

Abstract
BACKGROUND & AIMS
Innate immune activation has been postulated as a central mechanism for disease progression from hepatic steatosis to steatohepatitis in obesity-related fatty liver disease. Arginase 2 competes with inducible nitric oxide synthase (iNOS) for its substrate and the balance between these two enzymes plays a crucial role in regulating immune responses and macrophage activation. Our aim was to test the hypothesis that arginase 2 deficiency in mice favors progression from isolated hepatic steatosis, induced by high fat feeding to steatohepatitis.
METHODS
Arginase 2-knockout (Arg2−/−) mice were studied for changes in liver histology and metabolic phenotype at baseline and after a short term course (7 week) feeding with a high fat (HFAT) diet. In additional experiments, Arg2−/− mice received tail vein injections of liposome-encapsulated clodronate (CLOD) over a three-week period to selectively deplete liver macrophages.
RESULTS
Unexpectedly, Arg2−/− mice showed profound changes in their livers at baseline characterized by significant steatosis as demonstrated with histological and biochemical analysis. These changes were independent of systemic metabolic parameters and associated with marked increase mRNA levels of genes involved in hepatic de novo lipogenesis. Liver injury and inflammation were present with elevated serum ALT, marked infiltration of F4/80 positive cells, and increased mRNA levels of inflammatory genes. HFAT feeding exacerbated these changes. Macrophage depletion after CLOD injection significantly attenuated lipid deposition and normalized lipogenic mRNA profile of livers from Arg2−/− mice.
CONCLUSIONS
This study identifies arginase 2 as novel link between innate immune responses, hepatic lipid deposition, and liver injury.
Keywords: nonalcoholic fatty liver disease, steatohepatitis, arginase 2, macrophage activation, innate immunity, inflammation, liver fibrosis
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is currently the most common form of chronic liver disease affecting both adults and children, and is strongly associated with obesity and insulin resistance [1, 2]. One in three adults and one in ten children or adolescents in the United States have hepatic steatosis, a stage within the spectrum of NAFLD, that is characterized by triglyceride accumulation in liver cells and follows a benign non-progressive clinical course [3, 4]. Nonalcoholic steatohepatitis (NASH) is defined as lipid accumulation with evidence of cellular damage, inflammation and varying degrees of scarring or fibrosis [5]. NASH is a serious condition, as approximately 25% of these patients progress to cirrhosis and its known complications that include portal hypertension, liver failure and hepatocellular carcinoma [6–8].
NAFLD pathogenesis, and specifically disease progression to NASH, has been the subject of intense investigation over the past decade, as both diagnostic and prognostic markers and treatment options for patients with this disease remain limited [9]. A multi-hit model of NAFLD pathogenesis has emerged in recent years in which intrahepatic lipid accumulation in the setting of insulin resistance is postulated to be the first hit, which sets the stage for multiple “second hits” resulting in inflammation, hepatocellular damage and eventually fibrosis [10]. Growing evidence supports the concept that innate immune activation and increased oxidative stress in the liver are key “second hits” involved in disease progression to NASH. Activation of liver resident macrophages or Kupffer cells, at least in part by an increase in exposure to gut-derived bacterial wall components such as endotoxin, result in an increased production of pro-inflammatory cytokines as well as inducible nitric oxide synthase (iNOS) and has been proposed to play a pivotal role in this process [11, 12]. Expression of iNOS in cell types such as macrophages and hepatocytes is induced by proinflammatory cytokines [13], and iNOS has been implicated in the pathophysiology of NAFLD [14–16]. Mammals express two isoforms of arginase, an enzyme than competes with iNOS for its substrate, argninine, designated as types 1 and 2 [17–19]. Arginase 1 is mainly expressed in hepatocytes, and mice with a disruption of arginase 1 gene die soon after birth. Arginase 2 is poorly expressed in hepatocytes, and most highly expressed in kidney, prostate, and immune cells such as monocyte/ macrophages [17, 19]. An imbalance in iNOS/arginase ratio in favor of the former may play a crucial role in regulating immune responses and macrophage activation towards a classically “M1” activated, pro-inflammatory state [20–23]. The aim of our study was to test the hypothesis that arginase 2 deficiency in mice would result in progression from isolated hepatic steatosis typically seen in diet induced obesity (DIO) to steatohepatitis. Our data identify a novel link between Kupffer cell activation and hepatic steatosis and suggest that in the presence of arginase 2 deficiency, altered immune responses precedes, and is responsible of lipid deposition in hepatocytes mainly by activation of de novo lipogenesis pathways in the liver. These findings have important implications for the pathogenesis of NAFLD and potential development of novel treatment strategies for patients with this condition.
METHODS
Animal studies
The experimental protocol has been approved by the Institutional Animal Care and Use Committee at Cleveland Clinic. Male C57BL/6 wild-type (WT) were purchased from Jackson Laboratory. The generation of Arg2−/− mice has been described previously [24]. Arg2−/− mice are viable and indistinguishable from WT mice. Mice were fed either a diet consisting of 5% fat (TD 2918, Harlan Laboratories, Madison, WI) or a high fat (HFAT), western type diet (consisting of 42% of Kcal from fat, TD88137, Harlan Laboratories, Madison, WI). Total body weight was recorded on a weekly basis. At indicated time points, plasma and liver tissue were collected and weighed after an overnight fast as described previously [25].
Analyses of plasma and liver metabolic mediators
Blood was collected from Arg2−/− and WT mice after an overnight fast by cardiac puncture. Blood was spun at 2000 RPM for 15 min at 4°C, plasma drawn from the top layer, argon overlaid and stored at −80°C. Plasma assays of insulin and glucose were performed using commercially available mouse insulin ELISA (ALPCO Diagnostics, Salem, NH) and glucose assay (Cayman Chemical, Ann Arbor, MI) kits. Plasma and liver triglyceride and free fatty acid levels were measured with triglyceride-GPO liquid reagent (Pointe Scientific, Inc, Canton, MI), and free fatty acid quantification (BioVision, Mountain View, CA) kits according to manufacturers’ instructions. Serum alanine aminotransferase (ALT) concentrations were measured and expressed as international units per liter (Clinical Laboratory Services, Cleveland Clinic Foundation, Cleveland, OH).
