

Abstract
Purpose
Oxidative stress is a major factor involved in retinal pigment epithelium (RPE) apoptosis that underlies AMD. Drusen, extracellular lipid- and protein-containing deposits, are strongly associated with the development of AMD. Cell-derived microparticles (MPs) are small membrane-bound vesicles shed from cells. The purpose of this study was to determine if oxidative stress drives MP release from RPE cells, to assess whether these MPs carry membrane complement regulatory proteins (mCRPs: CD46, CD55, and CD59), and to evaluate the effects of a thiol antioxidant on oxidative stress–induced MP release.
Methods
Retinal pigment epithelium cells isolated from human donor eyes were cultured and treated with hydrogen peroxide (H2O2) to induce oxidative stress. Isolated MPs were fixed for transmission electron microscopy or processed for component analysis by flow cytometry, Western blot analysis, and confocal microscopy.
Results
Transmission electron microscopy showed that MPs ranged in diameter from 100 to 1000 nm. H2O2 treatment led to time- and dose-dependent elevations in MPs with externalized phosphatidylserine and phosphatidylethanolamine, known markers of MPs. These increases were strongly correlated to RPE apoptosis. Oxidative stress significantly increased the release of mCRP-positive MPs, which were prevented by a thiol antioxidant, N-acetylcysteine amide (NACA).
Conclusions
This is the first evidence that oxidative stress induces cultured human RPE cells to release MPs that carry mCRPs on their surface. The levels of released MPs are strongly correlated with RPE apoptosis. N-acetylcysteine amide prevents oxidative stress–induced effects. Our findings indicate that oxidative stress reduces mCRPs on the RPE surface through releasing MPs.
Keywords: microparticles, N-acetylcysteine amide, membrane complement regulatory proteins, AMD
Age-related macular degeneration (AMD) is the leading cause of blindness in the elderly affecting tens of millions of people worldwide, and is the most common cause of vision loss in the elderly in the United States. The dry form of AMD accounts for 80% to 90% of all AMD cases.1–4 The hallmarks of dry AMD are the appearance of drusen, and apoptosis of the retinal pigment epithelium (RPE).5–9 Drusen are extracellular lipid- and protein-containing deposits that accumulate mainly between the RPE and Bruch's membrane. Drusen are a hallmark of aging and early AMD, as well as a risk factor for developing late AMD.10–14 Although understanding of the mechanisms of AMD has increased, there is neither a cure nor means of prevention for AMD. Therefore, there is a critical need to identify new mechanisms for AMD to develop preventive and therapeutic strategies for this age-related blinding disease.
The deposition of drusen between Bruch's membrane and the RPE layer impairs the RPE cell layer through obstruction of nutrient flow to and from the choroid. It is believed that drusen are formed from cellular materials originating from RPE cells. The suggested mechanisms for drusen formation include cellular budding during apoptosis5,15 and exosomal trafficking during autophagy.16 An additional possible mechanism that has yet to be explored is through the generation of microparticles (MPs) by RPE cells.
Cell-derived MPs are small membrane-bound vesicles that range in diameter from 100 to 1000 nm. They are released from activated, injured, and apoptotic parental cells into the extracellular space.17–21 Microparticles shed by other organ systems under pathologic conditions have been noted to contain parental cell components and are capable of potentiating the disease state.17,22–25
The retina is highly susceptible to oxidative stress, which is recognized as a major risk factor for AMD.13,26,27 Retinal pigment epithelium cells spend their life recycling the lipid-rich photoreceptor outer segments, which requires an abundance of antioxidants and repair systems to maintain homeostasis.4,13,26 Disruption of this balance may lead to RPE dysfunction, cell death, and inflammation.3,13,26–28
Complement activation is currently thought to play an important role in AMD development.1,4,29 Membrane complement regulatory proteins (mCRPs: CD46, CD55, and CD59) provide normal cells with a first line of defense against complement attack, and have been identified on human RPE cells in culture30–33 and in situ.30,33–36 Oxidative stress leads to the loss of mCRPs on RPE cells.31 In addition, CD46 is lost from RPE cells at the earliest stage of AMD and has been shown to be present in drusen.33,34,36 However, mechanisms leading to the loss of mCRPs on RPE cells under oxidative stress or during early AMD remain unknown. Therefore, we hypothesized that oxidative stress–induced loss of mCRPs on RPE cells is through the release of MPs that carry mCRPs.
N-acetylcysteine amide (NACA; also termed AD4), the amide form of N-acetylcysteine (NAC), is a thiol antioxidant with enhanced properties of lipophilicity, membrane permeability, and antioxidant capacity when compared with NAC.37 Emerging evidence provides strong support for NACA as a protective agent against oxidative stress under numerous pathologic conditions in vitro and in vivo, including a mouse model of retinal degeneration.38
In this study, we investigated if oxidative stress drives MP release from RPE cells, assessed whether these released MPs carry mCRPs, and evaluated the effects of NACA, a thiol antioxidant, on oxidative stress–induced MP release.
Materials and Methods
Materials
Cell culture dishes, flasks, Falcon Primaria 24-well plates (Becton-Dickinson, Inc., Lincoln Park, NJ, USA), and fetal bovine serum (FBS) were purchased from Fisher Scientific (Pittsburgh, PA, USA). Phenol red-free Dulbecco's modified Eagle's medium/F12 (DMEM/F12), penicillin G, streptomycin sulfate, hydrogen peroxide (H2O2), and NACA were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). Dulbecco's phosphate-buffered saline (D-PBS), Hanks' balanced salt solutions (HBSS), fungizone, and Dead Cell Apoptosis Kit with Annexin V Alexa Fluor 488 and propidium iodide (PI) were obtained from Life Technologies (Gaithersburg, MD, USA). Dispase-II was purchased from Roche Molecular Diagnostics (Indianapolis, IN, USA). Filters (pore size 1.2 μm) were purchased from Sterlitech Corporation (Kent, WA, USA). A cyclic RGD peptide, Cyclo(-Arg-Gly-Asp-D-Phe-Cys (cRGDfC; hereafter referred to as cRGD) was purchased from Bachem Americas, Inc. (Torrance, CA, USA). All nonconjugated primary antibodies, fluorescent conjugated primary and secondary antibodies, isotype control IgGs, fluorophores, and fluorescently labeled proteins used for this study were purchased from Life Technologies, Haematologic Technologies (Essex Junction, VT, USA), Molecular Targeting Technologies (West Chester, PA, USA), AbD Serotec (Raleigh, NC, USA), Sigma-Aldrich Corp., or Santa Cruz Biotechnology (Dallas, TX, USA; see Supplementary Tables S1, S2). All other reagents were purchased from Sigma-Aldrich Corp. or Gibco BRL Life Technologies (Gaithersburg, MD, USA).
Human RPE Cell Culture and Treatment
All experiments were performed in accordance with the provisions of the Declaration of Helsinki for the use of human tissue in research and approved by the University of Michigan institutional review board (Ann Arbor, MI, USA). Donor eyes with no ocular disease history (a 52-year-old male and a 61-year-old male Caucasian) were obtained from Eversight (former name: Midwest Eye-Banks, Ann Arbor, MI, USA). Human RPE cells were enzymatically isolated from donor eyes and cultured as previously described.26,39 Briefly, the neural retina was separated gently from the RPE monolayer, and the RPE sheets were removed from Bruch's membrane using 30- to 60-minute incubation with 2% dispase-II in HBSS, followed by a gentle stream of HBSS applied by a fire-polished Pasteur pipette. Isolated RPE sheets were grown in Falcon Primaria 24-well plates with phenol-free DMEM/F12 containing 20% FBS, penicillin G (100 U mL−1), streptomycin sulfate (100 μg mL−1), and amphotericin B (0.25 μg mL−1) at 37°C in a humidified incubator under 5% CO2. Cells were then subcultured into cell culture dishes and grown in phenol red-free DMEM/F12 containing 10% FBS with the same antibiotics as described above. In all experiments, simultaneous, parallel assays were performed on fourth to ninth passaged RPE cells seeded at the same time and density from the same parent cultures. All experiments were repeated at least three times.
