

Abstract
Purpose
We determined the phenotypic variation, disease progression, and potential modifiers of autosomal dominant retinal dystrophies caused by a splice site founder mutation, c.828+3A>T, in the PRPH2 gene.
Methods
A total of 62 individuals (19 families) harboring the PRPH2 c.828+3A>T mutation, had phenotype analysis by fundus appearance, electrophysiology, and visual fields. The PRPH2 haplotypes in trans were sequenced for potential modifying variants and generalized estimating equations (GEE) used for statistical analysis.
Results
Several distinct phenotypes caused by the PRPH2 c.828+3A>T mutation were observed and fell into two clinical categories: Group I (N = 44) with mild pattern dystrophies (PD) and Group II (N = 18) with more severe cone-rod dystrophy (CRD), retinitis pigmentosa (RP), and central areolar chorioretinal dystrophy (CACD). The PRPH2 Gln304-Lys310-Asp338 protein haplotype in trans was found in Group I only (29.6% vs. 0%), whereas the Glu304-Lys310-Gly338 haplotype was predominant in Group II (94.4% vs. 70.4%). Generalized estimating equations analysis for PD versus the CRD/CACD/RP phenotypes in individuals over 43 years alone with the PRPH2 haplotypes in trans and age as predictors, adjusted for correlation within families, confirmed a significant effect of haplotype on severity (P = 0.03) with an estimated odds ratio of 7.16 (95% confidence interval [CI] = [2.8, 18.4]).
Conclusions
The PRPH2 c.828+3A>T mutation results in multiple distinct phenotypes likely modified by protein haplotypes in trans; the odds of having the CACD/RP-like phenotype (versus the PD phenotype) are 7.16 times greater with a Glu304-Lys310-Gly338 haplotype in trans. Further functional studies of the modifying haplotypes in trans and PRPH2 splice variants may offer therapeutic targets.
Keywords: phenotype, genetic modifiers, retinal dystrophy
Mutations in the PRPH2 gene (OMIM:179605) are known to cause a wide range of autosomal dominant retinal dystrophies (adRD; phenotype MIM: 613105, 608133, 169150, 608161, 608133, 136880), but the source of this variation is poorly understood. We previously identified a PRPH2 (NM_000322.4) splice site mutation, c.828+3A>T, in numerous patients with retinal dystrophies.1 Haplotype analysis revealed that the mutation derives from a common ancestor and its prevalence is due to a founder effect.2 Individuals with the c.828+3A>T mutation express a PRPH2 splice variant that was not found in controls and is consistent with abnormal splicing. Clinical diagnoses in individuals with this mutation range from pattern dystrophies (PD) to retinitis pigmentosa (RP), cone-rod dystrophy (CRD), and/or unspecified macular dystrophy.2 This phenotypic variation is comparable to other mutations in PRPH2, for example, a 3-bp deletion of codon 153, and a P210R missense change3 that were reported to cause multiple phenotypes within the same nuclear families.4 Another PRPH2 mutation, R172W, also exhibits a founder effect but with similar phenotypes in most families,5,6 although intrafamilial variation also has been described in two families.7,8
In this study, we evaluated clinical records, fundus photographs, and visual function data to characterize the phenotype of the PRPH2 c.828+3A>T splice site mutation. Due to the founder effect, this cohort is particularly well suited for identifying possible causes of variation that are not in cis to the splice site mutation.
