changes in volume in response to temperature variations and can potentially display angle‐independent structural colors. Our results showed that the microstructure of the prepared hydrogels using the colloidal amorphous arrays as templates is not suitable for displaying brightly saturated structural colors due to the large contribution from incoherent multiple scattering of light to the optical properties. The small amount of CB introduced into the porous hydrogels suppressed multiple scattering, enabling clearer observations of the structural colors. This method is easily applicable to many other existing stimuli‐sensitive soft materials. We expect that such stimuli‐sensitive soft materials displaying bright, angle‐independent structural colors will have many useful applications, such as in (bio)chemical sensing technology and electronic paper.
Experimental Section
Methods and Materials: The silica particles (269 and 341 nm in diameter) used in this study were purchased from Nippon Shokubai Co. Ltd. The CB, kindly obtained from Tokai Carbon Co. Ltd., had an average particle diameter of 100 nm. N‐isopropylacrylamide, provided by Kohjin Co., Japan, was recrystallized from a toluene‐hexane mixed solvent and dried under vacuum prior to use. Water was purified using a Direct‐Q UV water purification system (Millipore Corp.). All other chemicals used in this study were purchased at the highest purity and were used as received.
Template fabrication: Silica particles (0.5 g) and ethanol (1 mL) were mixed in an ultrasonic bath at room temperature to obtain the suspensions. We prepared the colloidal amorphous arrays from the suspensions by evaporating ethanol at 25 °C in a constant temperature bath. To confirm the existence of the short‐range order, we employed two‐dimensional Fourier analyses of SEM images of the arrays.13b, 20–22 To add CB to the colloidal amorphous arrays, the amount of CB suspended with the silica particles in ethanol was varied from 0.01, 0.025, 0.05, 0.075, to 0.1 wt % with respect to the weight of the silica particles.
Preparation of gels: A solution of NIPA (6.788 g), N,N′‐methylene‐bis‐acrylamide (0.467 g) as a cross‐linker, and azobisisobutyronitrile (0.101 g) as an initiator, which was dissolved in dimethyl sulfoxide, was degassed under nitrogen for 20 min. The solution was injected into the colloidal amorphous array in a glass cell consisting of two glass plates separated by a Teflon spacer. The gelation was performed at 60 °C for 30 h. After the gelation, the sample was soaked in a 10 wt % NaOH aqueous solution at 4 °C to dissolve the silica component. The resulting porous gel membrane was washed with water to remove unreacted chemicals and NaOH. The CB confined in the polymer networks cannot be released from the hydrogels because the primary particle size of the CB is much larger than the average mesh size of the hydrogels.
Swelling behaviour: The porous disk hydrogel was held in equilibrium in water at each temperature in a glass cell connected to a circulating water temperature control system. The diameter of the porous disk hydrogel as a function of temperature was measured using a ruler.
Shrinking kinetics: The kinetics of the shrinking process of the homogeneous and porous disk hydrogels in water was monitored by measuring the diameter after suddenly changing the water temperature from 15 °C to 45 °C.
Scattering spectra: We performed angle‐resolved scattering spectrometry to characterize the colors under directional white light illumination using an optical fibre spectrometer (QE65000, Ocean Optics), light source (DH‐2000‐BAL, Ocean Optics), optical fibres (P400‐1‐SR, Ocean Optics), a reflection/backscattering probe (R400‐7‐UV‐VIS, Ocean Optics), and spectrometer operation software (SpectraSuite, Ocean Optics). The incident angle relative to the normal angle of the planar surface of the samples and the measurement angle θ were varied from 0° to 45°.
Scanning electron microscopy: The arrangement of the silica colloidal particles in the arrays was investigated using a scanning electron microscope (SEM, Hitachi, Miniscope TM3000). The samples were coated with a 10 nm Au‐Pd layer using a magnetron sputtering apparatus (MSP‐1S), and the images were obtained with the SEM at 15 kV.
Digital camera observation: Photographs showing the colors of the samples were acquired using a digital camera.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Movie_S1
Movie_S2
miscellaneous_information
Acknowledgements
This study was supported by a Grant‐in‐Aid for Scientific Research (grant number 22107012) in the innovative area of “Fusion Materials” (area number 2206) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and by The Canon Foundation. NIPA and CB were kindly provided by Kohjin Co. Ltd. and Tokai Carbon Co. Ltd., respectively.
References
- 1.
- 1a. Takeoka Y., J. Mater. Chem. C 2013, 1, 6059; [Google Scholar]
- 1b. Takeoka Y., Seki T., Langmuir 2006, 22, 10223. [DOI] [PubMed] [Google Scholar]
- 2. Arsenault A. C., Puzzo D. P., Manners I., Ozin G. A., Nat. Photonics 2007, 1, 468. [Google Scholar]
- 3.