For H2O2 treatment, RPE cells were treated with 500 μM H2O2 for 2 to 24 hours or 50 to 2000 μM H2O2 for 16 hours in serum-free and phenol-free DMEM/F12. Cells without H2O2 treatment serve as controls. Prior to experiments, cells were switched to MP-free medium to avoid contamination from endogenous MPs present in the serum. In some cases, RPE cells were preincubated with 1 mM NACA for 8 hours prior to H2O2 exposure.
Microparticle Isolation and Characterization
Microparticle-free media, prepared by filtration through 0.1-μm filters, were used for all experiments. In each experiment, MPs were simultaneously isolated from vehicle- and H2O2-treated RPE cells by differential centrifugation40 and microfiltration as follows. After cell conditioned media were collected, cells were incubated with 0.05% trypsin/0.53 mM EDTA for 5 minutes, after which trypsin was neutralized with MP-free complete media. Cell suspensions were collected and pooled with the conditioned media that were collected prior to trypsinization. After centrifuging for 5 minutes at 500g at 4°C, cells were collected for flow cytometry analysis. Supernatants were collected and centrifuged at 1500g for 15 minute at 4°C to remove cell debris. Each of the supernatants was collected and passed through a 1.2-μm filter to remove any larger extracellular vesicles, such as apoptotic bodies. Supernatants were then centrifuged at 20,000g for 30 minutes at 4°C. The pellets were resuspended, washed in D-PBS, and centrifuged for a total of three times. Isolated MPs were then processed for transmission electron microscopy (TEM), flow cytometry, Western blot analysis, or confocal microscopy as described below.
Transmission Electron Microscopy
Isolated MPs were fixed with 4% paraformaldehyde for 1 hour, washed in D-PBS, and centrifuged at 20,000g for 30 minutes, after which the pellet was resuspended in water and negative stained with 1% uranyl acetate for 1 minute. Samples were imaged with an AMT camera (Advanced Microscopy Techniques, Woburn, MA, USA) on a Philips CM-100 (Philips, Andover, MA, USA) or JEOL JEM 1400 TEM (JEOL, Peabody, MA, USA) at the University of Michigan Microscopy and Image Analysis Core Facility.
ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA) was used to measure microparticle size with the global scale bar set based on the TEM image scale bar. Microparticles were distinguished as circular objects repelling the uranyl acetate stain and measured across their diameter.
Flow Cytometry
Isolated MPs were stained with the following antibody-fluorophores in varying combinations with compensation and IgG controls used where necessary: annexin V-FITC, annexin V-PE, PI, CD46-APC, CD55-PE, CD59-APC, Milk fat globule-epidermal growth factor (EGF) factor 8 (MFG-E8)-FITC, and duramycin-FITC (Supplementary Table S1). Controls for IgG1 and IgG2a conjugated to APC were used. In some cases, MPs were exposed to 16 μM, 100-fold excess compared with MFG-E8, cRGD for 30 minutes prior to staining with MFG-E8-FITC. Annexin V and PI staining was performed at room temperature for 15 minutes per the manufacturer's instructions while all other staining was performed on ice for 1 hour. Samples were run on a LSR II flow cytometer (BD Biosciences, San Jose, CA, USA; Becton Dickinson) equipped with 450, 488, and 633 nm lasers with a side-scatter threshold set to 750. Acquisition was performed with BD FACSDiva software. The injection port was wiped and water was run through the cytometer between samples to minimize cross-contamination of samples. FlowJo version 10 (FlowJo, LLC, Ashland, OR, USA) was used to analyze and quantify data.
Confocal Microscopy
Ten microliters of MFG-E8-FITC stained and washed samples for flow cytometry, prior to being diluted for flow cytometry, were pipetted onto a standard slide, coverslipped, and sealed with nail polish. Samples were imaged on a Leica SP5 confocal microscope (Leica Microsystems CMS GmbH, Wetzlar, Hesse, Germany) using a ×63 oil immersion lens, ×10 digital magnification, and a 488-nm laser.
Cell Death Detection
Flow Cytometry of Cell Death.
Retinal pigment epithelial apoptosis and necrosis were evaluated by Dead Cell Apoptosis Kit with Annexin V Alexa Fluor 488 and PI (Life Technologies) by flow cytometry, using the same setup mentioned above, according to procedures outlined by the manufacturer. FlowJo version 10 was used to analyze and quantify data.
TUNEL Assay.
Retinal pigment epithelial cells grown on sterile coverslips were treated with 0 to 2000 μM H2O2 for 16 hours. The coverslips were washed in PBS and stained with PI (0.15 mM) for 15 minutes at room temperature. After three washes, coverslips were fixed and subjected to TUNEL assay using the cell death detection kit (In Situ Cell Death Detection Kit, Cat#: 11684817910; Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer's protocol. Finally, the coverslips were washed three times with PBS, mounted on slides using VECTASHIELD antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). Cells were viewed with an epifluorescence microscope (model E800; Nikon, Melville, NY, USA). Digital images were collected with a cooled charge-coupled device (CCD) camera and the allied software (ACT; Nikon). Percentage of TUNEL-positive and PI-positive cells was quantified with ImageJ software using the cell counter plug-in. Representative images were obtained by merging single images of DAPI (blue), PI (red), and TUNEL (green) from the same field of view using ImageJ software.
Cellular GSH Measurement
Glutathione (GSH), also known as γ-glutamyl cysteinyl glycine, is an important tripeptide thiol antioxidant. Its intracellular concentrations were measured by a Glutathione Cell-Based Detection Kit (Blue Fluorescence; Cat#:600360; Cayman Chemical Company, Ann Arbor, MI, USA) following procedures outlined by the manufacturer. Briefly, a cell-permeable dye (monochlorobimane) was used to react with cellular GSH to generate a highly blue fluorescent product that was measured with a LSR II flow cytometer using a 405 nm laser and 450/50 bandpass filter. Data were quantified using FlowJo version 10.
Western Blot Analysis
Western blot analysis was performed using the technique described previously41 with modifications. Briefly, intact cells or isolated MPs were lysed with RIPA Lysis Buffer System (Cat#: sc-24948; Santa Cruz Biotechnology) containing 150 mM NaCl, 25 mM Tris- HCl (pH 7.4), 2 mM EDTA, 1.0% Triton X-100, 1.0% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride (PMSF), and complete protease inhibitor cocktail. Lysed samples were centrifuged at 14,000g for 10 minutes at 4°C and the supernatants collected. The protein content of each sample was determined by a bicinchoninic acid (BCA) assay kit (Cat#: BCA1; Sigma-Aldrich Corp.), with bovine serum albumin used as a standard. Proteins of whole-cell lysates or of MP lysates were solubilized in Laemmli sample buffer (62.5 mM Tris [pH 6.8], 25% glycerol, 2% SDS, 0.01% bromophenol blue; Bio-Rad, Hercules, CA, USA) without β-mercaptoethanol (nonreducing conditions), heated at 95°C for 5 minutes, cooled on ice, and then applied to 4% to 20% linear gradient long-life TGX (Tris-Glycine eXtended) gels (Cat#: 4561094; Bio-Rad). After electrophoresis, proteins were transferred to a low fluorescence polyvinylidene difluoride (PVDF) membrane (Cat#: IPFL00010; EMD Millipore Immobilon PVDF Transfer Membranes; Billerica, MA, USA), and immunoblotted with primary antibody against CD46 (2 μg/mL), CD59 (2 μg/mL), CD63 (2 μg/mL), β-actin (1:3000 dilution), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1 μg/mL; Supplementary Table S2) overnight in the cold room with constant rocking. Primary antibodies were visualized with fluorescently conjugated chicken anti-mouse IgG (Alexa Fluor 488; Life Technologies), or goat anti-rabbit IgG (Alexa Fluor 647; Life Technologies) at a working concentration of 10 μg/mL, using a GE Typhoon FLA 9000 Gel Imaging Scanner (GE Healthcare, Piscataway, NJ, USA). For Alexa Fluor 488–conjugated secondary antibody, a laser with 473-nm wavelength was used for excitation and a greater than or equal to 510-nm long-pass (LPB) filter was used for emission acquisition. For Alexa Fluor 647–conjugated secondary antibody, a laser with 635-nm wavelength was used for excitation and a greater than or equal to 665-nm long-pass (LPR) filter was used for emission acquisition. Fluorescent Western blotting images were processed and merged with ImageJ software.