Peripherin/PRPH2 is a photoreceptor-specific membrane protein that homodimerizes with itself. Polymorphic protein haplotypes in trans were considered as modifiers of dominant diseases because the two alleles are expressed at the same time in the same tissues, translation is concurrent, and protein products may follow identical pathways and interact directly. Therefore, sequence variants in the PRPH2 gene in trans that code for polymorphic protein isoforms (haplotypes), Glu304-Lys310-Gly338 (G910-A929-G1013), Gln304-Arg310-Asp338 (C910-G929-A1013), and Gln304-Lys310-Asp338 (C910-A929-A1013), coded by three polymorphic cSNPs, rs390659 c.910G>C (GAG->CAG) p.Glu304Gln, rs425876, c.929A>G (AAG->AGG) p.Lys310Arg, and rs434102 c.1013G>A (GGC->GAC) p.Gly338Asp were considered as potential modifiers of the disease phenotype.2,9,10
We also considered 3 other candidate genes as modifiers, ROM1, GUCA1A, and NXNL1. ROM1 is a photoreceptor-specific gene that encodes a rod outer-membrane protein localized to the photoreceptor outer segment disk membranes. It has 35% homology to PRPH2 and interacts with PRPH2-forming heterotetramers.11 Mutations in this gene alone have not been shown conclusively to cause any retinal disease, although a digenic form of RP with a PRPH2 mutation has been reported.12 Therefore, ROM1 was considered as a strong candidate modifier gene, particularly in individuals with the retinitis pigmentosa phenotype. The GUCA1A gene encodes a photoreceptor-specific guanylate cyclase-activator protein, a novel Ca(2+) binding protein that stimulates synthesis of cGMP in photoreceptors.13 Mutations in this gene are known to cause autosomal dominant cone dystrophy, making it a likely modifier gene.14,15 Another gene, NXNL1, encodes a thioredoxin-like protein 6, which is specifically expressed in photoreceptors and required for cone viability.16 We considered GUCA1A and NXNL1 as potential modifier genes, especially in those patients exhibiting predominant macular disease and cone loss.
Methods
Subjects and Clinical Studies
All subjects were ascertained in the United States after obtaining written informed consent and all procedures conformed to the Declaration of Helsinki and were approved by the Institutional Review Board (IRB) board of each respective institution as described previously.2 We studied 19 families (FAM) harboring the splice site mutation. Families 1 to 10 were ascertained by David Birch, PhD, at the Retina Foundation of the Southwest (RFSW; Dallas, TX, USA); Fam11 by Richard Ruiz, MD, at University of Texas Health Science Center (Houston, TX, USA); Fam12 by Jonathon Sears, MD, at Cole Eye Institute (Cleveland, OH, USA); Fam13, Fam15, and Fam17 to Fam19 by Edwin Stone, MD, PhD, at the University of Iowa (Iowa City, IA, USA); Fam14 at the University of Oregon (Eugene, OR, USA) by Richard Weleber, MD; and Fam16 by Gerald Fishman, MD, at the University of Illinois (Chicago, IL, USA). Screening for sequence variants was performed at the Molecular Ophthalmology Laboratory at the University of Iowa and the Laboratory for Molecular Diagnosis of Inherited Eye Diseases at the University of Texas Health Science Center.
Clinical findings were analyzed to determine the phenotype of the individuals and classify them into defined diagnostic groups. Fundus photographs were available for 62 individuals from 19 families with the splice site mutation. To maintain consistent standards, we used electrophysiology data from RFSW alone for analysis. Electroretinographic (ERG) data were available for 25 individuals, dark adaptometry for 24 individuals, and Humphrey visual fields for 10 individuals.
Visual Acuity
Best-corrected visual acuity was recorded using an Early Treatment Diabetic Retinopathy Study (ETDRS) chart or standard Snellen visual acuity charts.
Visual Fields
Visual fields were tested at RFSW using standard spot size 3, program 30-2 and/or 60-4 SITA Fast on a Humphrey field analyzer (Humphrey Instruments, Inc., San Leandro, CA, USA).
Electrophysiology
Patients were dilated using tropicamide 1% and phenylephrine 2.5% and then dark-adapted for 45 minutes before testing. Usually only the eye with the poorer visual acuity was tested. The ERG was recorded from the dilated, dark-adapted eye following the International Society for Clinical Electrophysiology of Vision (ISCEV) standards as described previously.17
Dark Adaptometry
Patients' eyes were dilated using tropicamide 1% and phenylephrine 2.5% and dark-adapted completely for 45 minutes before testing. Final dark-adapted thresholds were measured on a Goldmann-Weekers adaptometer according to standard protocols.18–20
Fundus Photographs
Fundus photographs were obtained from each site whenever available. Phenotypic classification was based on fundus findings available in 62 individuals. All individuals with macular and/or peripheral yellow, orange, or gray (dot and halo) deposits were grouped into various types of PD as described previously.21–23 Patients with macular pigmentation and atrophy preceding peripheral retinal changes were classified as having CRD.24 If the fundus appeared to have peripheral intraretinal bone spicules, mottling, and granularity of the RPE, and/or had optic nerve pallor and retinal vessel attenuation, patients were classified as having RP.25 Individuals with symmetric, sharply outlined areas of geographic atrophy of the RPE and choroid, approximately 3 to 4 disc diameters in size, and having a yellow-white appearance of the choroidal vessels, were classified as central areolar chorioretinal dystrophy (CACD).26
These diagnoses were combined into two groups to analyze the effect of modifier genes: Group I – milder phenotype with PD: The presence of yellow or pigmented lesions in the retina and/or retinal atrophy confined to the macula alone; and Group II – more severe phenotype with CRD, RP and CACD: These were characterized by photoreceptor loss, RPE, and choroidal atrophy extending outside the macula and/or presence of intraretinal bone spicules.