- 3a. Kang Y., Walish J. J., Gorishnyy T., Thomas E. L., Nat. Mater. 2007, 6, 957; [DOI] [PubMed] [Google Scholar]
- 3b. Yue Y. F., Kurokawa T., Haque M. A., Nakajima T., Nonoyama T., Li X. F., Kajiwara I., Gong J. P., Nat. Commun. 2014, 5, 4659. [DOI] [PubMed] [Google Scholar]
- 4. Matsubara K., Watanabe M., Takeoka Y., Angew. Chem. Int. Ed. 2007, 46, 1688; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1718. [Google Scholar]
- 5. Nakayama D., Takeoka Y., Watanabe M., Kataoka K., Angew. Chem. Int. Ed. 2003, 42, 4197; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4329. [Google Scholar]
- 6. Ueno K., Matsubara K., Watanabe M., Takeoka Y., Adv. Mater. 2007, 19, 2807. [Google Scholar]
- 7.
- 7a. Holtz J. H., Asher S. A., Nature 1997, 389, 829; [DOI] [PubMed] [Google Scholar]
- 7b. Zhang C. J., Losego M. D., Braun P. V., Chem. Mater. 2013, 25, 3239. [Google Scholar]
- 8. Takeoka Y., J. Mater. Chem. 2012, 22, 23299. [Google Scholar]
- 9. Marlow F., Muldarisnur, Sharifi P., Brinkmann R., Mendive C., Angew. Chem. Int. Ed. 2009, 48, 6212; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6328. [Google Scholar]
- 10.
- 10a. Prum R. O., Torres R. H., Williamson S., Dyck J., Nature 1998, 396, 28; [Google Scholar]
- 10b. Noh H., Liew S. F., Saranathan V., Mochrie S. G. J., Prum R. O., Dufresne E. R., Cao H., Adv. Mater. 2010, 22, 2871. [DOI] [PubMed] [Google Scholar]
- 11. Forster J. D., Noh H., Liew S. F., Saranathan V., Schreck C. F., Yang L., Park J. G., Prum R. O., Mochrie S. G. J., O’Hern C. S., Cao H., Dufresne E. R., Adv. Mater. 2010, 22, 2939. [DOI] [PubMed] [Google Scholar]
- 12. Liew S. F., Forster J., Noh H., Schreck C. F., Saranathan V., Lu X., Yang L., Prum R. O., O’Hern C. S., Dufresne E. R., Cao H., Opt. Express 2011, 19, 8208. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Takeoka Y., Honda M., Seki T., Ishii M., Nakamura H., ACS Appl. Mater. Interfaces 2009, 1, 982; [DOI] [PubMed] [Google Scholar]
- 13b. Harun‐Ur‐Rashid M., Bin Imran A., Seki T., Ishi M., Nakamura H., Takeoka Y., ChemPhysChem 2010, 11, 579. [DOI] [PubMed] [Google Scholar]
- 14. Gotoh Y., Suzuki H., Kumano N., Seki T., Katagiri K., Takeoka Y., New J. Chem. 2012, 36, 2171. [Google Scholar]
- 15. Hirotsu S., Hirokawa Y., Tanaka T., J. Chem. Phys. 1987, 87, 1392. [Google Scholar]
- 16. Takeoka Y., Watanabe M., Langmuir 2002, 18, 5977. [Google Scholar]
- 17. Li Y., Tanaka T., J. Chem. Phys. 1990, 92, 1365. [Google Scholar]
- 18. Takeoka Y., Watanabe M., Yoshida R., J. Am. Chem. Soc. 2003, 125, 13320. [DOI] [PubMed] [Google Scholar]
- 19. Shawkey M. D., Hill G. E., J. Exp. Biol. 2006, 209, 1245. [DOI] [PubMed] [Google Scholar]
- 20. Takeoka Y., Yoshioka S., Takano A., Arai S., Nueangnoraj K., Nishihara H., Teshima M., Ohtsuka Y., Seki T., Angew. Chem. Int. Ed. 2013, 52, 7261; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7402. [Google Scholar]
- 21. Takeoka Y., Yoshioka S., Teshima M., Takano A., Harun‐Ur‐Rashid M., Seki T., Sci. Rep. 2013, 3, 1–7; [DOI] [PMC free article] [PubMed] [Google Scholar]; Teshima M., Seki T., Kawano R., Takeuchi S., Yoshioka S., Takeoka Y., J. Mater. Chem. C 2015, 3, 769. [Google Scholar]
- 22. Hirashima R., Seki T., Katagiri K., Akuzawa Y., Torimotoa T., Takeoka Y., J. Mater. Chem. C 2014, 2, 344. [Google Scholar]
- 23. Dufresne E. R., Noh H., Saranathan V., Mochrie S. G. J., Cao H., Prum R. O., Soft Matter 2009, 5, 1792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Movie_S1
Movie_S2
miscellaneous_information