Statistical Analysis
Data are presented as mean ± SD, and analyzed using Graphpad Prism version 5 (GraphPad Software, Inc., San Diego, CA, USA). Student's t-test or 1-way ANOVA with Sidak's multiple comparisons test was used to compare groups. Correlation between PS-positive MPs versus RPE apoptosis was determined using Pearson's correlation test. Serum-free samples were used as negative controls, termed control in all plots, and subtracted out from the H2O2-treated samples when normalized to control is stated. P values less than 0.05 were considered as statistically significant.
Results
Characterization of RPE-Derived Microparticles
It is well established that H2O2 triggers RPE cell death by induction of the apoptotic signaling cascade through oxidative stress. To evaluate whether oxidative stress induces MP release from RPE cells, we treated cultured human RPE cells derived from a 52-year-old or a 61-year-old male Caucasian donor with 500 μM H2O2 or vehicle (control) for 16 hours with or without NACA (a thiol antioxidant). Microparticles released from RPE cells were characterized by TEM, the gold standard for vesicle size determination, and by flow cytometry, the most common method for analysis of MPs.17,42 Figure 1A shows representative TEM images and Figure 1B shows a quantitative histogram displaying the distribution of MP sizes in all four groups. As shown in Figure 1, MPs released from RPE cells are heterogeneous and range in diameter from 100 to 1000 nm with no statistical difference in size among the four groups.
Figure 1.

Characterization of RPE-derived microparticles by TEM. (A) Representative images of MPs isolated from control and 500 μM H2O2-treated human RPE cells derived from a 52-year-old male Caucasian donor with and without pretreatment with 1 mM NACA. Microparticles were stained with uranyl acetate prior to being imaged on a JEOL JEM-1400 TEM. Images were taken at ×54,700 magnification; scale bars: 400 nm. (B) Histograms of MP size distribution from four groups (control, control + NACA, H2O2, H2O2 + NACA). Microparticle size was determined on the basis of TEM images using ImageJ software.
To characterize MPs by flow cytometry, we first used fluorescent beads of known sizes (0.5 and 1 μm) and confirmed that a LSR II flow cytometer detected events within the size range of MPs (Fig. 2A). A gate encompassing the bead populations was set and maintained across all experiments. The majority of events in MP samples isolated from both control and H2O2-treated cells with or without NACA fell within the gate in a similar pattern to the distribution of the one-half micron beads, suggesting that we detected the MPs visualized with TEM. Total microparticles based on light-scattering measurements were significantly increased in H2O2-treated samples. This increase was reversed by pretreatment of RPE cells with NACA, a thiol antioxidant (Fig. 2B; P < 0.05).
Figure 2.

Detection of RPE-derived microparticles by flow cytometry. (A) A cocktail of 0.5- and 1-μm beads were run on a BD LSR II flow cytometer with a side-scatter threshold set to 750. The majority of events from samples obtained from control and 500 μM H2O2-treated RPE cells with or without NACA pretreatment fell within the gate built around the bead scatter profile. (B) Quantification of event counts per milliliter, which fell within the set gate for each of four groups as indicated. Data are presented as mean ± SD (n = 5–11). *P < 0.05 compared with all other groups.
A hallmark of MPs is phosphatidylserine (PS) externalization, which results from the loss of asymmetry of the cell membrane during apoptosis.20,22,43 Samples obtained from both control and H2O2-treated RPE cells demonstrated positive annexin V staining, suggesting that PS is exposed on the surface of MPs (Fig. 3A; upper panels). MFG-E8 has been suggested to bind PS more effectively than annexin V.44,45 Therefore, we used fluorescently (FITC) labeled MFG-E8 to stain PS on MPs (Fig. 3A; lower panels). As shown in Fig. 3A, MFG-E8 was superior to annexin V for the staining of PS on MPs, especially for the MPs released from control RPE cells. Fig. 3B shows summarized data by using two different PS-binding proteins, annexin V, and MFG-E8, to label MPs isolated from control and H2O2 (500 μM for 16 hours)-treated human RPE cells. Compared with annexin V, MFG-E8 detected more PS-positive MPs from control RPE cells (4-fold increase; P < 0.001; Fig. 3B). Despite this, H2O2 treatment still significantly increased the number of PS-positive MPs detected by either annexin V (6-fold increase, P < 0.001) or MFG-E8 (1.78-fold increase, P < 0.002). Due to the fact that MFG-E8 can also bind to integrin receptors (αVβ3 or αVβ5), we used a cyclic RGD peptide to block αVβ3 or αVβ5 integrin receptors possibly present on the MPs. Preincubation with cyclic RGD did not affect the percentage of PS-positive MPs, as measured by MFG-E8-FITC (Fig. 3C), suggesting that the increase in MFG-E8 binding to the surface of MPs was due to increased affinity for PS and not an additional binding site.
Figure 3.

Hydrogen peroxide increases presence of phosphatidylserine on the MP surface. (A) Representative scatter plots of MPs from control and 500 μM H2O2-treated RPE cells. Microparticles were stained with either annexin V or MFG-E8 to measure phosphatidylserine-positive (PS+) MPs. (B) Quantification of PS+ MPs measured by two PS-binding proteins, annexin V and MFG-E8. (C) Quantification of MFG-E8 binding to MPs from H2O2-treated RPE cells in the absence (MFG-E8) of or in the presence (MFG-E8 + cRGD) of cyclic RGD. Data are presented as mean ± SD (n = 3). ***P < 0.0001 compared with control annxein V; #P < 0.002 compared with control MFG-E8.
In addition to characterization of MPs by TEM and flow cytometry, we performed Western blot analysis to test whether CD63, an exosomal marker, was present on the MPs. As shown in Supplementary Figure S1, CD63 was detected in whole-cell lysates, but not in the lysates prepared from MPs, therefore ruling out contamination with exosomes.
Taken together, our data have demonstrated that the small particles released and prepared under our experimental conditions are MPs (100–1000 nm), but not exosomes (30–100 nm).
Oxidative Stress Induces Microparticle Release in a Dose- and Time-Dependent Manner
To further investigate the role of oxidative stress in MP release from RPE cells we performed time- and dose-dependent studies. Treatment with H2O2 causes both dose- and time-dependent increases in the percent of PS-positive MPs, as measured by MFG-E8-FITC, with peaks at 200 μM and 16 hours (Figs. 4A, 4B).
Figure 4.

Oxidative stress induces MP release in a dose- and time-dependent manner. Microparticles were isolated from control or RPE cells treated with 500 μM H2O2 for 2 to 24 hours (A, C) or 50 to 2000 μM H2O2 for 16 hours (B, D), and stained for PS with MFG-E8-FITC (A, B) or PE with duramycin-FITC (C, D). Data are presented as mean ± SD (n = 3).
Another aminophospholipid suggested to be present on the surface of MPs is phosphatidylethanolamine (PE), which can be targeted by duramycin.46 To see whether PE is externalized at the surface of MPs released from RPE, we stained isolated MPs from both control and H2O2-treated RPE cells with duramycin-FITC. Similar to PS, H2O2 induces PE-positive MP release in a dose- and time-dependent manner with peaks at 500 μM and 16 hours (Figs. 4C, 4D).
As H2O2 is able to drive both cellular apoptosis and necrosis, we performed TUNEL assay to confirm that MPs are being released primarily from apoptotic RPE cells (Supplementary Fig. S2). At 16 hours, serum free (control) and 50 μM H2O2 showed no signs of TUNEL- (green) or PI- (red) positive cells. Minor apoptosis (10%) was detected at 100 and 200 μM H2O2 with no necrosis. At 500 μM H2O2 there was significant apoptosis (80%) with minor necrosis (7%). There were increasing levels of necrosis in the 1000 μM H2O2 (35%) and 2000 μM H2O2 (59%) treatment groups. As 500 μM H2O2 for 16 hours constantly released higher MPs, we used 500 μM H2O2 for 16 hours for most of the data presented in this study.
