

Abstract
Heme-HNO species are crucial intermediates in several biological processes. To date, no well-defined Fe heme-HNO model compounds have been reported. Hydride attack on the cationic ferric [(OEP)Fe(NO)(5-MeIm)]OTf (OEP = octaethylporphy-rinato dianion) generates the Fe-HNO product that has been characterized by IR and 1H NMR spectroscopy. Results of DFT calculations reveal a direct attack of the hydride on the N atom of the coordinated ferric nitrosyl.
HNO is the conjugate acid of the product of one-electron reduction of NO. HNO elicits biological responses such as vasodilation and cardioprotection, but unlike NO, readily dimerizes generating N2O and water. 1 It is also present as a heme ligand in heme-HNO intermediates in important biological processes such as NO detoxification by fungal cytochrome P450 nitric oxide reductase (P450nor),2,3 and in the reaction cycles of cyt c nitrite reductase (ccNiR)4,5 and hydroxylamine oxidoreductase.6 Outstanding work by Farmer has resulted in the spectroscopic characterization of several heme protein-HNO adducts.7 Coordination compounds with HNO ligands have been reviewed.1,8,9
Heme-HNO intermediates in biology may be generated either from proton attack at reduced nitrosyl moieties such as proposed in ccNiR,4,5 or from hydride attack at the ferric nitrosyl center in P450nor.2 Elegant work by Ryan,10 Meyer,11 and others have shown that (por)Fe-HNO species are likely intermediates during the electrochemical reductions of ferrous-NO porphyrins in the presence of protons to yield Fe-NH2OH derivatives and NH3. A similar inference of an Fe-HNO species was based on UV-vis spectroscopy,12 but no spectral signals to verify the presence of bound HNO were obtained. The related anionic five-coordinate [(TFPPBr8)Fe(NO)]− (TFPPBr8 = octabromo[tetrakis(pentafluorophenyl)] porphyrinato dianion) complex has been isolated,13 and its crystal structure determined.14
Although nucleophilic attack by hydride (H−) at a ferric-NO moiety is a key step in NO detoxification by fungal P450nor en route to hyponitrite and N2O formation, there are no well-defined examples of this nucleophilic reaction type in Fe heme models. In fact, there are only two known examples of hydride attack at metal-coordinated nitrosyls. In 2001, Sellman reported hydride attack on [(py(by)S4)RuNO]+ to give the Ru-HNO product,15 and in 2004 we reported hydride attack on [(TTP)Ru(NO)(1-MeIm)]+ to give the Ru-HNO derivative.16 The latter was, prior to our current work, the only heme model-HNO compound reported for any metal. Indeed, despite the importance of heme-HNO intermediates in biology, it is surprising that there are no reports of well-characterized Fe heme model-HNO compounds. Some DFT calculations on (porphine)Fe-HNO species have have been reported.17,18 In this paper, we report the first experimental generation of a heme model-HNO complex from hydride attack at a ferric nitrosyl moiety, and utilize DFT calculations to probe the reaction pathway to form the heme-model-HNO adduct.
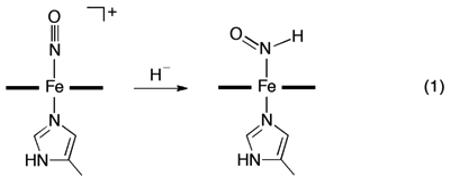
Our selection of the ferric nitrosyl reagent [(OEP)Fe(NO)(5-MeIm)]OTf (υNO 1895 cm−1; KBr pellet) was based on our observation that, unlike most ferric nitrosyls, this cationic precursor could be prepared in analytically pure form (Supporting Information). The molecular structure of the cation is shown in Figure 1, and confirms the identity of this formally {FeNO}6 precursor with a near-linear FeNO moiety. Hydride attack on this ferric nitrosyl cation was monitored by both IR and 1H NMR spectroscopy, and both point to the formation of the ferrous (OEP)Fe(HNO)(5-MeIm) product as shown in eq 1. Addition of 1.5 equiv. of the hydride reagent [NBu4]BH4 to a CHCl3 (1.5 mL) solution of [(OEP)Fe(NO)(5-MeIm)]OTf (9.5 mg, 0.011 mmol) at −20 °C resulted in a decrease of the precursor υNO band in the IR spectrum at 1912 cm−1 with the concomitant formation of a medium intensity band at 1383 cm−1 (Figure 2a).