Molecular Studies
DNA extraction and screening by SSCP or bidirectional fluorescent di-deoxy sequencing of the genes, PRPH2 (NM_000322.4), ROM1 (NM_000327.3), GUCA1A (NM_000409.3), and NXNL1 (NM_138454.1) were performed using standard conditions as described previously.1,27 PRPH2 haplotypes in trans were determined by subtraction of the known c.828+3A>T haplotype from the genotype determined via sequencing. Primers used in the study are available in Supplementary Table S1.
Statistical Analysis
Linear regression was used to analyze the relation between age and visual function parameters in patients with the splice site PRPH2 mutation. The best-fit values of the slope and intercept, along with their standard errors and 95% confidence intervals (CIs), were calculated. Associations between the two phenotype groups and the three protein haplotypes in trans were tested by Fisher's exact probability test. To confirm that significant associations were not due to age differences or familial clustering of genotypes, generalized estimating equation (GEE) models28 were fit using the “geepack” R package.29 These regressions modeled phenotype group as a function of haplotype and age via a logistic link, and robustly accounted for genetic correlation within families. An exchangeable correlation structure was specified for each familial cluster, but it is important to note that GEE models are robust even if the correlation structure is misspecified. For all haplotype analyses, we excluded all individuals younger than 43 years, taking into consideration the fact that the youngest age of an individual with the more severe phenotype in our cohort was 43 years.
Results
The presenting symptoms of the individuals with the c.828+3A>T splice site mutation include reduced, altered, or distorted central vision, sensitivity to bright lights, and reduced night vision.
Four distinct phenotypes were observed consistent with PDs, CRD, RP, or CACD with extensive intra- and interfamilial variation. Three types of PD, adult-onset vitelliform macular dystrophy (AVMD), butterfly-shaped macular dystrophy (BMD), and adult-onset flavimaculatus-like macular dystrophy (AOFMD) of three individuals in their 40s are shown in Figures 1A through 1C. Individuals having AVMD and BMD exhibited a milder phenotype at age 40 than the individual with AOFMD, who showed more severe ERG and field loss at age 28 years. A fluorescein angiogram shows hyperfluorescent lesions that are not visible by ophthalmoscope (Fig. 1C, inset). The absence of “dark choroid” distinguishes these from Stargardt's and fundus flavimaculatus caused by mutations in the ABCA4 gene.30 Individuals with more extensive vision loss, as in CRD, CACD, and RP phenotypes, are shown in Figures 2A through 2C.
Figure 1.

Fundus photographs, ERG, and Humphrey visual fields of three PD patients. (A) Adult-onset foveomacular vitelliform-like PD: 46-year-old female (Fam7-5790) showing lipofuscin deposits in the macula, normal rod and cone responses, very minimal field loss. (B) Butterfly-like PD: 49-year-old female (Fam1-214) showing lipofusin deposits in the macula and in the periphery, decreased rod and cone responses, moderate peripheral field loss. (C) Flavimaculatus-like PD: 28-year-old female (Fam2-4957) showing peripheral deposits (seen better in fluorescein inset), decreased rod responses but normal cone responses, mild peripheral field loss.
Figure 2.

Fundus photographs, ERG, and visual fields in more severe retinal dystrophies. (A) Cone-rod dystrophy in a 58-year-old female (Fam1-212) showing photoreceptor and RPE atrophy, reduced rod and cone responses with central field loss, and relative preservation of the peripheral field. (B) Central areolar chorioretinal dystrophy in a 56-year-old male (Fam1-309) showing geographic atrophy of photoreceptors, RPE, and choriocapillaries atrophy, with sparing of peripheral retina with corresponding Goldmann III4e visual field. (C) Retinitis pigmentosa in a 60-year-old female (Fam8-6138) showing pigmentary changes in the periphery with a generalized loss of photoreceptors and RPE, loss of rod function more than cone, and peripheral visual field loss with relative central sparing.