Microparticle Release Correlates With RPE Apoptosis
In other organ systems, cellular apoptosis leads to the release of MPs.17,18,24 Furthermore, oxidative stress induces RPE apoptosis. To test whether MP release correlates with RPE apoptosis, we evaluated both RPE apoptosis, by annexin V and PI double staining, and released PS-positive MPs, by MFG-E8 staining, over time. As shown in Fig. 5, released PS-positive MPs strongly correlated with RPE apoptosis (r = 0.9326, P < 0.05).
Figure 5.
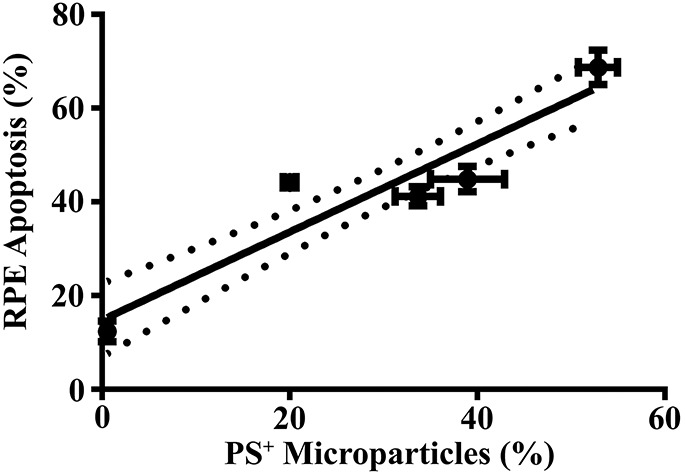
Microparticle release correlates with RPE cell apoptosis. Retinal pigment epithelial cells were exposed to 500 μM H2O2 for 2, 4, 8, 16, or 24 hours. Microparticles were isolated from H2O2-treated RPE cells, stained for PS with MFG-E8-FITC. Retinal pigment epithelial cell apoptosis was measured by annexin V-FITC and PI staining. Phosphatidylserine-positive MPs (%) were plotted on the x-axis, and the RPE apoptosis (%) was plotted on the y-axis for different time points (2–24 hours). Correlation coefficient (r) = 0.9326. Data are presented as mean ± SD (n = 3). P < 0.05.
Oxidative Stress Alters the Abundance of Membrane Complement Regulatory Proteins
Under normal physiological conditions, RPE cells express at least three mCRPs: CD46, CD55, and CD59. Following induction of oxidative stress in RPE cells, membrane abundance of these proteins is reduced.31 We assessed whether MPs may be one of the mechanisms by which mCRPs are shed from RPE cells under oxidative stress. Flow cytometric analysis showed that exposure of human RPE cells (derived from a 52-year-old male Caucasian donor) to 500 μM H2O2 for 16 hours significantly increased the percent of MPs staining positive for mCRPs: CD46 (9-fold, P < 0.0001), CD55 (6-fold, P < 0.002), and CD59 (5-fold, P < 0.0001), compared with controls (Fig. 6A). In addition, RPE cells with staining positive for mCRPs were significantly reduced (Fig. 6B), suggesting the loss of mCRPs from RPE cell surface is associated with the generation of mCRP-positive MPs. Similar results were obtained in experiments performed in a human RPE cell line, adult retinal pigment epithelium (ARPE)-19 cells (Supplementary Fig. S3), and cultured human RPE cells derived from another donor (a 61-year-old male Caucasian donor; Supplementary Fig. S4).
Figure 6.

Oxidative stress alters the abundance of membrane complement regulatory proteins on MP and RPE cells. Microparticles (A) and RPE cells (B) from control and 500 μM H2O2-treated RPE cells were stained with fluorescently labeled antibodies to each of three common membrane complement regulatory proteins (mCRPs): CD46, CD55, or CD59, and analyzed by flow cytometry. Data are presented as mean ± SD (n = 3–6). **P < 0.002, ***P < 0.0001, compared with control.
N-Acetylcysteine Amide Attenuates Oxidative Stress–Induced Effects on Microparticles and on RPE Cells
N-acetylcysteine amide can protect ARPE-19 cells against oxidative stress–induced death and decrease in cellular GSH levels.38,47 To determine if NACA can protect cultured human RPE cells against H2O2-induced MP release, cell death, and decrease in cellular GSH levels, we pretreated RPE cells with the thiol antioxidant NACA and analyzed cell apoptosis, the released MPs, and cellular GSH levels by flow cytometry. Pretreatment of RPE cells with NACA significantly attenuated 500 μM H2O2-induced increase in PS-positive MPs (Fig. 7A), which was further confirmed by confocal microscopy (Fig. 7B). In addition, NACA pretreatment prevented the elevation of mCRP-positive MPs in 500 μM H2O2-treated samples (Figs. 7C–E; P < 0.0001, compared with H2O2 without NACA). Furthermore, the observed NACA effect on MPs was associated with significant attenuation of RPE cell apoptosis (Fig. 8A; P < 0.0001) and with restoration of cellular GSH to control levels (Fig. 8B; P < 0.0001). N-acetylcysteine amide also significantly prevented RPE cells from loss of mCRPs induced by 500 μM H2O2 (Figs. 8C–E; P < 0.0001).
Figure 7.

N-Acetylcysteine amide attenuates H2O2-induced effects on RPE-derived MPs. (A) Microparticles isolated from control and 500 μM H2O2-treated RPE cells with N-acetylcysteine amide (+ NACA) or without NACA (−NACA) pretreatment were stained with MFG-E8-FITC and quantified by flow cytometry. (B) Representative images taken with ×63 oil immersion lens and ×10 digital zoom. Scale bars: 2.5 μm. (C–E) Microparticles were stained for membrane complement regulatory proteins CD46 (C), CD55 (D), and CD59 (E). Data are presented as mean ± SD (n = 3–6). ***P < 0.0001 compared with controls in the absence of NACA (−NACA); #P < 0.001 compared with H2O2-treated groups in the absence of NACA (−NACA).
Figure 8.

N-Acetylcysteine amide attenuates H2O2-induced apoptosis, decrease in cellular GSH levels, and loss of membrane complement regulatory proteins in RPE cells. Control and 500 μM H2O2-treated RPE cells with or without pretreatment of NACA were stained with annexin V-Alexa Fluor 488 and PI for apoptosis assay (A), stained with monochlorobimane (a cell-permeable dye) reacting with cellular GSH (B), or stained with fluorescently-labeled antibodies to CD46 (C), CD55 (D), or CD59 (E). Data are presented as mean ± SD (n = 3–5). **P < 0.01, ***P < 0.0001 compared with controls −NACA; #P < 0.0001 compared with H2O2-treated groups −NACA.
To confirm our flow cytometry findings presented in Figures 6 through 8, we performed fluorescent Western blot analysis. A single antibody blotting with either CD46 or CD59 specific antibody (Supplementary Table S2) detected a single band with molecular weight of approximately 60 kDa for CD46 or approximately 20 kDa for CD59 in cultured human RPE cells (data not shown), confirming the findings by others.32 By using multiplexed fluorescent Western blot analysis with low autofluorescence Immobilon-FL PVDF membrane (Cat#: IPFL00010; EMD Millipore), we directly visualized and compared the expression levels of CD46, CD59, GAPDH, and β-actin in the same blots without stripping. Stripping and reprobing of the blots can cause erroneous results due to the effect of stripping on target proteins.48 As shown in Figure 9, CD46 antibody detected a single band of approximately 60 kDa, and CD59 antibody detected a single band of approximately 20 kDa in whole-cell lysates and MP lysates. CD59 signal is much stronger than that of CD46 in the same multiplex blots. Compared with equal amount of whole-cell lysates (lanes labeled with 5 μg), both CD46 and CD59 are enriched in MPs in control, and H2O2-treated samples with or without NACA. H2O2 treatment increases the amount of CD46 and CD59 in MPs and decreases the amount of CD46 and CD59 in RPE cells, compared with control (left panel). Pretreatment with NACA decreases the amount of CD46 and CD59 in MPs and increases the amount of CD46 and CD59 in RPE cells, compared with H2O2 treatment alone (right panel). Therefore, our Western blot analysis confirms our flow cytometry findings: H2O2 induces the loss of CD46 and CD59 from RPE cells with the concomitant gain of CD46 and CD59 in MPs, all of which are attenuated by NACA.