Histopathology and immunostaining
Mouse tissue was diced into 5 × 5-mm sections, immersion-fixed in PBS containing 4% paraformaldehyde for 24 h at 4°C, and embedded in paraffin. Four micrometer sections were mounted on glass slides. Hematoxylin and eosin (H&E) stained liver specimens were evaluated by light microscopy for histopathological scoring by a hepatopathologist (BGP). Steatosis, inflammation, and ballooning were scored based on NAFLD activity score [26]. Presence of macrophage infiltration was assessed by immunohistochemical staining for F4/80. Paraffin-embedded liver sections were deparaffinized and antigen retrieval using 10 mM sodium citrate buffer was performed. Sections were incubated with primary antibody overnight at 4°C (1:50 dilution, Abd Serotec, Oxford). Subsequently, sections were incubated with a biotinylated anti-rat IgG secondary antibody (Vector Laboratories, Burlingame, CA) and a Vectastain ABC Elite Kit according to manufacturer’s instructions (Vector Labs). Sections were developed with ImmPACT DAB peroxidase substrate (Vector Labs) and counterstained with hematoxylin.
Oil Red O staining
Assessment of hepatic steatosis was performed by staining with Oil Red O (ORO), a fat-soluble diazo dye. Frozen liver sections (10 μm thick) were mounted on glass slides. ORO stock solution was prepared by mixing 300 mg ORO (Sigma-Aldrich, St. Louis, MO) and 100 ml 2-propanol, 99% (Fisher Scientific, Pittsburg, PA). A working solution of 1.5:1 ORO stock solution:distilled water was then prepared. Liver slides were stained with ORO working solution for 12 minutes, after which they were washed in distilled water twice for 20 seconds and rinsed in running tap water for 10 minutes. Finally, slides were stained with hematoxylin for 45 seconds and then washed in distilled water.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from liver using RNeasy Mini kit (Qiagen, Valencia, CA). One μg RNA was reverse transcribed using random decamers and MMLV-reverse transcriptase (Applied Biosystems). Genes assayed for mRNA expression levels included inflammatory cytokines, markers of hepatic stellate cell activation, hepatic insulin signaling, and mediators of lipid trafficking and metabolism (see supplementary table 1 for primer sequences). The fold change over control samples was calculated using CT, ΔCT, and ΔΔCT using MaxPro software (Agilent, Santa Clara, CA). 18S ribosomal RNA was used as an endogenous control.
Immunoblot analysis
Immunoblot analysis were performed as previously described.[27] Anti-Arginase 2 (ARG2, Santa Cruz, Santa Cruz, CA, USA), anti-fatty acid synthase (FAS, GeneTex, Irvine, CA, USA), and anit-stearoyl-CoA desaturase 1 (SCD-1, Cell Signaling Technology, Boston, MA, USA) antibodies were used in combination with appropriate peroxidase-conjugated secondary antibodies. Protein load was verified with an α-tubulin antibody (dilution 1:10,000) (Hybridomabank, University of Iowa) (kindly provided by M. Kaulich). Bands were visualized with the enhanced chemiluminescence reagent and digitized using a CCD camera (ChemiDoc®, Biorad, Hercules, CA, USA). Expression intensity was quantified by ImageLab (Biorad).
Liver-specific macrophage depletion
Macrophage depletion studies were done in Arg2−/− and wild type mice using liposome-encapsulated clodronate injection as is a validated method for in vivo depletion of tissue-specific macrophages [28, 29]. Mice received four tail vein injections of 100 uL/10 g body weight of 1 mg/ml PBS or clodronate (CLOD)-containing liposomes over a three-week period. Mice were sacrificed 24 hours after the fourth injection, at which time plasma, and liver tissue was collected.
Statistical analysis
All data were expressed as the mean ± S.D. unless otherwise indicated. Differences between groups were compared by an ANOVA analysis followed by a post hoc Bonferroni test to correct for multiple comparisons. Differences were considered to be statistically significant at p < 0.05.
RESULTS
Arg2−/− mice develop spontaneous liver injury and hepatic steatosis
Arg2−/− and WT mice showed similar trends in weight gain for up to 14 weeks of age (Fig. 1A) and no evidence of systemic metabolic derangements with plasma assays of fasting triglycerides, free fatty acids, glucose and insulin (Fig. 1B). However, serum levels of alanine aminotransferase (ALT) were elevated in Arg2−/− mice compared to WT animals (110 vs. 35 U/L, p <0.05; Fig. 1D) suggesting the presence of liver injury. Indeed, H&E and ORO stained liver sections revealed notable inflammation and lipid deposition in Arg2−/− mice compared to WT (Fig. 1C). This was associated with increased hepatic triglyceride levels (Arg2−/− 4.9 vs. WT 2.6 mg TG/g liver, p <0.05; Fig. 1E).
Figure 1. Spontaneous liver injury and hepatic steatosis is present in 14 weeks old Arg2−/− mice.
