

Abstract
Fast-scan cyclic voltammetry (FSCV) has attracted attention for studying in vivo neurotransmission due to its subsecond temporal resolution, selectivity, and sensitivity. Traditional FSCV measurements use background subtraction to isolate changes in the local electrochemical environment, providing detailed information on fluctuations in the concentration of electroactive species. This background subtraction removes information about constant or slowly changing concentrations. However, determination of background concentrations is still important for understanding functioning brain tissue. For example, neural activity is known to consume oxygen and produce carbon dioxide which affects local levels of oxygen and pH. Here, we present a microfabricated microelectrode array which uses FSCV to detect the absolute levels of oxygen and pH in vitro. The sensor is a collector-generator electrode array with carbon microelectrodes spaced 5 μm apart. In this work, a periodic potential step is applied at the generator producing transient local changes in the electrochemical environment. The collector electrode continuously performs FSCV enabling these induced changes in concentration to be recorded with the sensitivity and selectivity of FSCV. A negative potential step applied at the generator produces a transient local pH shift at the collector. The generator-induced pH signal is detected using FSCV at the collector and correlated to absolute solution pH by post-calibration of the anodic peak position. In addition, in oxygenated solutions a negative potential step at the generator produces hydrogen peroxide by reducing oxygen. Hydrogen peroxide is detected with FSCV at the collector electrode and the magnitude of the oxidative peak is proportional to absolute oxygen concentrations. Oxygen interference on the pH signal is minimal and can be accounted for with a post-calibration.
Graphical Abstract
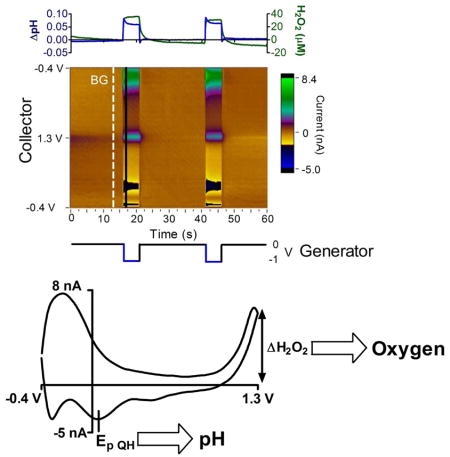
Electrochemical microsensors have seen widespread use for direct in vivo measurements due to their inexpensive cost, ability to monitor continuously, and high spatio-temporal resolution. These advantages are well suited for studying the dynamic chemical environment of the brain. The brain is 2% of body weight but consumes up to 25% of metabolic energy and 20% of cardiovascular output in higher-order mammals to support neural activity.1 Neural activity is fueled by the cellular metabolism of glucose which consumes oxygen and alters pH by producing carbon dioxide. Thus, pH and oxygen levels in the brain are correlated with metabolism and are highly regulated for optimal brain function.1,2,3 For this reason, many pH and oxygen sensors have been developed for use in the brain. Oxygen levels are 17–80 μM4, heterogeneous, and condition-dependent; while pH is typically 7.4 but ranges from 6.8 to 7.8 at extremes.1,5
There are many techniques for direct in vivo pH measurement including fiber-optic chemical sensors (FOCS)6 and potentiometric probes5. The traditional glass pH electrode measures the potential difference between a reference and an internal electrode encased in a pH-sensitive glass bulb. However, pH glass electrodes are too bulky for many in vivo uses5, are prone to instability and drift7, and need to be frequently calibrated. Furthermore, miniaturized pH glass electrodes8 are fragile which is problematic for animal behavioral experiments. Other electrochemical pH sensors are based on pH-sensitive materials such as metal oxides9, polymers5, and carbon10–14. Carbon-based pH sensors have the advantages of a large potential window, versatile chemistry, wide commercial availability, and low cost.15 Unmodified carbon surfaces have been used as pH sensors10,11,16,17 which utilize the presence of oxygen-containing surface groups for their sensitivity.