Figure 9.

Fluorescent Western blot analysis of CD46 and CD59 proteins in RPE cells and RPE-derived MPs. Human RPE cells were treated vehicle (control; lanes labeled with C), 500 μM H2O2 (lanes labeled with H), or 500 μM H2O2 with NACA pretreatment (lanes labeled with N+H) for 16 hours. Cells and MPs were harvested and isolated after treatment. Intact cells or isolated MPs were lysed and proteins were measured. Proteins of whole-cell lysates or of MP lysates were subjected to Western blot analysis of membrane complement regulatory proteins (mCRPs) CD46 and CD59. The anti-CD46, anti-CD59 or anti–β-actin primary antibody was detected with Alexa Fluor 488–conjugated chicken anti-mouse IgG, and protein bands were pseudocolored green. The primary antibody against GAPDH was detected with Alexa Fluor 647–conjugated goat anti-rabbit IgG, and single bands were pseudocolored red. Shown is an overlay of green (CD46, CD59, and β-actin) and red (GAPDH) images. GAPDH and β-actin serve as loading controls for whole cell lysates. M indicates protein size marker.
It's well-known that GAPDH and β-actin serve as loading controls for whole-cell lysates. However, whether GAPDH and β-actin can serve as loading controls for MP lysates is unknown. As shown in Figure 9, as expected, both GAPDH (red) and β-actin (green) are detected in whole-cell lysates, but only GAPDH (red) is detectable in MPs. Compared with equal amount of whole-cell lysates (lanes labeled with 5 μg), GAPDH expression levels in MPs (MP lysates) are much lower than those in RPE cells (whole-cell lysates). Due to lack of known loading controls for MP lysates, normalization and quantification were not attempted.
It is important to note that our flow cytometry analyzed CD46 and CD59 protein expression on the surface of RPE cells and of MPs, while our Western blot analysis detected CD46 and CD59 protein expression in whole cells and whole MPs.
To test if a lower concentration of H2O2 could cause similar effects as those induced by 500 μM H2O2 described above, we used 200 μM H2O2 that induced 10% RPE apoptosis with no necrosis (Supplementary Fig. S2). Similar to 500 μM, 200 μM H2O2 also significantly increased PS-positive MP and CD59-positive MP release, both of which were reversed by NACA pretreatment (Supplementary Figs. S5A, S5D). However, unlike 500 μM H2O2, 200 μM H2O2 had no statistically significant effect on CD46- or CD55-positive MP release (Supplementary Figs. S5B, S5C).
Discussion
Here, we find that 500 μM H2O2 treatment produced a marked increase in released MPs by human RPE cells. This MP increase was accompanied by increases in RPE apoptosis and loss of mCRPs (CD46, CD55, and CD59) on the RPE cell surface. The released MPs exposed PS and PE, and carried mCRPs on their surface. Moreover, we demonstrate for the first time in any cell types that NACA, a thiol antioxidant, prevented the H2O2-enhanced release of mCRP-positive MPs, and loss of mCRPs on the RPE cell surface.
Oxidative stress from reactive oxygen species is a major factor involved in the RPE death that underlies AMD. Previously, we and others demonstrated that oxidative stress induces RPE apoptosis and degeneration.49–54 Now we show that oxidative stress also induces RPE to release MPs that carry mCRPs.
Cell-derived extracellular vesicles are generally classified into three main types: MPs, exosomes, and apoptotic bodies. These groups are based primarily on their size and presumed biogenetic pathways. Microparticles (also termed ectosomes or microvesicles), formed by membrane blebbing, are a heterogeneous population of small vesicles of 100 to 1000 nm in diameter17–20; exosomes refer to smaller vesicles of 50 to 100 nm in diameter, generated by exocytosis of multivesicular bodies55; and apoptotic bodies are vesicles of 1000 to 5000 nm in diameter, and released as blebs from cells undergoing late apoptosis.56,57 They are released into the extracellular environment by various types of cells, including epithelial cells.58,59
Several research groups have investigated RPE-derived exosomes,16,33,60–64 and ARPE-19 blebs.65,66 However, a full manuscript investigating RPE-derived MPs has not published yet. In this study, we used both TEM and flow cytometry to characterize human RPE cells derived from two donors (52 years, 61 years) and ARPE-19 cells (19 years). To our knowledge, this is the first study to investigate MPs from RPE cells derived from older donors (52 years, 61 years), which could be more relevant to AMD. We show for the first time that MP sized vesicles are released from RPE cells under normal and oxidative stress conditions by TEM (Fig. 1), flow cytometry (Figs. 2–5, 6A, 7A, 7C–E), and confocal microscopy (Fig. 7B). Apoptotic bodies were eliminated by passing samples through a 1.2-μm filter. The centrifugal force (20,000g) we used to isolate MPs is not sufficient to collect exosomes, which require a centrifugal force of 100,000g.17,60 Furthermore, exosomes are not detected by flow cytometry because their size is generally less than 100 nm,18 below the detection limit of our flow cytometer which could not detect 0.1-μm beads. Moreover, CD63, an exosomal marker, was not detected by Western blot analysis, indicating no contamination with exosomes in our MP population (Supplementary Fig. S1).
Two key events in early AMD are the appearance of drusen5–7 and RPE apoptosis.8,9 The importance of the RPE in drusen formation is evidenced by the fact that membranous debris, drusen-like deposits, and drusen build up on apical and basolateral sides of the RPE monolayer,5–7,67,68 and drusen are composed of materials found in RPE cells.5,15,28,69 The strong correlation between RPE apoptosis and PS-positive MP shedding (Fig. 5) suggests that MPs released from the apoptotic RPE cells may be early events in subclinical drusen, carrying with them the cellular components commonly found in drusen.
In this study, we found that the increase in PS-, PE-, and mCRP-positive MPs was accompanied by the increase in apoptosis and loss of mCRPs (CD46, CD55, CD59) from 500 μM H2O2-treated RPE cells by flow cytometry, suggesting that the decreased mCRPs on the RPE surface result from increased shedding of mCRP-positive MPs. Our Western blot analysis confirms the flow cytometry findings for CD46 and CD59. Although we constantly found that CD55 was present on RPE cell surface and on MPs by flow cytometry (Figs. 6, 7D, 8D), we were unable to detect CD55 expression in lysates prepared from intact cells (whole-cell lysates) or lysates prepared from MPs by Western blot analysis, using two commercially available antibodies against CD55 (data not shown). One CD55 antibody we used was mouse anti-human CD55 monoclonal antibody from EMD Millipore (Cat#: CBL511; Lot#: 26017014), and another was rabbit anti-human CD55 monoclonal antibody (EPR66890, Cat#: ab133684; Lot#: GR93375-13; Abcam, Cambridge, MA, USA). Consistent with our results, Yang et al.32 did not detect CD55 expression in both native and cultured human RPE cells by Western blot analysis using a different commercially available antibody, although they did detect CD55 expression on RPE surface by flow cytometry. Thurman et al.31 reported that 1 mM H2O2 reduced the surface expression of CD55 (also known as decay accelerating factor [DAF]) and CD59 in ARPE-19 cells using flow cytometry, but no Western blot analysis of CD46, CD55, and CD59 was reported in their study. The reasons for not detecting CD55 protein expression by Western blot analysis are not clear. One possible explanation could be that CD55 antibodies commercially developed for Western blot analysis are not suitable for CD55 antigen in the RPE cells due to its unique posttranslational modifications. Additional investigations will be required to test this hypothesis in the future studies. Interestingly, Ebrahimi et al.33 found that cellular CD46 and CD59 proteins were decreased in ARPE-19 cells treated with oxidized low-density lipoproteins and that CD59 and CD46 along with an exosomal marker CD63 were detected in culture supernatant. They suggested that the decreased levels of CD46 and CD59 were in part due to the release of exosomes and apoptotic particles.33 Two proteomic studies were performed on extracellular vesicles derived from ARPE-19 cells. One type of extracellular vesicles (ARPE-19 membrane blebs) was obtained by centrifuging conditioned medium for 15 minutes at 100g,65 whereas another type of extracellular vesicles (exosomes) was obtained by centrifuging conditioned medium for 1 hour at 100,000g.60 Both proteomic studies did not demonstrate the detection of CD46, CD55, and CD59 on these vesicles. Although there are no standard procedures available to isolate different types of extracellular vesicles (mainly MPs, exosomes, and apoptotic bodies), the most common protocol for isolation of MPs consists of differential centrifugation with final centrifugal force of 20,000g.17,42 In this study, we followed this most common protocol to isolate and analyze MPs from cultured RPE cells derived from donor eyes as well as ARPE-19 cells. We detected PS- and PE-positive MPs, and found the presence of CD46, CD55, and CD59 on the surface of isolated MPs, suggesting the CD46 found in drusen during early AMD33,36 or CD59 detected in subretinal space33 could originate from RPE cells through the release of MPs.