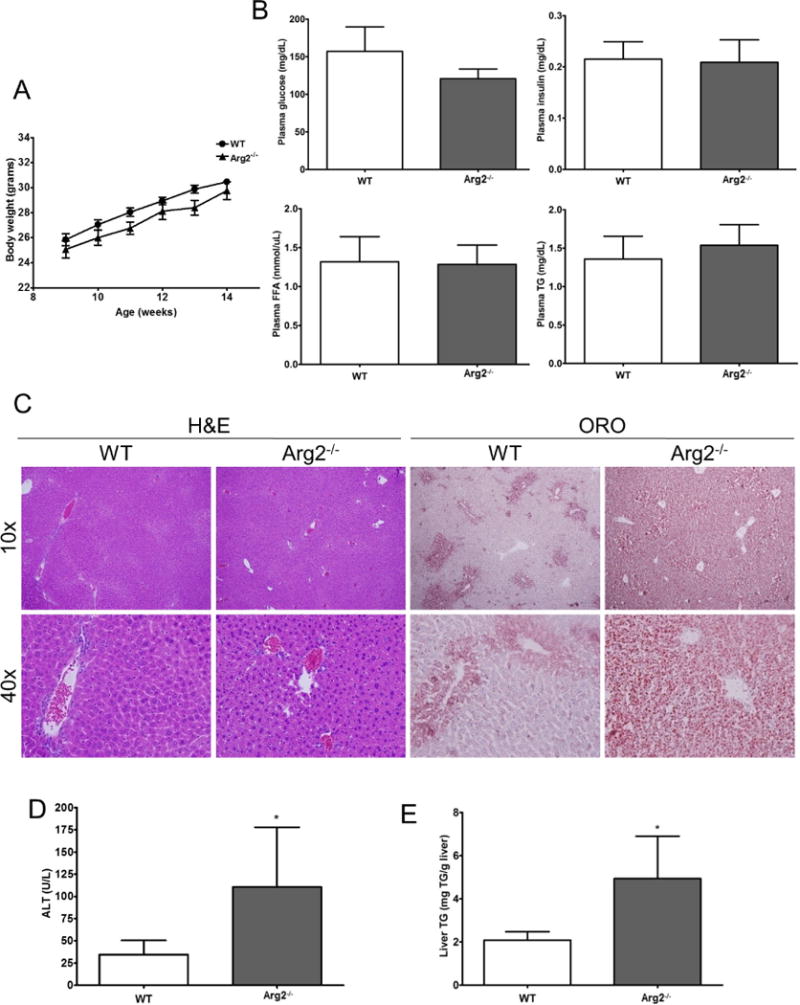
Body weight (A) along with several metabolic parameters – plasma glucose level, plasma insulin level, levels of free fatty acids in plasma, plasma triglycerides – are comparable in Arg2−/− mice and WT mice fed with normal chow (B). However, analysis of liver histology and assessment of hepatic steatosis with Oil Red O staining uncovered the development of liver injury and hepatic steatosis (C) (10× and 40×). In line with this ALT, levels were significantly increased in Arg2−/− mice when compared to WT mice (D). The amount of liver triglycerides was also increased in Arg2−/− (Arg2−/− 4.9 vs. WT 2.6 mg TG/g liver, p <0.05, E). Mean ± S.E.M. are shown. *p<0.05.
De novo lipogenesis (DNL) is enhanced in Arg2−/− mice
To further explore this phenotype of hepatic steatosis in Arg2−/− mice, the contributions of distinct processes involved in lipid trafficking and metabolism were characterized by qRT-PCR. Fatty acid uptake into and transport within the liver was assayed by CD36 or fatty acid translocase, fatty acid transport proteins (FATP) and fatty acid binding proteins (FABPs). Hepatic FABP1 mRNA expression was increased twofold, while FABP4, FATP2 and FATP5 were modestly decreased in Arg2−/− mice compared to WT animals (Fig. 2A). Fatty acid synthesis and de novo lipogenesis (DNL) were assessed by measuring mRNA levels of acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), stearoyl-CoA desturase 1 (SCD-1), the transcription factor sterol regulatory element binding protein 1c (SREBP-1c), liver X receptor alpha (LXR-α), ATP citrate lyase (ACYL), and elongation of long chain fatty acids 2, 5 and 6 (Elovl2, 5, 6). We were able to show that hepatic mRNA expression of several enzymes involved in DNL were significantly increased in Arg2−/− mice, with SREBP1c and SCD-1 expression increased three- and two-fold over WT controls, respectively (Fig. 2B). A significant increase was also documented for mRNA levels of LXR-α, ACLY, and Elovl2. Fatty acid oxidation, assessed by measuring mRNA levels of carnitine palmitoyl-transferase 1 (CPT-1), and acyl-coenzyme A dehydrogenase, C-4 to C-12 straight chain (ACADM), did not appear to contribute to hepatic lipid accumulation within Arg2−/− mice (Fig. 2C). Markers for triglyceride export; microsomal triglyceride transfer protein (MTTP), an enzyme that facilitates VLDL production and its transport out of the liver, as well as phosphatidylethanolamine N-methyltransferase (PEMT), were significantly increased in livers of Arg2−/− mice when compared to WT mice (Fig. 2D). As the most prominent changes were observed in the DNL pathway we assessed protein levels of FAS and SCD-1 in liver lysates. In line with the aforementioned results we found a significant increase of both proteins in livers of Arg2−/− mice when compared to wild type mice (Fig. 2E). Taken together our findings suggest that arginase 2 deficiency is associated with the development of hepatic lipid accumulation and involves up-regulated DNL within the liver.
Figure 2. Hepatic lipid metabolism is altered in Arg2−/−.
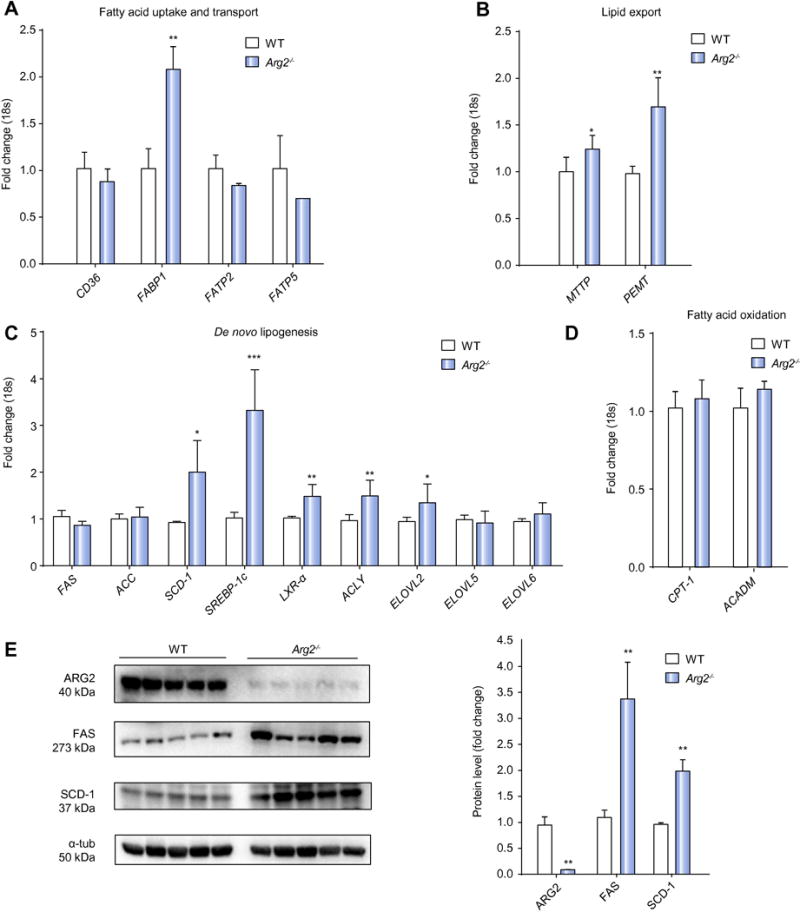
Among markers for fatty acid uptake and transport within the liver (CD36, fatty acid transport proteins (FATP) fatty acid binding proteins (FABPs) we found a significant increase in levels of FABP1 (A). Fatty acid synthesis and de novo lipogenesis (DNL) assessed by quantifying mRNA levels of fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), stearoyl-CoA desturase 1 (SCD-1), the transcription factor sterol regulatory element binding protein 1c (SREBP-1c), liver X receptor alpha (LXR-α), ATP citrate lyase (ACYL), and elongation of long chain fatty acids 2, 5 and 6 (Elovl2, 5, 6). Significant increases were detected for mRNA levels of SREBP1c, SCD-1, LXR-α, ACLY, and Elovl2 in Arg2−/− mice when compared to WT mice (B). Fatty acid oxidation, assessed