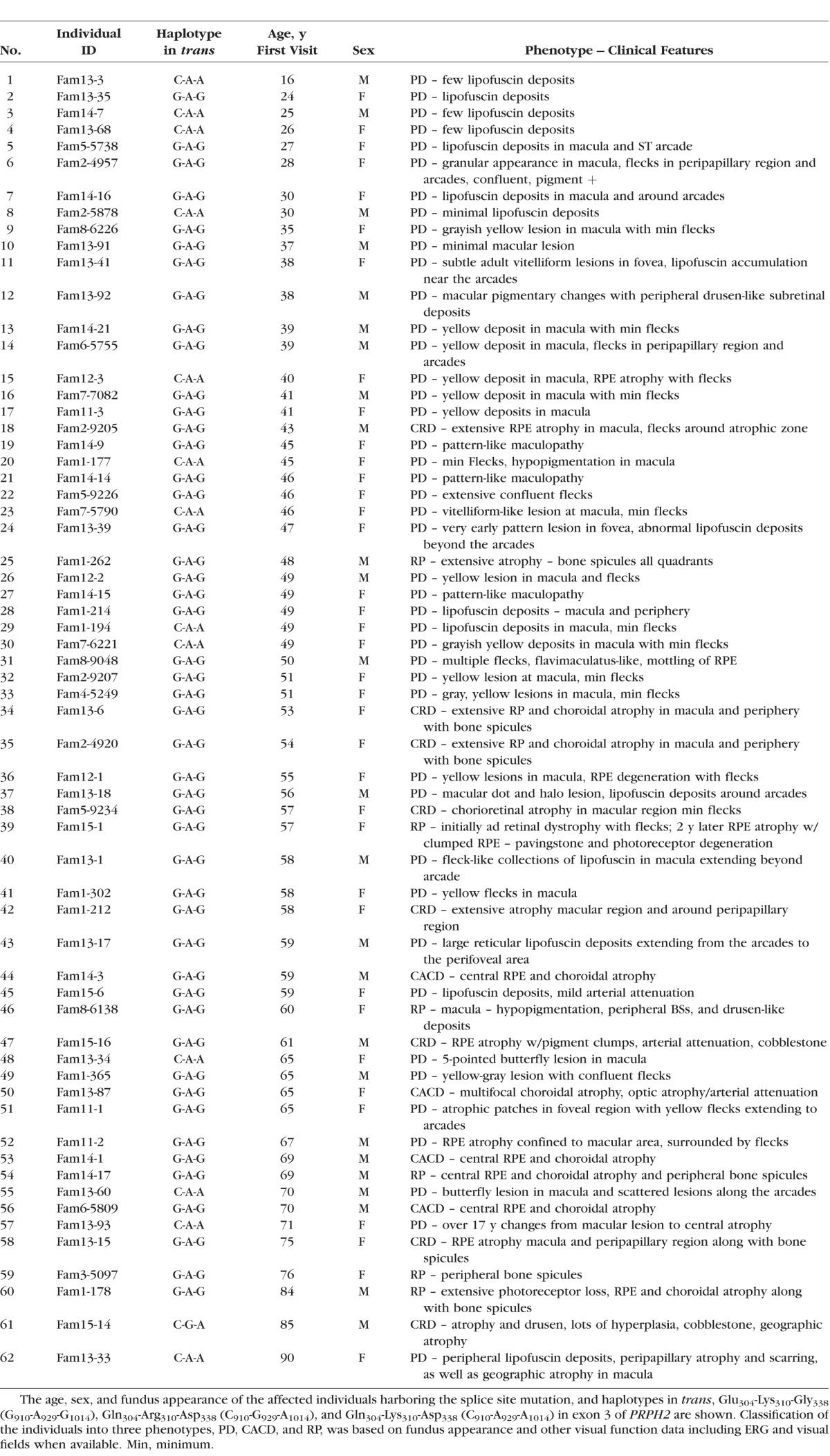
The fundus features of 62 individuals, along with their diagnoses, age, and sex, and the PRPH2 mini-haplotypes in trans, are shown in Table 1 and pedigrees for 14 families are available in the Supplementary Material. Of the individuals, 44 were graded as Group I and had the clinical diagnosis of PD, and 18 were graded as Group II and had CRD, RP, or CACD.