Figure 1.
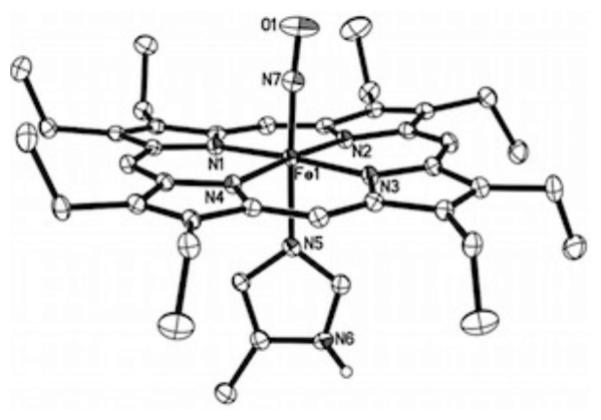
Ordered molecular structure of the cation of [(OEP)Fe(NO)(5-MeIm)]OTf, with thermal ellipsoids shown at 50% probability. The hydrogen atoms (except for the imidazole N6 proton) and the anion have been omitted for clarity. The imidazole N6 proton is hydrogen-bonded to an O atom of the triflate anion, with N6…O(OTf) = 2.880(2) Å (not shown). Selected bond lengths (Å) and angles (deg): Fe1–N7 = 1.6437(16), N7–O1 = 1.152(2), Fe1–N5 = 1.9823(15), Fe1–N(por) = 2.0039(14)– 2.0119(14), ∠Fe1–N7–O1 = 175.38(16).
Figure 2.
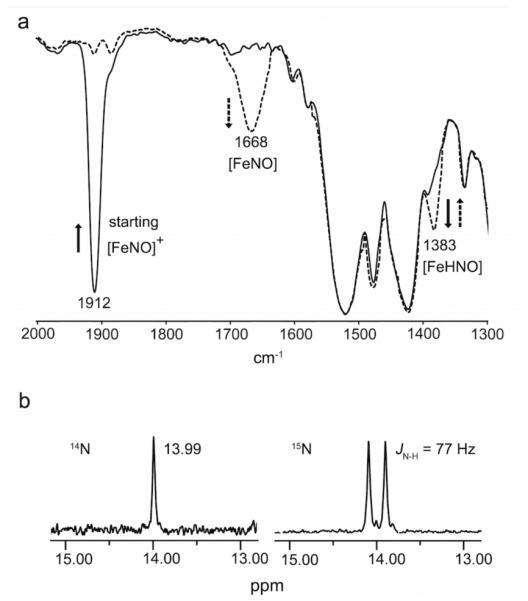
Spectroscopic characterization of the bound HNO ligand in (OEP)Fe(HNO)(5-MeIm). (a) IR spectrum showing formation of the υNO 1383 cm−1 band (dashed line) upon hydride addition to the cationic precursor (υNO 1912 cm−1). The new 1383 cm−1 band slowly converts to the band at 1668 cm−1. (b) 1H NMR spectrum showing formation of the HNO ligand at δ = 13.99 ppm (left) and the JNH coupling for the H15NO derivative (right).
 |
Employing the 15N-labeled precursor [(OEP)Fe(15NO)(5-MeIm)]OTf (υ15NO 1874 cm−1) for the reaction shifted this isotope-sensitive band from 1383 cm−1 in the unlabeled product to 1360 cm−1 (Figure S1; Supporting Information), confirming the assignment of this new band to υNO of the product. Addition of PPh3 as an HNO trap19 to the product mixture at –20 °C resulted in the generation of HN=PPh3 (m/z 278.1101) and O=PPh3 (m/z 279.0937) as determined by ESI mass spectrometry, confirming the presence of HNO in the product of eq. 1. The υNO band at 1383 cm−1 for this ferrous porphyrin derivative is in the range observed for non-porphyrin metal-HNO complexes (1335-1493 cm−1)8 and is similar to the υNO determined for Mb(HNO) (1385 cm−1).20 This new 1383 cm−1 band assigned to (OEP)Fe(HNO)(5-MeIm) slowly converts, even at −20 °C, to the υNO band at 1668 cm−1 assigned to the known five-coordinate (OEP)Fe(NO), with an overall yield of the latter ferrous nitrosyl, based on the precursor ferric nitrosyl cation (υNO 1912 cm−1; Figure 2a), of ~85% as judged by IR spectroscopy.