by measuring mRNA levels of carnitine palmitoyl-transferase 1 (CPT-1), and acyl-coenzyme A dehydrogenase, C-4 to C-12 straight chain (ACADM) was not found to be different in Arg2−/− and WT mice (C). Markers for triglyceride export - microsomal triglyceride transfer protein (MTTP) and phosphatidylethanolamine N-methyltransferase (PEMT) – were significantly increased in livers of Arg2−/− when compared to WT mice (D). In assessing ARG2 levels in liver lysates we were able to document an almost complete absence of the protein in ARG2 knockout mice (E). Protein quantifications of FAS (273kDa) and SCD-1 (37kDa) revealed significantly increased levels in Arg2−/− mice when compared to wild type mice (E). Mean ± S.E.M. are shown. *p<0.05. **p<0.01. ***p<0.001
Arg2−/− mice show evidence of spontaneous inflammation with M1 polarized macrophages
Due to the potential role of arginase 2 deficiency in immune responses, we next focused on inflammatory signaling and found a marked infiltration of F4/80 positive cells in the livers of Arg2−/− when compared to WT mice (supplemental Fig. 1A). Analysis of mRNA levels revealed an increased expression predominantly in F4/80 (six-fold), in CD11c (three-fold), in IL1-β (six-fold), and in iNOS (four-fold) in Arg2−/− mice when compared to WT mice, indicating that predominantly M1 polarized proinflammatory macrophages are present in livers of Arg2−/− mice (supplemental Fig. 1B).
Liver-specific macrophage depletion results in reversal of hepatic steatosis and fibrosis in Arg2−/− mice
The findings of spontaneous hepatic steatosis and hepatic macrophage activation in the livers of Arg2−/− mice in conjunction with the fact that arginase 2 is normally poorly expressed in hepatocytes but present in monocyte/macrophages, lead us to explore the hypothesis that selective suppression of macrophages in the liver of Arg2−/− would result in the correction of the abnormal hepatic lipid accumulation found in these animals. To test this hypothesis we selectively depleted macrophages in the liver by intravenous injection of liposome-encapsulated clodronate (CLOD). During this process we did not observe differences in total body and liver weight between the CLOD-treated and PBS control treated Arg2−/− mice (Fig. 3A, B). Depletion of liver macrophages after four injections of liposome-encapsulated CLOD was confirmed by F4/80 immunohistochemistry (Fig. 3A). Oil-Red O staining of frozen liver sections revealed a notable attenuation of lipid deposition in the CLOD-treated mice compared to their PBS-treated counterparts (Fig. 3A). This corresponded to a decrease in liver triglyceride levels in the CLOD group compared to the PBS group (Fig. 3C). Furthermore, when expression levels of genes involved in lipid metabolism were examined by qRT-PCR, key enzymatic mediators of DNL and lipid export from the liver were found to be down-regulated in CLOD-treated mice when compared to PBS controls (Fig. 3D). Analysis of epididymal adipose tissue did not reveal significant differences in morphology and only showed a slight and non-significant reduction in macrophage infiltration (Fig. 3E, F). As expected, we found a depletion of macrophages in livers of WT mice injected with CLOD when compared to WT mice injected with PBS. While body weight, liver weight, epidermal adipose tissue weight, levels of liver triglycerides, and serum triglycerides did not differ in WT mice injected with CLOD when compared to mice injected with PBS, mRNA levels of F4/80 and arginase 2 were significantly decreased in WT mice injected with CLOD (supplemental Fig. 2). Taken together, these findings strongly suggest that arginase 2-deficient macrophages play an important role in the accumulation of triglycerides in hepatocytes of Arg2−/− mice.
Figure 3. Reversal of hepatic steatosis and inflammation in Arg2−/− mice after liver-specific macrophage depletion.