Techniques to measure in vivo oxygen include blood oxygen-level dependent functional magnetic resonance imaging (BOLD fMRI)18, electron paramagnetic resonance (EPR), fiber optics chemical sensors (FOCS)6, and polarographic microelectrodes.3 Polarographic electrodes measure the reduction of oxygen on noble metals (Pt or Au) or carbon. Metal electrodes, such as the Pt Clark electrode, require a protective, gas-selective membrane for oxygen sensing. In contrast, carbon electrodes are less prone to surface fouling and can function without a protective membrane.19
Fast-scan cyclic voltammetry (FSCV) with carbon fiber microelectrodes (CFMs) has been used to measure fluctuations in pH16,20–22 and oxygen23–26 in vivo. CFMs are small enough (5–10 μm diameter) to cause minimal tissue damage, provide high spatial resolution, and have been used in freely-moving experiments.27,28 FSCV at CFMs has also been widely used to study neurotransmission29 since it has sub-second time resolution, high selectivity21, and sensitivity.30 Selectivity is achieved by identification of characteristic cyclic voltammograms (CV) specific to the redox of the electroactive analytes. In vivo, the individual components of a chemical mixture can be resolved with statistical methods such as principal component regression analysis (PCR).21 Despite these advantages, FSCV has been hitherto restricted to differential measurements on top of an unknown background, or absolute, concentration. This means that while FSCV at a CFM is very sensitive to fluctuations in analytes, the absolute concentrations in solution remains unknown. This limitation arises because FSCV measurements are taken by applying a triangle wave at a high scan rate (ν > 100 V/s). This high scan rate generates a large background current from charging of the electrical double layer. Additionally, the CV contains Faradaic current from the redox of electroactive species in solution. To isolate changes in the Faradaic current, i.e. to take differential measurements, the background current at a chosen time is digitally subtracted from the current signal at all other times. The subtracted CVs are then stacked sequentially with time to resolve changes in concentration over time. This background subtraction also removes the Faradaic contribution from constant or slowly-changing concentrations and thereby limits FSCV to differential measurements. The only exception is fast-scan controlled adsorption voltammetry, which has been recently introduced, to determine absolute concentrations.31,32
This report describes a microfabricated collector-generator sensor that expands on differential-only FSCV measurements by adding absolute pH and oxygen sensing. The additional sensing capability does not preclude differential FSCV measurements of analytes in parallel, such as catecholamines, oxygen, pH. Absolute pH and oxygen sensing is enabled by a collector-generator design which has many applications in electroanalysis.33 The electrodes are made of pyrolyzed photoresist (PPF), a material that has similar electrochemical properties to glassy carbon15,34,35 and to carbon fiber microelectrodes.36,37 Microfabricated silicon-based sensors can be batch fabricated with high-reproducibility and are mechanically robust for use in freely-moving experiments.38,39 In this work, the sensor is a collector-generator comprised of three band electrodes with 5 μm spacing between each band. A square wave is periodically applied to the generator, the two outer band electrodes which are electrically connected to each other, to electrochemically induce concentration changes in the local environment. The collector, the center band electrode, continually performs FSCV to detect the induced changes with high selectivity and sensitivity. This approach allows FSCV determination of the background or absolute concentrations of electroactive analytes, as demonstrated for dopamine.40 For absolute pH sensing, the potential of the generator is manipulated to change the protonation of generator surface groups and induce a local pH change. The induced pH change diffuses to the nearby collector where it is detected with FSCV as a characteristic pH CV. The anodic peak of the pH CV can then be used to determine absolute solution pH with a postcalibration. Similarly, the potential of the generator can be stepped to a reducing potential to reduce oxygen and generate hydrogen peroxide. The hydrogen peroxide diffuses to the adjacent FSCV collector and is subsequently oxidized.41 The collected hydrogen peroxide concentration can be used to determine absolute oxygen concentration. This work presents an in vitro, proof-of-concept study for simultaneous absolute pH and oxygen measurements with FSCV.
EXPERIMENTAL SECTION
Chemicals
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and used as received unless specified otherwise. Electrode calibrations and experiments were done in PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM NaH2PO4, 2 mM K2H2PO4). Solutions were buffered to the desired pH (6.5 – 8) and measured with an Accumet AB-15 pH meter (Hudson, MA). Oxygen concentrations were prepared by bubbling proportional mixtures of air and nitrogen gas (Airgas Inc, Radnor PA) into solution for a minimum of 15 minutes. Oxygen concentrations were calculated from a Henry’s Law solubility equation that factors in the effect of ion concentrations and temperature.42 Gas lines were throttled with two rota-meters (Dwyer, Michigan City, IN) and then mixed and fed into separate flasks with an 8-way valve perfusion system to bubble solutions in parallel (AutoMate Scientific, Berkeley CA).
Fabrication of Microelectrode Arrays
Microelectrode arrays of PPF were fabricated using a similar process as our previous research40 and are detailed elsewhere.43 Briefly, an insulating layer of LPCVD silicon nitride was deposited on fused silicon wafers followed by lithographic patterning of the electrode trace design with AZ 1518 (AZ Electronic Materials, Branchburg, NJ) photoresist. The photoresist was pyrolyzed to PPF by heating to 1000 °C in a tube furnace under a forming gas atmosphere (95% N2, 5% H2). The PPF electrodes were insulated with a layer of PECVD Silicon Nitride. The second PECVD nitride layer is selectively etched to expose the electroactive area and connection pads. Individual electrodes are released from the wafer with a dicing blade tool. Wire connections are made to the carbon connection pads first with silver conductive epoxy (MG Chemicals, Ontario, B.C.) followed by a 15-minute epoxy (Bob Smith Industries, Atascadero, CA) for mechanical strength. The design of the collector-generator electrode is shown in Figure 1 and is comprised of an inner band electrode, 8 μm × 100 μm, surrounded by two interconnected band electrodes of the same size with 5 μm spacing between the band electrodes. Sensor performance is dependent on its geometry and could be further optimized.
Figure 1.

Optical micrograph and schematic depicting the geometry of the microfabricated microelectrode sensor. (a) Bright field microscopy image of the electroactive part of the sensor, scale bar (white arrow) is 100 μm. Three parallel band electrodes are 100 × 8 × 0.4 μm each, separated by 5 μm. (b) Cross-sectional schematic of the electrode dimensions and the gap between them. Electrode function is labeled underneath.
Instrumentation and Software
During experiments, the collector continually performs FSCV with a triangular potential waveform operated at 400 V/s from −0.4 V to 1.3 V and back to −0.4 V vs. Ag/AgCl that is repeated every 100 ms. The potential is held at −0.4 V between application of the waveforms. The triangle waveform was both generated and recorded with an ADC/DAC card PCI-6363 (National Instruments). At the generator, a square wave potential is applied with 20 seconds at 0 V vs. Ag/AgCl and then a potential step to either 0.8 V or −1 V for 5 seconds. The square wave potential was generated with a function generator and recorded with the PCI-6363 card. The square wave was synchronized to apply the potential step during the FSCV holding time. Both square wave and FSCV output waveforms were filtered through a 2 kHz low pass filter to remove digitization steps.