Monitoring the hydride addition to the ferric nitrosyl cation (eq. 1) in CDCl3 at −20 °C by 1H NMR spectroscopy revealed the appearance of a new peak at 13.99 ppm assigned to the bound HNO of the product of eq. 1 (Figure 2b). When the 15N-labeled nitrosyl cation precursor is used for the reaction, the new peak splits into a doublet with a JNH coupling of 77 Hz. The downfield 1H NMR chemical shift of 13.99 ppm for the bound HNO ligand in (OEP)Fe(HNO)(5-MeIm) is close to those determined for ferrous heme globin-HNO adducts (14.63-15.53 ppm) by Farmer and coworkers.7 The magnitude of the JNH coupling constant (77 Hz) is typical for N-coordinated HNO ligands and consistent with the direct attachment of the proton to the N atom of the HNO moiety.21 Examination of the 1H NMR spectrum (Figure 2b) revealed the presence of a minor (~9% of the HNO signal) species with δ = 13.91 ppm and JNH 74 Hz, which we tentatively assign as a rotational isomer. We note that similar minor signals have been observed in some heme protein-HNO adducts.7 Consistent with the IR spectral results, the (OEP)Fe(HNO)(5-MeIm) compound decomposes slowly even at –20 °C, and we have identified H2 as a byproduct of this decomposition based on its characteristic 1H NMR chemical shift22 at 4.62 ppm in CDCl3. The 1H signal for the bound Fe-HNO persists in the product mixture for at least 2-4 hours at −20 °C, and integrates to ~11% yield at this temperature. The H2 product is also present in the headspace after the reaction as determined by 1H NMR spectroscopy and gas chromatography (Supporting Information, Figure S3), consistent with a previous report by Ryan.10
Importantly, our results represent the first experimental demonstration of hydride attack on a coordinated Fe heme nitrosyl to give a heme model-HNO compound, and the first direct spectroscopic characterization of a bound HNO ligand in an Fe heme model.
We investigated the mechanism of hydride attack on the ferric nitrosyl cation (eq. 1) using density functional theory (DFT) calculations. Three different DFT methods were used (Supporting Information), and all three methods showed basically the same trends. The mPW1PW91 method was previously found to yield excellent predictions of mechanistic properties,23-25 hence results from this method are shown in Scheme 1 and discussed here.
Scheme 1.

DFT-calculated N-pathway for hydride addition to the [(P)Fe(NO)(5-MeIm)]+ cation.
For the isolated nitrosyl cation [(P)Fe(NO)(5-MeIm)]+ (reactant 1; R1 in Scheme 1; P = unsubstituted porphine macrocycle), the nitrosyl N-atom has a large positive charge of 0.705e, whereas the O-atom has a charge of – 0.040e. This suggests that the N-atom shall be more easily attacked (than the O-atom) by the incoming negatively charged hydride from BH4− (reactant 2; R2) to form the HNO rather than the NOH product. Interestingly, we find that for the first intermediate formed from the intermolecular interaction between R1 and R2, all trials that placed the hydride closer to the nitrosyl N of R1 than the O atom, to form a potential I-1N intermediate, resulted in the same structure as the intermediate I-1O (i.e., I-1N = I-1O; Scheme 1),26 with the distance between the hydride (H) to be transferred and the O-atom (2.738 Å) being shorter than that for the nitrosyl N-atom (3.353 Å). This is probably due to repulsion between the strong negative charge of BH4− and the porphine ring if they are too close. This result is reminiscent of that obtained from quantum chemical investigations of hydride attack on the NO moiety in the heme protein cyt P450nor.3
Since both the N and O atoms of the nitrosyl moiety in the first intermediate I-1N/O may, in principle, accept the transferred hydride from borohydride, we investigated both the N- and O-pathways for hydride addition to the bound nitrosyl of the common intermediate I-1N/O (Supporting Information, Scheme S1). Relative electronic energy (ΔESCF), zero-point energy corrected electronic energy (ΔESCF+ZPE), enthalpy (ΔH), and Gibbs free energy (ΔG) results with respect to reactants for both the N- and O-pathways are shown in the Supporting Information Table S8. All the energy results show the same trend with the formation of the HNO complex (i.e., N-pathway) having a much lower kinetic barrier and more favorable thermodynamic driving force.27 Optimized structures of the lowest energy conformations of key species for the N-pathway are shown in Scheme 1, as are selected bond lengths and angles for the complexes.