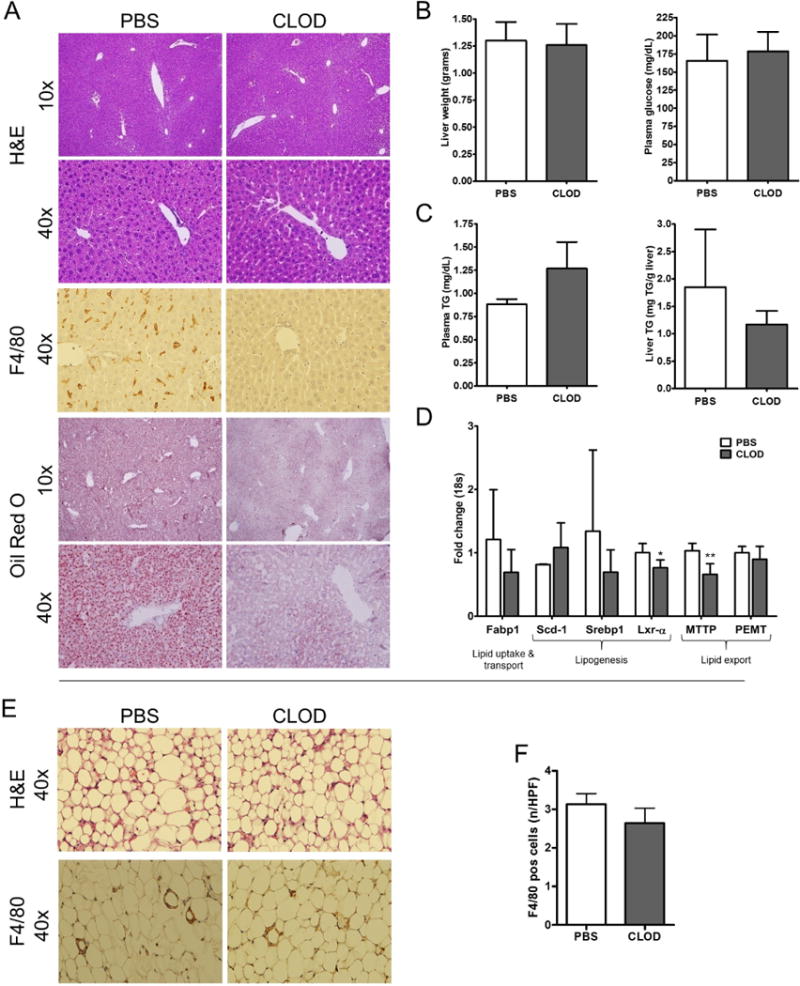
Selectively depleted macrophages in the liver by intravenous injection of liposome-encapsulated clodronate (CLOD) did not alter liver weight and metabolic status – assessed via plasma glucose levels - between the CLOD-treated and PBS control treated Arg2−/− mice (A). Depletion of liver macrophages after four injections of liposome-encapsulated CLOD was confirmed by F4/80 immunohistochemistry (B). Oil-Red O staining revealed a notable attenuation of lipid deposition in the CLOD-treated mice compared to their PBS-treated counterparts (C). This corresponded to a decrease in liver triglyceride levels in the CLOD group compared to the PBS group (C). Expression levels of genes involved in DNL and lipid export from the liver were found to be reduced in CLOD-treated mouse livers compared to PBS controls (D). No differences were observed in gross morphology of adipose tissue samples of mice treated with CLOD (E). Macrophage infiltration assessed via F4/80 immunohistochemistry only showed a slight reduction in mice treated with CLOD when compared to mice injected with PBS (E, F). Microphotographs are displayed in 10× and 40× magnifications. Mean ± S.E.M. are shown. *p<0.05, **p<0.01. ***p<0.001.
Arginase 2 deficiency results in increase susceptibility to high fat diet induced liver injury
Our data revealed that arginase 2 deficiency results in spontaneous development of steatosis and inflammation. Next, to test our initial hypothesis that arginase 2 deficiency in mice would result in progression from isolated hepatic steatosis induced by high fat (HFAT) feeding to steatohepatitis, we examined the effects of short-term HFAT diet feeding on Arg2−/− and WT animals (Fig. 4A). Gross examination of the livers from Arg2−/− animals showed hepatomegaly that translated to an increased liver/body weight ratio compared to WT mice on the HFAT diet (Fig. 4B). Arg2−/− mice on HFAT diet showed significant elevation of serum ALT levels that were 4–5-fold higher than the normal levels and significantly higher than those observed in the WT animals on the HFAT diet (Fig. 4B). Histological evaluation of liver sections revealed that, while WT mice developed as expected hepatocellular steatosis to a mild degree (in average 30%, grade 1), upon short-term HFAT feeding, hepatocytes of Arg2−/− mice exhibited steatosis to a severe degree (average 85%, grade 3). Moreover, arginase 2 deficiency resulted in marked exacerbation of lobular inflammation with up to four spots of inflammatory foci per 20× magnification while livers of WT mice only exhibited one or two inflammatory foci per field of view (Fig. 4C, D). Hepatocellular ballooning was neither diagnosed in livers of Arg2−/− mice nor WT mice. However, livers of Arg2−/− showed numerous hepatocytes with multiple small droplet steatosis which made the distinction of balloon hepatocytes a significant challenge (Fig. 4C, D). This histological finding translated into increased mRNA levels of inflammatory marker (F4/80, TNF-α, and iNOS) in Arg2−/− mice fed with high fat diet when compared to WT mice (Fig. 4E). Taken together our data identifies arginase 2 as a key mediator of increased susceptibility to high fat diet induced hepatic steatosis and liver damage.
Figure 4. HFAT feeding exacerbates the high fat diet induced liver injury in Arg2−/− mice.
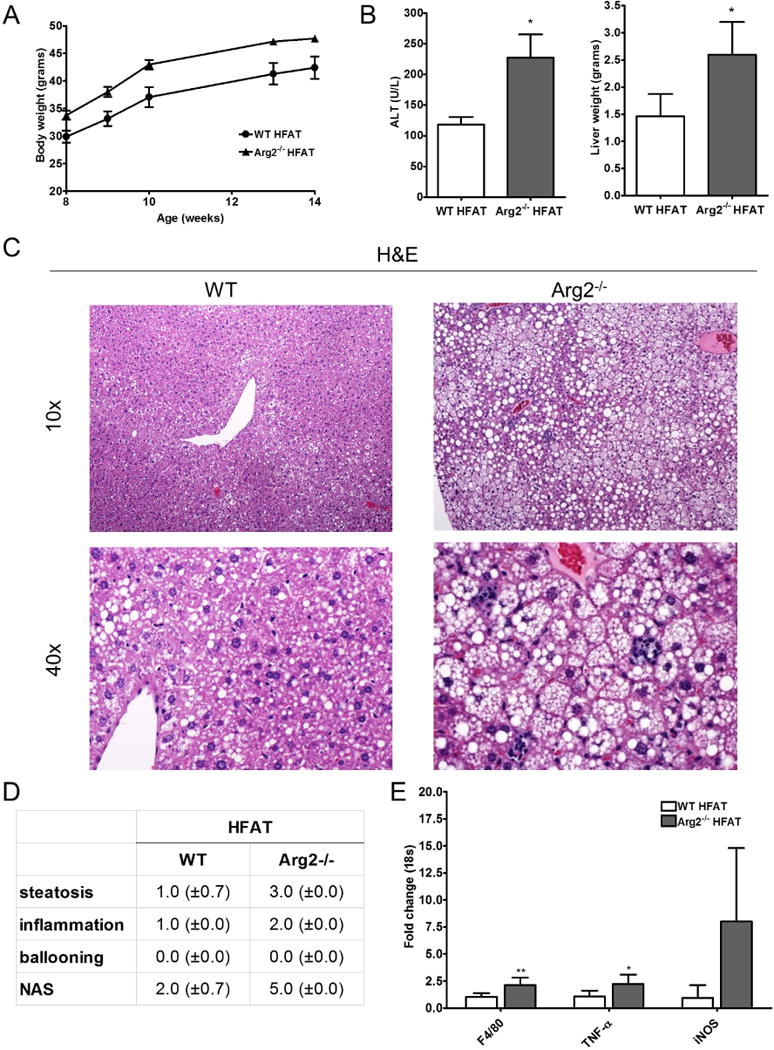
Within a short term feeding period with high fat (HFAT) diet for 6 weeks, Arg2−/− and WT animals gained significant weight (A). Livers of Arg2−/− mice weighed significantly more than those of WT mice after 6 weeks of high fat diet and ALT serum levels were significantly elevated in Arg2−/− mice on HFAT diet (B). Histological analysis of liver samples of revealed that Arg2−/− mice develop more steatosis than WT mice (C). More importantly, Arg2−/− mice showed increased lobular increased lobular inflammation which translated into an increase in the NAFLD-activity score (NAS). In line with this, global marker for macrophages (F4/80), as well as, markers for inflammatory macrophage polarization (TNF- α and iNOS) were increased in Arg2−/− mice when compared to WT mice on HFAT diet (E). Microphotographs are displayed in 10× and 40× magnifications. Mean ± S.E.M. are shown. *p<0.05, **p<0.01.