FSCV data was processed through the High Definition Cyclic Voltammetry (HDCV) analysis program.35 Each file was digitally filtered (fast Fourier transform, time cutoff 1.3 Hz, DP cutoff 3 kHz) before further analysis and plotting in GraphPad Prism (GraphPad Software, San Diego, CA, USA). Data are reported as mean ± standard deviation. Limit of detection was calculated by multiplying the standard deviation of the noise in a blank signal by three and dividing by the slope of the calibration curve. Principal component regression with residual analysis (PCR) is a chemometric method to determine concentrations from multivariate data and was done through HDCV.35,44 PCR requires building a training set for expected analytes over a relevant concentration range. Principal components (PCs) are calculated from the training set and a regression is done to relate concentrations to PC distances. Concentrations in an unknown sample can be predicted by projecting the sample onto the PCs and comparing distances. The quantity Q is the sum of squares of the residual values for each CV sample while Qα is the threshold for significance of information not accounted for by the PCs. Statistical significance was determined from residual analysis at the 95% level.
Electrochemical Experiments
Flow-injection analysis experiments were done with a syringe pump (Harvard Apparatus, Holliston, MA) operating at 1 mL/min using PEEK tubing (Sigma-Aldrich) connected to a pneumatically controlled 6-port injection valve (Rheodyne, Rohnert Park, CA). Boluses were introduced with an injection loop into the electrochemical cell. Potentials are reported versus an Ag/AgCl reference electrode. Before application of FSCV or square waveforms, both electrodes were conditioned with the FSCV waveform (400 V/s triangle scan from −0.4 V to 1.3 V and back to −0.4 V) for 15 minutes at 60 Hz and 15 minutes at 10 Hz. This conditioning step is known to increase electrode sensitivity to catecholamines by introducing oxygen-containing functional groups on the electrode surface.36,45,46
RESULTS & DISCUSSION
Response of Carbon Microelectrodes to pH Changes
In FSCV, the rapid scan rate generates a background current with faradaic and non-faradaic components arising from charging of the electrical double layer. In addition, there is a surface-confined redox couple which is pH sensitive.47 At a pH of 7.4, the position of the pH sensitive oxidation peak, Epa (Figure 2A top), is ~+0.15 V on the forward anodic scan and the position of the reduction peak, Epc, is ~−0.3 V on the reverse cathodic scan (Figure 2A bottom). Precise localization of Epa and Epc on the background is difficult at high scan rates (ν = 400 V/s) because of the large overall current. One way to enhance the features at Epa and Epc is to introduce small changes in pH, which slightly shifts the peaks, and evaluate the difference between the two signals.47 For example, Figure 2B is a characteristic background-subtracted CV for a basic pH change of +0.25 pH units introduced by changing the pH of the bulk solution. Figure 2B results from subtracting the FSCV signal before the change in pH, in this case pH 7.4 (Figure 2A black line), from the signal after a basic shift, in this case to 7.65 pH (Figure 2A dashed line). Note that since the FSCV signal at 7.65 pH (Figure 2A dashed line) is smaller in amplitude relative to the 7.4 pH (Figure 2A black line), the subtracted voltammogram (Figure 2B) from 7.65–7.4 pH has a flipped sign convention about the x-axis. Conversely, Figure 2C is a representative background-subtracted CV for an acidic pH shift of −0.25 pH units, taken by subtracting the FSCV signal at pH 7.4 (Figure 2A black line) from the signal at pH 7.15 (Figure 2A grey line). The peaks labeled Q and QH reflect changes in the surface faradaic processes and so their positions, Ep Q and Ep QH, can be used to approximate Epc and Epa (Figure 2A).
Figure 2.

Characteristic FSCV signals in response to pH changes. (A) CVs of the background currents in various pHs, arrows indicate scan direction. Background peak positions, Epa and Epc, and their response to pH are displayed in the top and bottom inset respectively. Epa and Epc, are approximated by background subtracted peak position, Ep QH and Ep Q, as labeled. (B) A background subtracted CV for a basic change (+0.25 pH units) is generated by subtracting the CV at pH 7.4 (A, black line) from pH 7.15 (A, dashed line). (C) An acidic shift (−0.25 pH units) is found by subtracting pH 7.4 (A, black line) from pH 7.65 (A, grey line). QH, Q peaks, and scan direction are labeled.
The surface groups responsible for the Q and QH peaks are hypothesized to be a hydroquinone moiety on the carbon surface.16,47,48 The Q peak results from the quinone to hydroquinone reduction reaction and the QH peak from the oxidation of hydroquinone to quinone (Equation 1).
| (1) |
In the physiologic pH range (pH 7.2–7.6), the height of the QH peak has a linear relationship to the size of the pH change and is used to quantify in vivo pH fluctuations with an in vitro post-calibration.16,20 However, the height of the QH peak is a differential measurement that cannot be used to determine the absolute pH. Despite this, determination of absolute pH should still be possible by recording Ep QH and Ep Q. As discussed above, Ep QH and Ep Q can be accurately resolved from the large background signal by introducing a small pH change. However, this is not feasible on demand at a single microelectrode in vivo. In this work, a small pH shift is electrochemically produced by modulating the potential of the nearby generator. This local pH shift enables Ep QH and Ep Q to be recorded at the collector.