Hydride attack along the N-pathway (Scheme 1) proceeds via the transition state complex TS-1N, in which the hydride to be transferred is positioned between the nitrosyl N-atom and boron. The geometric parameters (Table S11; Supporting Information) show that this TS-1N geometry is more similar to the reactants than the products, suggesting an early transition state for a facile reaction as observed experimentally. Charge analysis (Table S12) for TS-1N shows that both the boron atom and the hydride to be transferred possess negative charges, –0.671e and –0.073e respectively, to help repel each other to break the B–H bond in the borohydride. The B– H bond cleavage generates the second intermediate I-2N, followed by the BH3 molecule dissociating completely to form the products P-1N and P2.
In contrast, reaction along the O-pathway (bottom of Scheme S1, Supporting Information) generates the first transition state TS-1O, for which charge analysis shows opposite charges for the boron atom (−0.237e) and the hydride to be transferred (0.243e). This results in an attraction between B and H and hinders the bond-breaking process needed for the hydride addition reaction to proceed via the O-pathway.
Further evidence for the formation of the Fe-HNO rather than the Fe–NOH product comes from calculations of the spectral properties of these adducts (Table 1). The NMR properties were calculated using the B3LYP method with solvent effects (Supporting Information), similar to the approach used previously to study 1H NMR chemical shifts in various organometallic complexes.28 Compared to the experimental values for (OEP)Fe(HNO)(5-MeIm), our results for (P)Fe(HNO)(5-MeIm) (P-1N in Scheme 1) only have errors of 0.08 ppm (1H NMR) and 6 cm−1 (IR). In contrast, the calculations for the various (P)Fe(NOH)(5-MeIm) conformations yield much greater errors of ~2 ppm and/or up to 400 cm−1.
Table 1.
1H NMR chemical shifts (in ppm) and NO vibrational stretching frequencies (cm−1) for the bound HNO/NOH ligands
| system | δ H | υ NO | |
|---|---|---|---|
| (OEP)Fe(HNO)(5-MeIm) | expt | 13.99 | 1383 |
| (P)Fe(HNO)(5-MeIm) P-1N | calc | 13.91 | 1375 |
| (P)Fe(NOHDown)(5-MeIm) P-1O (trans)a | calc | 11.92 | 1005 |
| (P)Fe(NOHDown)(5-MeIm) P-1O (cis)a | calc | 11.36 | 992 |
| (P)Fe(NOHUp)(5-MeIm) | calc | 13.54 | 955 |
NO trans/cis with respect to the Me substituent of 5-MeIm. As this trans/cis effect is relatively small, only the trans isomer for the NOHup conformation was studied here. The Down/Up conformations are for the H pointing to and away from the porphyrin ring, respectively.
In summary, this report represents the first experimental demonstration of hydride attack on a coordinated nitrosyl of a synthetic ferric-NO porphyrin and the first spectral characterization of a bound HNO group in an Fe heme model. DFT calculations are consistent with a direct hydride attack at the nitrosyl N-atom in the precursor ferric nitrosyl porphyrin. Experiments to determine the fundamental chemistry patterns of heme model Fe-HNO moieties are currently underway.