In addition to potentially driving or participating in drusen formation, our data indicate a possible effect of MP shedding on the retinal cells, the loss of mCRPs in the RPE layer may lead to enhanced complement attack and further cell death. Numerous studies point to deregulation of the complement cascade in AMD.4,33,34,36,70 Loss of mCRPs leads to RPE cells being vulnerable to complement attack. Here, we observed 500 μM H2O2-induced loss of CD46, CD55, and CD59 on the surface of cultured human RPE cells at two key points: activity of C3 convertase and membrane attack complex (MAC) formation. CD46 and CD55 block the complement cascade at C3 activation, while CD59 inhibits MAC assembly.71 Consistent with our results obtained by 500 μM H2O2, previous studies have shown that 1 mM H2O2 treatment reduced the surface expression of mCRPs on ARPE-19 cells by flow cytometry, and sensitized ARPE-19 cells to complement-sufficient serum-mediated complement activation and disruption of the barrier function.31,72 A lower concentration of H2O2 (200 μM) also induced PS-positive MP and CD59-positive MP releases that were prevented by NACA pretreatment (Supplementary Figs. S5A, S5D). Although some CD46- or CD55-positive MP release was detectable in control samples, 200 μM H2O2 had no significant effect on their release (Supplementary Figs. S5B, S5C). The mechanisms for this are unknown. Retinal pigment epithelial cells in vivo probably encounter a complex and varying mix of low concentrations of oxidative stress stimuli that might have additive and/or synergistic effects on MP release. CD46 and CD55 inhibit complement attack by blocking C3 activation while CD59 prevents complement attack by the inhibition of the MAC. We speculate that there could be some sorting differences associated with these two different levels of protection and our data suggest that MP release may be a deliberate and coordinated process instead of a random release of membrane-bound vesicles.
Reduction in the capacity to cope with oxidative demands results in RPE apoptosis that is thought to be an early event in the development of AMD.8,9 N-acetylcysteine amide, a thiol antioxidant, has been demonstrated to have the ability to penetrate the cell membrane better than other antioxidants such as NAC, vitamin E, and carotenoids and is less toxic when compared with NAC.73–76 One of the key endogenous thiol antioxidants is GSH, which is depleted during oxidative stress. N-acetylcysteine amide has been shown to restore cellular GSH and to have metal (e.g., Cu2+, Fe2+) chelating capacity, eventually scavenging free radicals.74,75,77,78 In ARPE-19 cells exposed to oxidative stress, NACA has been shown to prevent cell death through restoration of cellular GSH levels as well as glutathione peroxidase activity.38,47 Furthermore, NACA pretreatment of ARPE-19 cells protected against oxidative stress–induced lipid peroxidation, cell permeability, and decrease in transepithelial electrical resistance.38,47 We used NACA to prime RPE cells against the oxidative stress induced by H2O2. Cells pretreated with NACA had significantly attenuated apoptosis, restored cellular GSH levels, and decreased loss of mCRP proteins on the RPE surface, which was associated with the generation of fewer PS- and mCRP-positive MPs (Figs. 7–9).
Consistent with our results demonstrating NACA's protection of cultured human RPE cells against oxidative stress–induced apoptosis, NACA was shown to protect ARPE-19 cells against tert-butyl hydroperoxide–induced or methamphetamine-induced oxidative stress and cell death.38,47 Intraperitoneal injection of NACA prevented oxidative damage, photoreceptor cell death, and RPE cell damage in a light-induced toxicity mouse model.38 Furthermore, oral administration of NACA drastically reduced inflammation, matrix metalloproteinase-9 activity, clinical signs, and protected axons from demylination damage in mouse studies.73 Therefore, NACA has potential to prevent or slow oxidative stress-related diseases, including AMD.
Microparticle research is still in the early stages.79,80 The role of RPE-derived MPs in AMD is not known. A preliminary account of MPs isolated from ARPE-19 cells under oxidative stress has been reported.81 They found that ARPE-19 cells can uptake ARPE-19–derived MPs, which is partially dependent on CD36. ARPE-19–derived MPs also decreased RPE cell viability, and induced RPE cell-cycle arrest at G0/G1 phase associated with the increases of the senescence-associated β-galactosidase activity. The findings by Tahiri et al.81 further support our findings that RPE-derived MPs could play an important role in AMD pathogenesis. Microparticles of the cardiovascular system were reported to promote oxidative stress and organ dysfunction in vitro,82 implicating that MPs released by RPE cells under oxidative stress may act within a positive feedback mechanism that could accelerate RPE apoptosis and AMD progression. Further biophysical and functional evaluation of MPs would strengthen our findings.
The plasma membrane budding/blebbing has been proposed as one of the mechanisms for the generation of MPs in other cell systems,55,83 although the exact mechanisms for generating MPs are still lacking in any systems.83 Based on our findings that RPE-derived MPs exposed PS (Figs. 3–5, 7A) and PE (Figs. 4C, 4D), carried enriched membrane proteins mCRPs (Figs. 6, 7, 9) and few cytoplasmic protein GAPDH (Fig. 9), and did not contain CD63 (Supplementary Fig. S1), we propose that RPE-derived MPs could come from budding/blebbing, and then detaching directly from the plasma membrane of RPE cell surface, similar to other cell systems.55,83
In conclusion, this study demonstrates for the first time that oxidative stress induces human RPE cells to release PS-, PE-, and mCRP-positive MPs. The levels of released MPs are strongly correlated with RPE apoptosis. All of the effects induced by oxidative stress are prevented by NACA. Our data support the concept that RPE-derived MPs can carry components found in drusen, and suggest that oxidative stress could trigger or participate in drusen formation by releasing MPs from the RPE. Our study also describes a novel role for NACA in the prevention of mCRP-positive MP release, which might have broad therapeutic implications, particularly in diseases associated with loss of mCRPs on the cell surface, and with enhanced release of MPs.
Supplementary Material
Acknowledgments
The authors thank Michael Pihalja (the University of Michigan Flow Cytometry Core) for assistance with flow cytometry during the early phases of microparticle analysis, and Jeffrey Harrison and Dotty Sorenson (the University of Michigan Microscopy and Image Analysis Laboratory) for assistance with electron microscopy. The authors thank Eversight (Ann Arbor, MI, USA) for assistance in obtaining human eye tissues.
Supported by the National Institutes of Health Core Grant P30EY007003 (Bethesda, MD, USA), a special assistance stipend from Eversight (Ann Arbor, MI, USA), and the University of Michigan Start-Up Funds (Ann Arbor, MI, USA).