Operation of the Sensor
The sensor functions as a collector-generator assembly with the inner electrode serving as the collector and the outer electrode as the generator (Figure 1). A square wave potential is applied to the generator to consume or produce electroactive chemical species during the potential step. At the collector, FSCV is utilized to detect changes in the concentration of electroactive species induced by the generator.
Initial experiments were performed in deoxygenated PBS solution with a pH of 7.4 (Figure 3). The set of background-subtracted cyclic voltammograms (CVs) measured at the collector electrode are displayed as a color plot (Figure 3A, 3C). The generator electrode potential is shown below the color plot while PCR analysis of relative pH change at the collector is above (Figure 3A, 3C). Individual background-subtracted CVs at times of interest, highlighted by the black lines on Figure 3A, 3C, are plotted in Figure 3B, 3D. Figure 3A shows that stepping the generator electrode from 0 V to a +0.8 V potential produces electrochemical changes at the collector electrode. The background-subtracted CV has the signature shape of an acidic pH shift, indicating that the electrochemical changes are from a localized pH shift. The characteristic Q and QH peaks are visible and labeled in Figure 3B, 3D. Figure 3E illustrates a possible mechanism for the generated pH signals. In this figure, the triple band electrode is displayed in a half cross section with the respective applied waveforms underneath. The initial potential step at the generator electrode is color-coded with the initial direction of the redox reaction. In Figure 3E, the green positive potential step from 0 V to +0.8 V in corresponds to the green arrow showing the oxidative deprotonation of surface groups on the generator. Deprotonation increases the local H+ concentration. The H+ diffuses to collector spanning the 5 μm gap in approximately 12 ms (Einstein relation49 x2 = 2Dt, where DH+ = 1.0 × 10−9 m2/s)50 and results in a pulsatile acidic shift at the collector. After 5 seconds at +0.8 V, the generator potential is returned to 0 V which re-protonates the surface groups on the generator, consuming local H+ causing a local pH shift in a basic direction before returning to baseline. These potential-dependent pH shifts are quantified using principal component regression (PCR) with a pH training set. The resulting magnitude of observed pH shift is plotted above the color plot (Figure 3A). PCR accuracy can be visualized as a color plot of the residual currents (see Figure 3F). The lack of features suggests that all relevant information contained in the color plot is described by the pH training set. Furthermore, there are no significant current contributions at the 95% significance level (Figure 3F above).
Figure 3.

FSCV signal from the collector electrode during operation of the sensor in deoxygenated pH 7.4 PBS solution. (A) Background-subtracted color plot over two square wave cycles from 0 to +0.8 V vs. Ag/AgCl applied to the generator electrodes (potential is below). Background is the white dashed line, labeled BG. PCR of relative pH vs. time is above. (B) Background-subtracted CV taken from the color plot (A, black line – white dashed line) taken immediately after a generator electrode potential step to +0.8 V. Q and QH peaks are labeled. (C) Same as A, but the generator electrode cycles from 0 to −1 V. (D) Same as B, but taken from color plot C. (E) Schematic of sensor operation. FSCV is continuously applied to the collector electrode to monitor changes in pH. The generator electrode influences local pH when its potential is changed between 0 V and +0.8 V (A) or 0 and −1 V (B). Local pH shifts are generated from surface groups on the generator electrode, labeled Q and QH. The direction of redox is color coded green-blue for oxidation-reduction and is coordinated with the direction of the generator electrode potential step. (F) Residual color plot of (A) with pH used in the PCR training set. Above is a residual Q trace with a dashed line marking Qα of 95% significance.
The amount of H+ generated (Ngen) in Figure 3A can be quantified by integrating the generator current over the positive potential step (supplementary Figure 1A) to get the total charge (Qgen). Faraday’s law (Q = nNF) is then used to calculate moles of H+ generated (Ngen = 2.71 femtomoles), where n = 2 is the number of electrodes in the oxidation of surface groups and F = 96,485 C/mol is Faraday’s constant. The surface coverage of quinone on the generator, Γ = 126 pmol/cm2, can be estimated from Γ= Ngen/A where A = 2.16 × 10−5 cm2 is the area of the generator. This value is similar to edge plane pyrolytic graphite (Γ = 170 pmol/cm2)15 and less than a monolayer coverage of hydroquinone (Γ~500 pmol/cm2).51 The amount of H+ collected (Ncoll) in Figure 3A is quantified by integrating the oxidation peak of the pH shift over the 5 second potential step (Figure 3A green box) to get the total charge (Qcoll). Faraday’s Law is then used to calculate moles of H+ collected (Ncoll = 0.065 femtomoles). The collection efficiency, Ncoll/Ngen, amounts to a 2.39%. This value is lower than typical triple band electrodes52 and could result from (1) loss of Ngen to phosphate buffering and (2) short time spent at an oxidizing potential during FSCV.
Another possible source of H+ from the generator at +0.8V is water oxidation to H+ and oxygen at Eo = 1.01 V vs. Ag/AgCl.53 As a control, the experiment was run with the collector monitoring oxygen changes using FSCV.37 No detectable oxygen production was measured when stepping the generator to +0.8 V (data not shown) so water oxidation is unlikely to be the source of the pH shift. In addition, the pH signal (Figure 3A above) and generator current (supplemental Figure 1A) at +0.8 V rapidly decays back to baseline which suggests the origin of the pH shift is not from continuous redox of solution species.