DISCUSSION
The principal findings of this study relate to the role of arginase 2 in the development of hepatic steatosis and the progression to steatohepatitis. The results demonstrate that arginase 2 deficiency leads to spontaneous development of hepatic steatosis that is independent of weight gain and systemic metabolic changes and associated with increased hepatic de novo lipogenesis and liver Kupffer cell/macrophage activation towards a pro-inflammatory M1 phenotype. Selective depletion of Kupffer cells in Arg2−/− mice results in protection from hepatic steatosis and liver inflammation in these animals, while arginase 2 deficiency favors progression from isolated hepatic steatosis induced by high fat feeding to steatohepatitis. This study identifies arginase 2 as a novel link between innate immune responses, hepatic lipid deposition, and liver injury.
Within the multi-hit model of NAFLD pathogenesis, growing evidence supports the concept that innate immune activation in the liver is a key “second hit” involved in disease progression to NASH. Activation of liver resident macrophages or Kupffer cells resulting in an increased production of pro-inflammatory cytokines as well as inducible nitric oxide synthase (iNOS) has been proposed to play a pivotal role in this process [13]. Indeed, the liver microenvironment harbors numerous potential stimuli for macrophage activation, including both pathogen-associated and damage-associated molecular patterns (PAMPs and DAMPS, respectively) [11]. Sources of PAMPs are structural motifs of proteins, lipids, and nucleic acids originating from invading microorganisms, while DAMPS are derived from injured, dying or malignantly transformed host cells [30]. Among the many exogenous danger molecules that are likely to play a role in liver disease, LPS has been extensively implicated in the pathogenesis of NAFLD. Evidence for the pivotal role of LPS in NAFLD development and progression includes increased circulating LPS levels in NAFLD patients, and increased sensitivity to LPS in experimental NAFLD models [31, 32]. In alcoholic liver disease, direct alcohol toxicity disrupts the intestinal epithelium impairing its barrier function, which leads to leaking of LPS from the gut lumen to the portal circulation [33]. In NAFLD, increased levels of and sensitivity to LPS have been proposed to result from impaired clearance by fatty hepatocytes and altered toll-like receptor 4 (TLR4) signaling, leading to insufficient control of danger recognition [30]. A recent report showed that patients with NAFLD do in fact, have both small intestinal bacterial overgrowth and increased intestinal permeability [34]. This increase in intestinal permeability may be a result of quantitative alterations in gut microbiota, hyperinsulinemia and increased circulating levels of inflammatory cytokines, or more likely a combination of all three [35]. Gut microbiota has also been shown to affect fat storage and energy metabolism via increased intestinal permeability leading to endotoxemia with subsequent triggering of inflammation and metabolic derangements [35]. For instance, microbiota has been shown to promote an increase in hepatic triglyceride content via upregulation of de novo fatty acid synthesis [36].
Mammals express two isoforms of arginase, an enzyme than competes with iNOS for its substrate, arginine, designated as types 1 and 2. Arginase 1 is mainly express in hepatocytes and it is the isoform that participates in the urea cycle [17]. Arginase 2 is poorly expressed in hepatocytes, and most highly expressed in kidney, prostate, and immune cells such as monocyte/ macrophages [17]. An imbalance in iNOS / arginase ratio in favor of the former have been postulated to play a crucial role in regulating immune responses and macrophage activation. Our data supports this concept by demonstrating that arginase 2 deficiency results in activation of Kupffer cells towards a classically “M1” activated, pro-inflammatory state. Unexpectedly, these changes in the livers of Arg2−/− were associated with the spontaneous development of hepatic steatosis. The increase deposition of lipids in the liver where independent of changes in body weight and systemic metabolic changes. The interactions between fatty acid metabolism, insulin resistance, dyslipidemia and hepatic triglyceride content that contribute to hepatic steatosis in NAFLD are complex, and involve different organs and cell types [37]. Obese subjects with NAFLD show evidence of increased lipolysis in adipose tissue and subsequent increase in rate of free fatty acid (FFA) delivery to and uptake by liver [37].
Given our findings of unaltered circulating FFA and FFA uptake by liver in Arg2−/− mice, impaired lipid metabolism resulting in de novo lipogenensis (DNL) within the liver was suspected as the mechanism leading to intrahepatic triglyceride accumulation in Arg2−/− mice. Indeed, increase in DNL is found in different experimental models of NAFLD as well as in patients with NAFLD [38]. The mechanisms involved in the activation of DNL in obese animals and humans remain incompletely understood, but increased plasma glucose and insulin levels in the context of a state of systemic insulin resistance is thought to be an important stimuli for DNL via activation of key transcription factors SREBP-1c and ChREBP [39]. In contrast with normal subjects in whom DNL contributes only marginally to total intrahepatic triglyceride production and FAs incorporated into VLDL, in human subjects with NAFLD, DNL accounts for as much as 23% of FAs within intrahepatic triglycerides and those secreted in VLDL [37]. The observed increase in DNL in Arg2−/− mouse livers was not associated with evidence of insulin resistance which lead us to further explore the potential mechanism triggering de novo lipid synthesis in the livers of these mice.