Disclosure: K.A. Carver, None; D. Yang, None
References
- 1. Leung E,, Landa G. Update on current and future novel therapies for dry age-related macular degeneration. Expert Rev Clin Pharmacol. 2013; 6: 565–579. [DOI] [PubMed] [Google Scholar]
- 2. Fritsche LG,, Fariss RN,, Stambolian D,, Abecasis GR,, Curcio CA,, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014; 15: 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ambati J,, Atkinson JP,, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013; 13: 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bowes Rickman C Farsiu S, Toth CA, Klingeborn M. Dry age-related macular degeneration: mechanisms, therapeutic targets, and imaging. Invest Ophthalmol Vis Sci. 2013; 54: ORSF68–ORSF80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burns RP,, Feeney-Burns L. Clinico-morphologic correlations of drusen of Bruch's membrane. Trans Am Ophthalmol Soc. 1980; 78: 206–225. [PMC free article] [PubMed] [Google Scholar]
- 6. Curcio CA,, Millican CL. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch Ophthalmol. 1999; 117: 329–339. [DOI] [PubMed] [Google Scholar]
- 7. Abdelsalam A,, Del Priore L,, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999; 44: 1–29. [DOI] [PubMed] [Google Scholar]
- 8. Dunaief JL,, Dentchev T,, Ying GS,, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002; 120: 1435–1442. [DOI] [PubMed] [Google Scholar]
- 9. Del Priore LV,, Kuo YH,, Tezel TH. Age-related changes in human RPE cell density and apoptosis proportion in situ. Invest Ophthalmol Vis Sci. 2002; 43: 3312–3318. [PubMed] [Google Scholar]
- 10. Anderson DH,, Mullins RF,, Hageman GS,, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002; 134: 411–431. [DOI] [PubMed] [Google Scholar]
- 11. Anderson DH,, Talaga KC,, Rivest AJ,, Barron E,, Hageman GS,, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004; 78: 243–256. [DOI] [PubMed] [Google Scholar]
- 12. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004; 122: 598–614. [DOI] [PubMed] [Google Scholar]
- 13. Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012; 33: 418–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferris FL,, III, Wilkinson CP,, Bird A,, et al. Clinical classification of age-related macular degeneration. Ophthalmology. 2013; 120: 844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gouras P,, Ivert L,, Neuringer M,, Mattison JA. Topographic and age-related changes of the retinal epithelium and Bruch's membrane of rhesus monkeys. Graefes Arch Clin Exp Ophthalmol. 2010; 248: 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang AL,, Lukas TJ,, Yuan M,, Du N,, Tso MO,, Neufeld AH. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One. 2009; 4: e4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mause SF,, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res. 2010; 107: 1047–1057. [DOI] [PubMed] [Google Scholar]
- 18. Beyer C,, Pisetsky DS. The role of microparticles in the pathogenesis of rheumatic diseases. Nat Rev Rheumatol. 2010; 6: 21–29. [DOI] [PubMed] [Google Scholar]
- 19. Pisetsky DS,, Ullal AJ,, Gauley J,, Ning TC. Microparticles as mediators and biomarkers of rheumatic disease. Rheumatology (Oxford). 2012; 51: 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Loyer X,, Vion AC,, Tedgui A,, Boulanger CM. Microvesicles as cell-cell messengers in cardiovascular diseases. Circ Res. 2014; 114: 345–353. [DOI] [PubMed] [Google Scholar]
- 21. Voloshin T,, Fremder E,, Shaked Y. Small but mighty: microparticles as mediators of tumor progression. Cancer Microenviron. 2014; 7: 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roseblade A,, Luk F,, Rawling T,, Ung A,, Grau GE,, Bebawy M. Cell-derived microparticles: new targets in the therapeutic management of disease. J Pharm Pharm Sci. 2013; 16: 238–253. [DOI] [PubMed] [Google Scholar]
- 23. Burger D,, Montezano AC,, Nishigaki N,, He Y,, Carter A,, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arterioscler Thromb Vasc Biol. 2011; 31: 1898–1907. [DOI] [PubMed] [Google Scholar]
- 24. Jimenez JJ,, Jy W,, Mauro LM,, Soderland C,, Horstman LL,, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res. 2003; 109: 175–180. [DOI] [PubMed] [Google Scholar]
- 25. Chironi GN,, Boulanger CM,, Simon A,, Dignat-George F,, Freyssinet JM,, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res. 2009; 335: 143–151. [DOI] [PubMed] [Google Scholar]
- 26. Yang D,, Elner SG,, Bian ZM,, Till GO,, Petty HR,, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007; 85: 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Payne AJ,, Kaja S,, Naumchuk Y,, Kunjukunju N,, Koulen P. Antioxidant drug therapy approaches for neuroprotection in chronic diseases of the retina. Int J Mol Sci. 2014; 15: 1865–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Telander DG. Inflammation and age-related macular degeneration (AMD). Semin Ophthalmol. 2011; 26: 192–197. [DOI] [PubMed] [Google Scholar]
- 29. Carroll MV,, Sim RB. Complement in health and disease. Adv Drug Deliv Rev. 2011; 63: 965–975. [DOI] [PubMed] [Google Scholar]
- 30. McLaughlin BJ,, Fan W,, Zheng JJ,, et al. Novel role for a complement regulatory protein (CD46) in retinal pigment epithelial adhesion. Invest Ophthalmol Vis Sci. 2003; 44: 3669–3674. [DOI] [PubMed] [Google Scholar]
- 31. Thurman JM,, Renner B,, Kunchithapautham K,, et al. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J Biol Chem. 2009; 284: 16939–16947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang P,, Tyrrell J,, Han I,, Jaffe GJ. Expression and modulation of RPE cell membrane complement regulatory proteins. Invest Ophthalmol Vis Sci. 2009; 50: 3473–3481. [DOI] [PubMed] [Google Scholar]
- 33. Ebrahimi KB,, Fijalkowski N,, Cano M,, Handa JT. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J Pathol. 2013; 229: 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson LV,, Leitner WP,, Staples MK,, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001; 73: 887–896. [DOI] [PubMed] [Google Scholar]
- 35. Vogt SD,, Barnum SR,, Curcio CA,, Read RW. Distribution of complement anaphylatoxin receptors and membrane-bound regulators in normal human retina. Exp Eye Res. 2006; 83: 834–840. [DOI] [PubMed] [Google Scholar]
- 36. Vogt SD,, Curcio CA,, Wang L,, et al. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp Eye Res. 2011; 93: 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sunitha K,, Hemshekhar M,, Thushara RM,, et al. N-acetylcysteine amide: a derivative to fulfill the promises of N-acetylcysteine. Free Radic Res. 2013; 47: 357–367. [DOI] [PubMed] [Google Scholar]
- 38. Schimel AM,, Abraham L,, Cox D,, et al. N-acetylcysteine amide (NACA) prevents retinal degeneration by up-regulating reduced glutathione production and reversing lipid peroxidation. Am J Pathol. 2011; 178: 2032–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang D,, Swaminathan A,, Zhang X,, Hughes BA. Expression of Kir7.1 and a novel Kir7.1 splice variant in native human retinal pigment epithelium. Exp Eye Res. 2008; 86: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang JG,, Williams JC,, Davis BK,, et al. Monocytic microparticles activate endothelial cells in an IL-1β-dependent manner. Blood. 2011; 118: 2366–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang D,, Pan A,, Swaminathan A,, Kumar G,, Hughes BA. Expression and localization of the inwardly rectifying potassium channel Kir7.1 in native bovine retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003; 44: 3178–3185. [DOI] [PubMed] [Google Scholar]
- 42. Dinkla S,, Brock R,, Joosten I,, Bosman GJ. Gateway to understanding microparticles: standardized isolation and identification of plasma membrane-derived vesicles. Nanomedicine (Lond). 2013; 8: 1657–1668. [DOI] [PubMed] [Google Scholar]
- 43. Distler JH,, Pisetsky DS,, Huber LC,, Kalden JR,, Gay S,, Distler O. Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum. 2005; 52: 3337–3348. [DOI] [PubMed] [Google Scholar]