The mechanism for generating pH shifts was further examined by changing the potential waveform applied to the generator to a square wave with an initial potential of 0 V stepping to −1.0 V (Figure 3C). A negative potential step results in an initial basic pH shift at the collector electrode, verified by the CV (Figure 3D). This basic shift rapidly decays back to baseline which is consistent with protonation of reduced surface groups at the generator electrode (Figure 3E, blue arrow). This rapid decay suggests the basic shift does not originate from solution reactions such as water to H2 and OHEo = −1.05 V vs. Ag/AgCl53 and H+ to H2 Eo = −0.223 V vs. Ag/AgCl.53 The magnitude of the initial pH shift is larger for a negative potential step than for a positive step and so a negative potential step is used for further characterization of the sensing scheme. A possible explanation for the difference in magnitude is that there is a greater surface concentration of quinone-like species in the oxidized form than the reduced form on the generator surface when the potential is held at 0 V. Equilibrium is assumed to occur during the holding period, 20 s, since generator currents stabilize within seconds (supplementary Figure 1).
To verify the selectivity of the device to H+ compared to other cations, the response of four was collected in PBS and in PBS solutions where the concentration of NaCl and KCl were increased by 50%. The increased ion concentration caused no significant effect on Ep QH (one-way between subjects ANOVA, P = 0.965, F(2,9) = 0.035) or Ep Q, (P = 0.694, F(2,9) = 0.6937) as well as the overall CV shape (supplemental Figure 2). In addition, increasing the buffering capacity of the PBS solution decreases the magnitude of the response of the sensor (data not shown). These results further confirm the identity of the signal as changes in H+ concentration and demonstrate that it is unaffected by large changes in ionic concentrations.
pH Determination in Deoxygenated Buffer
The dependence of pH on Ep QH and Ep Q was investigated over a physiologically relevant pH range, pH 6.5–8.0 (Figure 4). The reported CVs were obtained by taking a CV before the potential step and subtracting it from a CV collected after the potential step from 0 V to −1 V, as described for Figure 3D. A black diagonal line highlights the shift in Ep QH with pH (Figure 4A). Ep QH and Ep Q were determined by averaging the peak positions for three consecutive pulses and using four sensors (n = 4) at each pH. These average peak positions are displayed as points in Figure 4B with error bars as standard deviations. The error bars in Figure 4B represent some variability between sensors. This sensor-to-sensor variability indicates that each sensor needs to be calibrated in order to accurately quantify pH. Ep QH exhibits a linear relationship vs. pH (solid black line 98.6 mV/pH, n = 4, r2 = 0.92) while Ep Q has a slight pH dependence that levels out at pH 7.25 and above. The magnitude of the QH peak is also pH dependent (Figure 4A). But because the magnitude of the QH peak is strongly dependent on the solution oxygen concentration (vide infra) and is not linear with pH, the peak position was used to quantify the pH.
Figure 4.

pH dependence of the collected FSCV signal. (A) Background subtracted CVs taken from the collector electrode after a negative potential step in deoxygenated PBS solutions of pH 6.5 to 8. Q and QH peaks are labeled. Ep QH vs. pH is loosely traced (black line). (B) Ep vs. pH plot for the QH and Q peaks. Points are averaged from four electrodes (n = 4) and bars are standard deviations. The QH peak has a slope of −98.6 mV/pH units (R2 = 0.92).
The redox processes at the QH and Q peaks are under kinetic control as evidenced by their broad peak separation (>300 mV). Under these conditions, the peak positions are controlled by the pH dependence of the one-electron formal potentials.54 Indeed, the pH dependence of the peaks indicates the order of the proton-electron transfer suggesting that the reactions occur in the following sequence (written in the order of an oxidation).
| (2) |
For a quinodal species at intermediate pH, E1o’ is independent of pH, and E2o’ has a 120 mV/pH dependence.54 This trend at pyrolyzed photoresist is different from that found with carbon fiber microelectrodes where the cathodic process held a 120 mV/pH dependence and the anodic process held no pH dependence.47 One explanation is that the FSCV waveform in this work has a higher scan rate (400 V/s vs. 300 V/s) and higher anodic limit (1.3 V vs. 1.0 V) than the previous research.47 These waveform modifications have changed the oxygen containing groups on the carbon surface which are known to be affected by anodic conditioning.55,36,45
Determination of Oxygen Concentrations
To further characterize the sensor, the signal response was investigated in solutions of varying oxygen concentration. Figure 5A and 5B displays the color plot of voltammograms collected at pH 7.5 with 250 or 50 μM oxygen respectively. The higher concentration is an air saturated solution,42 while the lower concentration is a typical value found deep in the mammalian brain.56 Below each color plot is the generator potential and above is the PCR analysis showing the local changes in pH and hydrogen peroxide concentration. The PCR analysis used pH and hydrogen peroxide training sets to predict the changes in concentrations reported by the collector. The training sets accurately describe the data (Figure 5A) as evidenced by a relatively featureless residual color plot and residual current contribution below 95% significance (Figure 5F). As in deoxygenated solution (Figure 3C), stepping the generator electrode from 0 to −1 V produces a basic pH shift which is confirmed by the signature pH CV (Figure 5C). In oxygenated solutions, the additional peak at 1.27 V is present from the oxidation of hydrogen peroxide (Figure 5C, labeled H2O2). Hydrogen peroxide irreversibly oxidizes in a 2 electron process above 1.0 V on the FSCV waveform.41 Its cyclic voltammogram is comparable to the signal collected for a change in the bulk concentration of hydrogen peroxide (Figure 5D). The collected hydrogen peroxide peak has a linear dependence on the oxygen concentration (n = 4 electrodes, 0.059 ± 0.001 nA/μM O2, r2 = 0.98, LOD 29±3.5 μM) and can be used as a measurement of the absolute oxygen concentrations.