Our data identify a novel link between Kupffer cell activation and hepatic steatosis by demonstrating that selective depletion of Kupffer cells in these mice resulted in a reversal of hepatic steatosis and normalization of the expression of genes involved in DNL. These results suggest that in the presence of arginase 2 deficiency, altered immune responses precede, and is responsible of lipid deposition in hepatocytes mainly by activation of de novo lipogenesis pathways in the liver.
The idea that steatosis is the necessary initial step preceding all other features of NAFLD, such as inflammation, has been recently challenged by studies demonstrating that inflammation can actually precede steatosis and even contribute to its development in both mouse models and in vitro studies. For instance, features of steatohepatitis examined after short-term high fat feeding (2, 4, 7 days) in APOE2 knock-in mice revealed prominent macrophage infiltration after just two days of HFD feeding, whereas lipid droplets were not visible until 4–7 days post-HFD feeding [40]. Furthermore, up-regulation of inflammatory genes preceded that of lipid metabolism genes, and expression of inflammatory genes was immediately reduced upon fenofibrate treatment, before that of lipid metabolism genes [40]. Our data supports this concept by identifying an experimental model in which altered immune activation appears as the initial mechanism leading to hepatic steatosis. These concepts have important implications for the understanding of NAFLD pathogenesis and potential development of novel treatment strategies for patients with this condition.
Finally our data demonstrates that arginase 2 deficiency increases the susceptibility of mice to fatty liver disease associated with DIO. Indeed, Arg2−/− mice on a high fat diet showed marked elevation of serum liver enzymes and more severe lipid deposition and features of liver damage. A central question in human NAFLD is why patients with similar degrees of obesity, insulin resistance and other risk factors for lipid overloading of the liver develop different degrees of liver involvement. Our results identify arginase 2 deficiency as a novel deleterious trait contributing to disease progression and liver injury and warrants future investigations of arginase 2 status in human NAFLD.
Supplementary Material
Supplemental Figure 1. Evidence of spontaneous inflammation in livers of Arg2−/− mice with M1 polarized macrophages. Immunofluorescence analysis for hepatic macrophages (F4/80) revealed markedly increased cells in liver section of Arg2−/− mice (A) (40×). Analysis of mRNA level showed a sixfold increase in F4/80 along with significant increases for CD11c, IL1-beta and iNOS indicating that infiltrating macrophages are predominantly M1 polarized. Mean ± S.E.M. are shown. *p<0.05.
Supplemental Figure 2. Clodronate injection in WT mice depletes hepatic macrophages but does not alter triglycerides or ARG2. Macrophage depletion studies were done in WT mice using liposome-encapsulated clodronate injection. Mice received four tail vein injections of 100 uL/10 g body weight of 1 mg/ml clodronate (CLOD) or PBS-containing liposomes over a three-week period, 24 hours after the fourth injection, mice were sacrificed. No gross morphologic changes were detected in liver and adipose tissue (A). No significant differences were observed in total body weight, liver weight, epidermal adipose tissue weight, levels of liver triglycerides and plasma triglycerides (B, C). An almost complete ablation of F4/80 positive macrophages was evident in liver samples of WT mice injected with clodronate when compared to those injected with PBS (D). In line with this, analysis of hepatic mRNA expression revealed significantly reduced levels of F4/80 and Arg2 (E).
Acknowledgments
This work was supported by NIH grants to AEF (DK076852, DK082451), SG, and SCE (HL103453, HL109250, HL60917).
ABBREVIATIONS
- ACC
acetyl-CoA carboxylase
- ACADM
acyl-coenzyme A dehydrogenase
- ALT
alanine aminotransferase
- Arg1
arginase 1
- Arg2
arginase 2
- CD11c
cluster of differentiation 11c
- CD36
cluster of differentiation 36 or fatty acid translocase
- CD68
cluster of differentiation 68
- CLOD
clodronate
- CPT1
carnitine palmitoyl-transferase 1
- DIO
diet induced obesity
- DAMP
damage-associated molecular pattern
- DNL
de novo lipogenesis
- F4/80
mouse macrophage marker
- FABP4
fatty acid binding protein 4
- FAS
fatty acid synthase
- FATP2/FATP5
fatty acid transport proteins 2/5
- HFAT
high fat
- IL1-beta
interleukin 1 beta
- IL-6
interleukin 6
- iNOS
inducible nitric oxide synthase
- LXR-a
liver X receptor alpha
- MTTP
microsomal triglyceride transfer protein
- NAFLD
Nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- ORO
Oil Red O
- PAMP
pathogen-associated molecular pattern
- PEMT
phosphatidylethanolamine N-methyltransferase
- SCD-1
stearoyl-CoA desturase 1
- SREBP-1
sterol regulatory element binding protein 1c
- TNF-alpha
tumor necrosis factor alpha
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures / Conflict of interest: None
Author Contributions:
study concept and design: LN, AW, SE, AF;
acquisition of data: LN, AW, DP, MB, AE, BP, SG;
analysis and interpretation of data: LN, AW, DP, SE, AF;
drafting of the manuscript: LN, AW, DP, SE, AF;
critical revision of the manuscript for important intellectual content: all authors;
statistical analysis: LN, AW, AF;
obtained funding: SG, SE, AF;
administrative, technical, or material support: MB, AE, BP;
study supervision: SE, AF.