Supplementary Material
ACKNOWLEDGMENT
G.B.R-A thanks the National Science Foundation (CHE-1213674) and Y.Z. thanks the National Institutes of Health (GM085774) for financial support. We thank Dr. Ralph Tanner (OU) for assistance with the GC measurements.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental, crystallographic data, computational details.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Miranda KM. Coord. Chem. Rev. 2005;249:433–455. [Google Scholar]
- 2.Daiber A, Shoun H, Ullrich V. J. Inorg. Biochem. 2005;99:185–193. doi: 10.1016/j.jinorgbio.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Kramos B, Menyhard DK, Olah J. J. Phys. Chem. B. 2012;116:872–885. doi: 10.1021/jp2080918. [DOI] [PubMed] [Google Scholar]
- 4.Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. J. Am. Chem. Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- 5.Bykov D, Neese F. Inorg. Chem. 2015;54:9303–9316. doi: 10.1021/acs.inorgchem.5b01506. [DOI] [PubMed] [Google Scholar]
- 6.Cabail MZ, Kostera J, Pacheco AA. Inorg. Chem. 2005;44:225–231. doi: 10.1021/ic048822a. [DOI] [PubMed] [Google Scholar]
- 7.Kumar MR, Pervitsky D, Chen L, Poulos T, Kundu S, Hargrove MS, Rivera EJ, Diaz A, Colon JL, Farmer PJ. Biochemistry. 2009;48:5018–5025. doi: 10.1021/bi900122r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farmer PJ, Sulc F. J. Inorg. Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Doctorovich F, Bikiel D, Pellegrino J, Suarez SA, Larsen A, Marti MA. Coord. Chem. Rev. 2011;255:2764–2784. [Google Scholar]
- 10.Choi I-K, Liu Y, Feng D, Paeng K-J, Ryan MD. Inorg. Chem. 1991;30:1832–1839. [Google Scholar]
- 11.Barley MH, Takeuchi KJ, Meyer TJ. J. Am. Chem. Soc. 1986;108:5876–5885. doi: 10.1021/ja00279a036. [DOI] [PubMed] [Google Scholar]
- 12.Goodrich LE, Roy S, Alp EE, Zhao JY, Hu MY, Lehnert N. Inorg. Chem. 2013;52:7766–7780. doi: 10.1021/ic400977h. [DOI] [PubMed] [Google Scholar]
- 13.Pellegrino J, Bari SE, Bikiel DE, Doctorovich F. J. Am. Chem. Soc. 2010;132:989–995. doi: 10.1021/ja905062w. [DOI] [PubMed] [Google Scholar]
- 14.Hu B, Li JF. Angew. Chem. Int. Ed. 2015;54:10579–10582. doi: 10.1002/anie.201505166. [DOI] [PubMed] [Google Scholar]
- 15.Sellmann D, Gottschalk-Gaudig T, Haussinger D, Heinemann FW, Hess BA. Chem. Eur. J. 2001;7:2099–2103. doi: 10.1002/1521-3765(20010518)7:10<2099::aid-chem2099>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Richter-Addo GB. J. Inorg. Biochem. 2004;98:1247–1250. doi: 10.1016/j.jinorgbio.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Linder DP, Rodgers KR. Inorg. Chem. 2005;44:8259–8264. doi: 10.1021/ic0504745. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y. J. Inorg. Biochem. 2013;118:191–200. doi: 10.1016/j.jinorgbio.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reisz JA, Zink CN, King SB. J. Am. Chem. Soc. 2011;133:11675–11685. doi: 10.1021/ja203652z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Immoos CE, Sulc F, Farmer PJ, Czarnecki K, Bocian DF, Levina A, Aitken JB, Armstrong RS, Lay PA. J. Am. Chem. Soc. 2005;127:814–815. doi: 10.1021/ja0433727. [DOI] [PubMed] [Google Scholar]
- 21.Southern JS, Green MT, Hillhouse GL, Guzei IA, Rheingold AL. Inorg. Chem. 2001;40:6039–6046. doi: 10.1021/ic010669m. [DOI] [PubMed] [Google Scholar]
- 22.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI. Organometallics. 2010;29:2176–2179. [Google Scholar]
- 23.Michael MA, Pizzella G, Yang L, Shi YL, Evangelou T, Burke DT, Zhang Y. J. Phys. Chem. Lett. 2014;5:1022–1026. doi: 10.1021/jz5002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Guo ZJ, You XZ. J. Am Chem. Soc. 2001;123:9378–9387. doi: 10.1021/ja0023938. [DOI] [PubMed] [Google Scholar]
- 25. The energy difference between perpendicular and parallel axial planes in computed (P)Fe(N(H)O)(ligand) is negligible,17 and only the parallel orientations are shown here.
- 26. The computed Fe-N and FeNO geometries of I-1N/O, namely [(P)Fe(NO)(5-MeIm)][BH4], more closely match the experimental [(OEP)Fe(NO)(5-MeIm)]OTf than the isolated R1.
- 27. Interestingly, these data are also consistent with results from a previous quantum chemical study of hydride attack on the nitrosyl of a ferric heme thiolate as a model for cyt P450nor.
- 28.Zhang Y, Lewis JC, Bergman RG, Ellman JA, Oldfield E. Organometallics. 2006;25:3515–3519. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.