Figure 5.

FSCV signal from the collector electrode during operation of the sensor in oxygenated PBS solutions. (A) Background-subtracted color plot over two generator electrode square wave cycles from 0 to −1 V vs. Ag/AgCl (potential is below) in 250 μM O2. Background is the white dashed line, labeled BG. Above is PCR of relative pH in blue and H2O2 in green. (B) Same as A, but 50 μM O2. (C) Background-subtracted cyclic voltammograms calculated from the color plots. The 250 μM O2 black line is from (A) color plot, black line – white dashed line, and the 50 μM light grey line is from (B). CVs from intermediate O2 concentrations are overlayed for comparison (color plots not shown). (D) Background-subtracted CV for a flow injection bolus of 200 μM H2O2. (E) Schematic of sensor operation. FSCV is continuously applied to the collector electrode. The generator electrode reduces O2 to H2O2 during a negative potential pulse. The blue arrow denotes O2 reduction and is color coordinated with the generator electrode potential. (F) Residual color plot of (A) with pH and H2O2 used in the PCR training set. Above is a residual Q trace with a dashed line marking Qα of 95% significance.
Figure 5E shows the proposed mechanism where the generator electrode potential step is color coded with the reduction reaction taking place. On carbon, the initial one-electron reduction is the rate limiting step, followed by protonation of the superoxide ion and the second reduction.57,58
| (3) |
The reduction of oxygen in aqueous media consumes both oxygen and protons (Equation 3).57 The depletion of protons from oxygen reduction increases the magnitude of the basic shift measured in an oxygen-dependent manner. In air-saturated solutions, the hydrogen peroxide concentration, magnitude of the basic pH shift, and reductive current at the generator increase with time before leveling off (blue and green lines, Figure 5A, supplementary Figure 1C), consistent with continuous reduction of oxygen from solution. This result contrasts with the pH shifts seen in deoxygenated buffer that quickly decayed with time (Figure 3) because they were limited by surface concentrations on the generator electrode. The two mechanisms for inducing a pH shift, constant consumption of H+ due to oxygen reduction and the pulsatile change in surface chemistry at the generator electrode, are both apparent at 50 μM oxygen (Figure 5B).
Variability between electrodes (n = 4) is minimal as evidenced by the error bars in Figure 6 and the small confidence interval of the oxygen signal sensitivity (0.059 ± 0.001 nA/μM O2). The stability of the pH and oxygen signals was investigated over three hours of continuous sensor operation in oxygenated PBS buffer after 30 minutes of conditioning. The magnitude of the Q and QH peaks in the collected FSCV signal decayed ~10% in the first hour before stabilizing for the remainder of the test. However, there was no significant change in Ep QH and Ep Q and so the absolute pH measurement was unaffected. The magnitude of the H2O2 peak, used for oxygen measurements, was unchanged over this time period (supplementary Figure 3).
Figure 6.

Interference of O2 on the pH signal Ep QH. Points are averages from four electrodes (n =4) and bars are standard deviation. The slope of the pH vs. Ep QH curve increases with O2 concentrations (−98.6 mV/pH units for 0 μM O2 r2=0.92, 131 mV/pH units for 250 μM O2 r2=0.97). O2 interference takes effect above pH 7.0.
Oxygen interference on the pH signal
The reduction of oxygen by the generator electrode increases the magnitude of the pH-shifts recorded at the collector electrode and generates hydrogen peroxide (Figure 5C). Oxygen reduction thus enhances pH signal amplitude and enables detection of background oxygen concentrations. Despite these benefits, oxygen reduction distorts the pH calibration slope in a negative direction at basic pH values (Figure 6). The calibration curve for pH in the presence of 250 μM oxygen has an increased slope of 131 ± 5 mV/pH unit compared to 98.6 ± 5 mV/pH unit for 0 μM. A possible explanation for the distortion is that Ep QH is sensitive to the size of the pH shift used to obtain it. When the generator electrode induces a local basic pH shift, Epa at the collector electrode is shifted negative and Ep QH measures the difference. At increased oxygen levels and decreased proton concentrations (basic pHs), the induced basic pH shift is greater (Figure 5C). These conditions subsequently shift Epa and thus Ep QH in an increasingly negative direction. Of course, the oxygen concentration can be determined from the magnitude of the hydrogen peroxide peak in the CV, enabling the contribution of oxygen in the pH signal to be accounted for in the pH calibration. In addition, the pH distortion due to oxygen is expected to be minimal in deep brain regions where oxygen concentrations are approximately 50 μM.3,4 In more oxygenated environments, such as arterial vessels (124–171 μm), the post-calibration pH curve must be performed with the appropriate oxygen concentration to accurately measure pH.
CONCLUSIONS
This proof–of-concept study demonstrates a microfabricated sensor that can simultaneously measure the absolute levels of pH and oxygen with FSCV. Absolute measurements are made by modulating the local electrochemical environment at the collector electrode by changing the potential of the adjacent generator electrode. For absolute pH measurements, the generator induces a local pH shift that is used to resolve the anodic peak position of the quinone/hydro-quinone-like couple on the collector. With calibration the position of this peak can be used to determine absolute pH. For absolute oxygen concentration, the generator electrode reduces oxygen creating hydrogen peroxide. The increase in local hydrogen peroxide concentration is detected by the collector and with calibration can be used to determine the absolute oxygen levels. Oxygen reduction interferes with the absolute pH signal but this can be accounted for since oxygen levels are simultaneously measured. Near term studies will focus on maximizing signal to noise and stability while also minimizing any interference from in vivo sources.