- 44. Shi J,, Shi Y,, Waehrens LN,, Rasmussen JT,, Heegaard CW,, Gilbert GE. Lactadherin detects early phosphatidylserine exposure on immortalized leukemia cells undergoing programmed cell death. Cytometry A. 2006; 69: 1193–1201. [DOI] [PubMed] [Google Scholar]
- 45. Hou J,, Fu Y,, Zhou J,, et al. Lactadherin functions as a probe for phosphatidylserine exposure and as an anticoagulant in the study of stored platelets. Vox Sang. 2011; 100: 187–195. [DOI] [PubMed] [Google Scholar]
- 46. Larson MC,, Woodliff JE,, Hillery CA,, Kearl TJ,, Zhao M. Phosphatidylethanolamine is externalized at the surface of microparticles. Biochim Biophys Acta. 2012; 1821: 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carey JW,, Tobwala S,, Zhang X,, et al. N-acetyl L-cysteine amide protects retinal pigment epithelium against methamphetamine-induced oxidative stress. J Biophys Chem. 2012; 3: 101–110. [Google Scholar]
- 48. Park J,, Mabuchi M,, Sharma A. Multiplexed fluorescent immunodetection using low autofluorescence Immobilon®-FL membrane. Methods Mol Biol. 2015; 1314: 195–205. [DOI] [PubMed] [Google Scholar]
- 49. Imamura Y,, Noda S,, Hashizume K,, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2006; 103: 11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fujihara M,, Nagai N,, Sussan TE,, Biswal S,, Handa JT. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS One. 2008; 3: e3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang D,, Elner SG,, Lin LR,, Reddy VN,, Petty HR,, Elner VM. Association of superoxide anions with retinal pigment epithelial cell apoptosis induced by mononuclear phagocytes. Invest Ophthalmol Vis Sci. 2009; 50: 4998–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang D,, Elner SG,, Chen X,, Field MG,, Petty HR,, Elner VM. MCP-1-activated monocytes induce apoptosis in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011; 52: 6026–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Field MG,, Yang D,, Bian ZM,, Petty HR,, Elner VM. Retinal flavoprotein fluorescence correlates with mitochondrial stress apoptosis, and chemokine expression. Exp Eye Res. 2011; 93: 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mao H,, Seo S,, Biswal MR,, et al. Mitochondrial oxidative stress in the retinal pigment epithelium leads to localized retinal degeneration. Invest Ophthalmol Vis Sci. 2014; 55: 4613–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. György B,, Szabó TG,, Pásztói M,, et al. Membrane vesicles current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011; 68: 2667–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hristov M,, Erl W,, Linder S,, Weber PC. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood. 2004; 104: 2761–2766. [DOI] [PubMed] [Google Scholar]
- 57. Berda-Haddad Y,, Robert S,, Salers P,, et al. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1α. Proc Natl Acad Sci U S A. 2011; 108: 20684–20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bastarache JA,, Fremont RD,, Kropski JA,, Bossert FR,, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2009; 297: L1035–L1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Press JZ,, Reyes M,, Pitteri SJ,, et al. Microparticles from ovarian carcinomas are shed into ascites and promote cell migration. Int J Gynecol Cancer. 2012; 22: 546–552. [DOI] [PubMed] [Google Scholar]
- 60. Biasutto L,, Chiechi A,, Couch R,, Liotta LA,, Espina V. Retinal pigment epithelium (RPE) exosomes contain signaling phosphoproteins affected by oxidative stress. Exp Cell Res. 2013; 319: 2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sreekumar PG,, Kannan R,, Kitamura M,, et al. αB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS One. 2010; 5: e12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gangalum RK,, Atanasov IC,, Zhou ZH,, Bhat SP. AlphaB-crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells. J Biol Chem. 2011; 286: 3261–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kang GY,, Bang JY,, Choi AJ,, et al. Exosomal proteins in the aqueous humor as novel biomarkers in patients with neovascular age-related macular degeneration. J Proteome Res. 2014; 13: 581–595. [DOI] [PubMed] [Google Scholar]
- 64. Locke CJ,, Congrove NR,, Dismuke WM,, Bowen TJ,, Stamer WD,, McKay BS. Controlled exosome release from the retinal pigment epithelium in situ. Exp Eye Res. 2014; 129: 1–4. [DOI] [PubMed] [Google Scholar]
- 65. Alcazar O,, Hawkridge AM,, Collier TS,, et al. Proteomics characterization of cell membrane blebs in human retinal pigment epithelium cells. Mol Cell Proteomics. 2009; 8: 2201–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Marin-Castaño ME,, Csaky KG,, Cousins SW. Nonlethal oxidant injury to human retinal pigment epithelium cells causes cell membrane blebbing but decreased MMP-2 activity. Invest Ophthalmol Vis Sci. 2005; 46: 3331–3340. [DOI] [PubMed] [Google Scholar]
- 67. Sarks JP,, Sarks SH,, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye (Lond). 1988; 2 (pt 5): 552–577. [DOI] [PubMed] [Google Scholar]
- 68. Curcio CA,, Messinger JD,, Sloan KR,, McGwin G,, Medeiros NE,, Spaide RF. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: morphology, prevalence, topography, and biogenesis model. Retina. 2013; 33: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Crabb JW,, Miyagi M,, Gu X,, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Anderson DH,, Radeke MJ,, Gallo NB,, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010; 29: 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim DD,, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006; 118: 127–136. [DOI] [PubMed] [Google Scholar]
- 72. Thurman JM,, Renner B,, Kunchithapautham K,, Holers VM,, Rohrer B. Aseptic injury to epithelial cells alters cell surface complement regulation in a tissue specific fashion. Adv Exp Med Biol. 2010; 664: 151–158. [DOI] [PubMed] [Google Scholar]
- 73. Offen D,, Gilgun-Sherki Y,, Barhum Y,, et al. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J Neurochem. 2004; 89: 1241–1251. [DOI] [PubMed] [Google Scholar]
- 74. Grinberg L,, Fibach E,, Amer J,, Atlas D. N-acetylcysteine amide a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med. 2005; 38: 136–145. [DOI] [PubMed] [Google Scholar]
- 75. Ates B,, Abraham L,, Ercal N. Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radic Res. 2008; 42: 372–377. [DOI] [PubMed] [Google Scholar]
- 76. Tobwala S,, Khayyat A,, Fan W,, Ercal N. Comparative evaluation of N-acetylcysteine and N-acetylcysteineamide in acetaminophen-induced hepatotoxicity in human hepatoma HepaRG cells. Exp Biol Med (Maywood). 2015; 240: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Penugonda S,, Mare S,, Lutz P,, Banks WA,, Ercal N. Potentiation of lead-induced cell death in PC12 cells by glutamate: protection by N-acetylcysteine amide (NACA) a novel thiol antioxidant. Toxicol Appl Pharmacol. 2006; 216: 197–205. [DOI] [PubMed] [Google Scholar]
- 78. Amer J,, Atlas D,, Fibach E. N-acetylcysteine amide (AD4) attenuates oxidative stress in beta-thalassemia blood cells. Biochim Biophys Acta. 2008; 1780: 249–255. [DOI] [PubMed] [Google Scholar]
- 79. Herring JM,, McMichael MA,, Smith SA. Microparticles in health and disease. J Vet Intern Med. 2013; 27: 1020–1033. [DOI] [PubMed] [Google Scholar]
- 80. Colombo M,, Raposo G,, Théry C. Biogenesis secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014; 30: 255–289. [DOI] [PubMed] [Google Scholar]
- 81. Tahiri H,, Yang C,, Duhamel F,, Chemtob S,, Hardy P. Retinal pigment epithelium cell-derived microparticles mediate oxidative stress–induced retinal cells dysfunction. Acta Ophthalmologica. 2013; 91( suppl S252). [Google Scholar]
- 82. Brodsky SV,, Zhang F,, Nasjletti A,, Goligorsky MS. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol. 2004; 286: H1910–H1915. [DOI] [PubMed] [Google Scholar]
- 83. Raposo G,, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.