References
- 1.Wieckowska A, Feldstein AE. Nonalcoholic fatty liver disease in the pediatric population: a review. Current opinion in pediatrics. 2005;17:636–641. doi: 10.1097/01.mop.0000172816.79637.c5. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P. Nonalcoholic fatty liver disease. The New England journal of medicine. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 3.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 4.Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–1393. doi: 10.1542/peds.2006-1212. [DOI] [PubMed] [Google Scholar]
- 5.Wieckowska A, Feldstein AE. Diagnosis of nonalcoholic fatty liver disease: invasive versus noninvasive. Seminars in liver disease. 2008;28:386–395. doi: 10.1055/s-0028-1091983. [DOI] [PubMed] [Google Scholar]
- 6.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 8.Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 9.Pascale A, Pais R, Ratziu V. An overview of nonalcoholic steatohepatitis: past, present and future directions. Journal of gastrointestinal and liver diseases: JGLD. 2010;19:415–423. [PubMed] [Google Scholar]
- 10.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 11.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nature reviews Gastroenterology & hepatology. 2013;10:627–636. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 12.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Digestive diseases and sciences. 2010;55:560–578. doi: 10.1007/s10620-009-1081-0. [DOI] [PubMed] [Google Scholar]
- 13.Taylor BS, Alarcon LH, Billiar TR. Inducible nitric oxide synthase in the liver: regulation and function. Biochemistry Biokhimiia. 1998;63:766–781. [PubMed] [Google Scholar]
- 14.Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clinica chimica acta; international journal of clinical chemistry. 2011;412:1297–1305. doi: 10.1016/j.cca.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Fujita K, Nozaki Y, Yoneda M, Wada K, Takahashi H, Kirikoshi H, et al. Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the choline-deficient, l-amino acid-defined diet-fed rat model. Alcoholism, clinical and experimental research. 2010;34(Suppl 1):S18–24. doi: 10.1111/j.1530-0277.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- 16.Aram G, Potter JJ, Liu X, Torbenson MS, Mezey E. Lack of inducible nitric oxide synthase leads to increased hepatic apoptosis and decreased fibrosis in mice after chronic carbon tetrachloride administration. Hepatology. 2008;47:2051–2058. doi: 10.1002/hep.22278. [DOI] [PubMed] [Google Scholar]
- 17.Munder M. Arginase: an emerging key player in the mammalian immune system. British journal of pharmacology. 2009;158:638–651. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotoh T, Sonoki T, Nagasaki A, Terada K, Takiguchi M, Mori M. Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS letters. 1996;395:119–122. doi: 10.1016/0014-5793(96)01015-0. [DOI] [PubMed] [Google Scholar]
- 19.Jenkinson CP, Grody WW, Cederbaum SD. Comparative properties of arginases. Comparative biochemistry and physiology Part B, Biochemistry & molecular biology. 1996;114:107–132. doi: 10.1016/0305-0491(95)02138-8. [DOI] [PubMed] [Google Scholar]
- 20.Mori M. Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. The Journal of nutrition. 2007;137:1616S–1620S. doi: 10.1093/jn/137.6.1616S. [DOI] [PubMed] [Google Scholar]
- 21.Gallardo-Soler A, Gomez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A, et al. Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-gamma/delta-mediated effect that links lipid metabolism and immunity. Molecular endocrinology. 2008;22:1394–1402. doi: 10.1210/me.2007-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Louis CA, Reichner JS, Henry WL, Jr, Mastrofrancesco B, Gotoh T, Mori M, et al. Distinct arginase isoforms expressed in primary and transformed macrophages: regulation by oxygen tension. The American journal of physiology. 1998;274:R775–782. doi: 10.1152/ajpregu.1998.274.3.R775. [DOI] [PubMed] [Google Scholar]
- 23.Salimuddin, Nagasaki A, Gotoh T, Isobe H, Mori M. Regulation of the genes for arginase isoforms and related enzymes in mouse macrophages by lipopolysaccharide. The American journal of physiology. 1999;277:E110–117. doi: 10.1152/ajpendo.1999.277.1.E110. [DOI] [PubMed] [Google Scholar]
- 24.Shi O, Morris SM, Jr, Zoghbi H, Porter CW, O’Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Molecular and cellular biology. 2001;21:811–813. doi: 10.1128/MCB.21.3.811-813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, et al. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. The Journal of biological chemistry. 2010;285:3428–3438. doi: 10.1074/jbc.M109.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 27.Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Rooijen N, Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods in molecular biology. 2010;605:189–203. doi: 10.1007/978-1-60327-360-2_13. [DOI] [PubMed] [Google Scholar]
- 29.van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. Journal of immunological methods. 1996;193:93–99. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 30.Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. Journal of hepatology. 2009;51:212–223. doi: 10.1016/j.jhep.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Konigsrainer A, et al. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. The Journal of nutrition. 2008;138:1452–1455. doi: 10.1093/jn/138.8.1452. [DOI] [PubMed] [Google Scholar]
- 32.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 34.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 35.Vanni E, Bugianesi E. The gut-liver axis in nonalcoholic fatty liver disease: Another pathway to insulin resistance? Hepatology. 2009;49:1790–1792. doi: 10.1002/hep.23036. [DOI] [PubMed] [Google Scholar]
- 36.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic Fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes, obesity & metabolism. 2010;12(Suppl 2):83–92. doi: 10.1111/j.1463-1326.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 40.Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, et al. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. Journal of hepatology. 2006;44:732–741. doi: 10.1016/j.jhep.2005.10.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Evidence of spontaneous inflammation in livers of Arg2−/− mice with M1 polarized macrophages. Immunofluorescence analysis for hepatic macrophages (F4/80) revealed markedly increased cells in liver section of Arg2−/− mice (A) (40×). Analysis of mRNA level showed a sixfold increase in F4/80 along with significant increases for CD11c, IL1-beta and iNOS indicating that infiltrating macrophages are predominantly M1 polarized. Mean ± S.E.M. are shown. *p<0.05.
Supplemental Figure 2. Clodronate injection in WT mice depletes hepatic macrophages but does not alter triglycerides or ARG2. Macrophage depletion studies were done in WT mice using liposome-encapsulated clodronate injection. Mice received four tail vein injections of 100 uL/10 g body weight of 1 mg/ml clodronate (CLOD) or PBS-containing liposomes over a three-week period, 24 hours after the fourth injection, mice were sacrificed. No gross morphologic changes were detected in liver and adipose tissue (A). No significant differences were observed in total body weight, liver weight, epidermal adipose tissue weight, levels of liver triglycerides and plasma triglycerides (B, C). An almost complete ablation of F4/80 positive macrophages was evident in liver samples of WT mice injected with clodronate when compared to those injected with PBS (D). In line with this, analysis of hepatic mRNA expression revealed significantly reduced levels of F4/80 and Arg2 (E).