Supplementary Material
Acknowledgments
The authors acknowledge financial support from NIH (DA032530 and NS015841). The authors thank Dr. Martin Edwards for helpful discussions.
Footnotes
Supplementary Figure 1 contains generator currents for Figure 3. Supplementary Figure 2 is a control experiment for ionic contributions to the signal. Supplemental Figure 3 examines drift of the pH and oxygen measurements over several hours. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kaila K, Ransom BR. pH and brain function. Wiley-Liss; New York: 1998. p. xiii.p. 688. [Google Scholar]
- 2.Heisler N. Acid-base regulation in animals. Elsevier; Sole distributors for the U.S.A. and Canada, Elsevier Science Pub. Co; Amsterdam; New York New York, NY, U.S.A: 1986. p. x.p. 492. [Google Scholar]
- 3.Ndubuizu O, LaManna JC. Antioxid Redox Sign. 2007;9:1207–1219. doi: 10.1089/ars.2007.1634. [DOI] [PubMed] [Google Scholar]
- 4.Feng ZC, Roberts EL, Sick TJ, Rosenthal M. Brain Res. 1988;445:280–288. doi: 10.1016/0006-8993(88)91190-0. [DOI] [PubMed] [Google Scholar]
- 5.Korostynska O, Arshak K, Gill E, Arshak A. Ieee Sens J. 2008;8:20–28. [Google Scholar]
- 6.Wang XD, Wolfbeis OS. Anal Chem. 2013;85:487–508. doi: 10.1021/ac303159b. [DOI] [PubMed] [Google Scholar]
- 7.Kristensen HB, Salomon A, Kokholm G. Anal Chem. 1991;63:A885–A891. [Google Scholar]
- 8.Fedirko N, Svichar N, Chesler M. J Neurophysiol. 2006;96:919–924. doi: 10.1152/jn.00258.2006. [DOI] [PubMed] [Google Scholar]
- 9.Johnson MD, Kao OE, Kipke DR. J Neurosci Meth. 2007;160:276–287. doi: 10.1016/j.jneumeth.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 10.Lu M, Compton RG. Analyst. 2014;139:4599–4605. doi: 10.1039/c4an00866a. [DOI] [PubMed] [Google Scholar]
- 11.Lu M, Compton RG. Analyst. 2014;139:2397–2403. doi: 10.1039/c4an00147h. [DOI] [PubMed] [Google Scholar]
- 12.Wildgoose GG, Pandurangappa M, Lawrence NS, Jiang L, Jones TGJ, Compton RG. Talanta. 2003;60:887–893. doi: 10.1016/S0039-9140(03)00150-4. [DOI] [PubMed] [Google Scholar]
- 13.Fierro S, Einaga Y. Top Appl Phys. 2015;121:295–318. [Google Scholar]
- 14.Jurmann G, Schiffrin DJ, Tammeveski K. Electrochimica Acta. 2007;53:390–399. [Google Scholar]
- 15.McCreery RL. Chem Rev. 2008;108:2646–2687. doi: 10.1021/cr068076m. [DOI] [PubMed] [Google Scholar]
- 16.Takmakov P, Zachek MK, Keithley RB, Bucher ES, McCarty GS, Wightman RM. Anal Chem. 2010;82:9892–9900. doi: 10.1021/ac102399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings VJ, Pearson P. Nature. 1975;256:31–31. [Google Scholar]
- 18.Buxton RB. Introduction to functional magnetic resonance imaging : principles and techniques. 2. Cambridge University Press; New York: 2009. p. xi.p. 457. [Google Scholar]
- 19.Bolger FB, McHugh SB, Bennett R, Li J, Ishiwari K, Francois J, Conway MW, Gilmour G, Bannerman DM, Fillenz M, Tricklebank M, Lowry JP. J Neurosci Meth. 2011;195:135–142. doi: 10.1016/j.jneumeth.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 20.Venton BJ, Michael DJ, Wightman RM. J Neurochem. 2003;84:373–381. doi: 10.1046/j.1471-4159.2003.01527.x. [DOI] [PubMed] [Google Scholar]
- 21.Heien MLAV, Johnson MA, Wightman RM. Anal Chem. 2004;76:5697–5704. doi: 10.1021/ac0491509. [DOI] [PubMed] [Google Scholar]
- 22.Makos MA, Omiatek DM, Ewing AG, Heien ML. Langmuir. 2010;26:10386–10391. doi: 10.1021/la100134r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmerman JB, Kennedy RT, Wightman RM. J Cereb Blood Flow Metab. 1992;12:629–637. doi: 10.1038/jcbfm.1992.87. [DOI] [PubMed] [Google Scholar]
- 24.Zimmerman JB, Wightman RM. Anal Chem. 1991;63:24–28. doi: 10.1021/ac00001a005. [DOI] [PubMed] [Google Scholar]
- 25.Bucher ES, Fox ME, Kim L, Kirkpatrick DC, Rodeberg NT, Belle AM, Wightman RM. J Cereb Blood Flow Metab. 2014;34:1128–1137. doi: 10.1038/jcbfm.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy RT, Jones SR, Wightman RM. Neuroscience. 1992;47:603–612. doi: 10.1016/0306-4522(92)90169-3. [DOI] [PubMed] [Google Scholar]
- 27.Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PE, Wassum KM, Wightman RM. Proc Natl Acad Sci U S A. 2005;102:10023–10028. doi: 10.1073/pnas.0504657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson DL, Wightman RM. Front Neuroeng. 2007;1:17–34. [Google Scholar]
- 29.Robinson DL, Hermans A, Seipel AT, Wightman RM. Chem Rev. 2008;108:2554–2584. doi: 10.1021/cr068081q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keithley RB, Takmakov P, Bucher ES, Belle AM, Owesson-White CA, Park J, Wightman RM. Anal Chem. 2011;83:3563–3571. doi: 10.1021/ac200143v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atcherley CW, Laude ND, Parent KL, Heien ML. Langmuir. 2013;29:14885–14892. doi: 10.1021/la402686s. [DOI] [PubMed] [Google Scholar]
- 32.Atcherley CW, Wood KM, Parent KL, Hashemi P, Heien ML. Chem Commun. 2015;51:2235–2238. doi: 10.1039/c4cc06165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes EO, Lewis GEM, Dale SEC, Marken F, Compton RG. Analyst. 2012;137:1068–1081. doi: 10.1039/c2an16174e. [DOI] [PubMed] [Google Scholar]
- 34.Ranganathan S, McCreery R, Majji SM, Madou M. J Electrochem Soc. 2000;147:277–282. [Google Scholar]
- 35.Bucher ES, Brooks K, Verber MD, Keithley RB, Owesson-White C, Carroll S, Takmakov P, McKinney CJ, Wightman RM. Anal Chem. 2013;85:10344–10353. doi: 10.1021/ac402263x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takmakov P, Zachek MK, Keithley RB, Walsh PL, Donley C, McCarty GS, Wightman RM. Anal Chem. 2010;82:2020–2028. doi: 10.1021/ac902753x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zachek MK, Takmakov P, Moody B, Wightman RM, McCarty GS. Anal Chem. 2009;81:6258–6265. doi: 10.1021/ac900790m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berenyi A, Somogyvari Z, Nagy AJ, Roux L, Long JD, Fujisawa S, Stark E, Leonardo A, Harris TD, Buzsaki G. Journal of Neurophysiology. 2014;111:1132–1149. doi: 10.1152/jn.00785.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan D, Rich D, Holtzman T, Ruther P, Dalley JW, Lopez A, Rossi MA, Barter JW, Salas-Meza D, Herwik S, Holzhammer T, Morizio J, Yin HH. Plos One. 2011;6 doi: 10.1371/journal.pone.0022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dengler AK, McCarty GS. J Electroanal Chem. 2013;693:28–33. doi: 10.1016/j.jelechem.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanford AL, Morton SW, Whitehouse KL, Oara HM, Lugo-Morales LZ, Roberts JG, Sombers LA. Anal Chem. 2010;82:5205–5210. doi: 10.1021/ac100536s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schumpe A, Adler I, Deckwer WD. Biotechnol Bioeng. 1978;20:145–150. [Google Scholar]
- 43.Zachek MK, Park J, Takmakov P, Wightman RM, McCarty GS. Analyst. 2010;135:1556–1563. doi: 10.1039/c0an00114g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keithley RB, Heien ML, Wightman RM. Trac-Trend Anal Chem. 2009;28:1127–1136. doi: 10.1016/j.trac.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heien MLAV, Phillips PEM, Stuber GD, Seipel AT, Wightman RM. Analyst. 2003;128:1413–1419. doi: 10.1039/b307024g. [DOI] [PubMed] [Google Scholar]
- 46.Bath BD, Michael DJ, Trafton BJ, Joseph JD, Runnels PL, Wightman RM. Anal Chem. 2000;72:5994–6002. doi: 10.1021/ac000849y. [DOI] [PubMed] [Google Scholar]
- 47.Runnels PL, Joseph JD, Logman MJ, Wightman RM. Anal Chem. 1999;71:2782–2789. doi: 10.1021/ac981279t. [DOI] [PubMed] [Google Scholar]
- 48.Karweik DH, Hu IF, Weng S, Kuwana T. Acs Sym Ser. 1985;288:582–595. [Google Scholar]
- 49.Einstein A. Ann Phys-Berlin. 1905;17:549–560. [Google Scholar]
- 50.Junge W, Mclaughlin S. Biochim Biophys Acta. 1987;890:1–5. doi: 10.1016/0005-2728(87)90061-2. [DOI] [PubMed] [Google Scholar]
- 51.Soriaga MP, Hubbard AT. J Am Chem Soc. 1982;104:3937–3945. [Google Scholar]
- 52.Fosset B, Amatore C, Bartelt J, Wightman RM. Anal Chem. 1991;63:1403–1408. [Google Scholar]
- 53.Bard AJ, Faulkner LR. Electrochemical methods : fundamentals and applications. 2. John Wiley; New York: 2001. p. xxi.p. 809. [Google Scholar]
- 54.Kawagoe KT, Garris PA, Wightman RM. J Electroanal Chem. 1993;359:193–207. [Google Scholar]
- 55.Engstrom RC, Strasser VA. Anal Chem. 1984;56:136–141. [Google Scholar]
- 56.LaManna JC. J Biomed Opt. 2007;12 doi: 10.1117/1.2804184. [DOI] [PubMed] [Google Scholar]
- 57.Taylor RJ, Humffray AA. J Electroanal Chem. 1975;64:85–94. [Google Scholar]
- 58.Tammeveski K, Kontturi K, Nichols RJ, Potter RJ, Schiffrin DJ. J Electroanal Chem. 2001;515:101–112. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.