Table 1.
Phenotype and Haplotypes of PRPH2 Gene in trans to the Splice Site Mutation
Age
The age range of our patients was 16 to 90 years, with majority in the 40- to 50-year range. The youngest individual examined, a 16-year-old male, had minimal yellow lesions in the peripheral retina and the oldest individual, a 90-year-old female, had geographic atrophy in the macula; both belonged to the same family. The majority of the individuals with the splice site mutation fell into the PD Group I (44), with ages ranging from the 20s to the 70s (Table 1). Individuals having a fundus appearance suggestive of CRD (8), RP (6), and CACD (4) were in their fifth to sixth decades of life (Table 1).
Visual Acuity
Visual acuity data were available for 46 individuals with the splice site mutation, and the best-corrected visual acuity of the better eye was used for plotting (Fig. 3A). Most of the PD patients retain fairly good visual function, with their acuity remaining above 20/40 (0.5) until 65 years of age (green symbols). There was a modest decrease of visual acuity in PD with age due to photoreceptor and RPE atrophy in the macular region (Fig. 3A, solid line). In contrast, patients with CACD and CRD exhibited significantly lower visual acuity (red symbols), with more than half of the patients over age 45 having VA below 20/100 (0.2). Retinitis pigmentosa patients had VA ranging from 20/40 to 20/400 (0.2 to <0.08).
Figure 3.

Visual function in patients with PD (solid green circles; solid regression line) and widespread dystrophy (open red circles). (A) Visual acuity (decimal) as a function of age. (B) Final dark-adapted threshold as a function of age. Dashed line is upper limit of normal. (C) Cone b-wave amplitude to ISCEV standard 30-Hz flicker. Lines connect repeated measurements in a patient. Solid regression is linear fit to amplitudes for PD. Dashed line is lower limit of normal (P < 0.05). (D) Rod b-wave amplitude to ISCEV standard flash. Lines connect repeated measurements in a patient. Solid regression is linear fit to amplitudes for PD. Dashed line is lower limit of normal (P < 0.05).
Visual Fields
Humphrey visual fields were available in 10 individuals. Individuals with PD had better visual fields than those with CRD, CACD, or RP (Figs. 1, 2). Individuals with vitelliform-like PD had the most well-preserved visual fields (Fig. 1A). Butterfly-type PD (Fig. 1B) and flavimaculatus-like PD (Fig. 1C) showed preserved central and mild to moderate peripheral field loss. The CRD and CACD patients had central and paracentral scotoma (Figs. 2A, 2B) and RP patients had primarily peripheral visual field loss progressing to loss of central visual fields as expected (Fig. 2C).
Dark Adaptometry
Final dark-adapted thresholds were available from 24 patients (Fig. 3B). The majority of patients with PD (green symbols) had thresholds near the upper limit of normal. Thresholds were elevated approximately 2 to 4 log units above normal in individuals with the RP, CRD, and CACD phenotypes (red symbols).
Electrophysiology
Electroretinographic data were available for 25 individuals, with many patients tested on multiple occasions. Summary ERG parameters are shown in Table 2 and Figure 3. In general, cone responses were affected less than rod responses. As shown in Figure 3C, patients with PD showed a gradual loss with age of cone amplitude to 30 Hz flicker. The solid green curve is the regression line for cone amplitudes as a function of age on the first visit only. Lines connecting green symbols show repetitive measures on the same individual, with mild decline evident between visits, especially after 40 years of age. In contrast, red symbols show that patients with CRD, CACD, and RP typically had smaller cone amplitudes than patients with PD. Cone b-wave implicit times showed greater delays in patients with more widespread disease than in patients with PD, where cone implicit times were only slightly delayed.
Table 2.
Mean and Standard Deviations of ERG Values for Affected (Aff) Individuals Harboring the c.828+3A>T Splice Site Mutation and Normal (Nor) Controls Are Shown
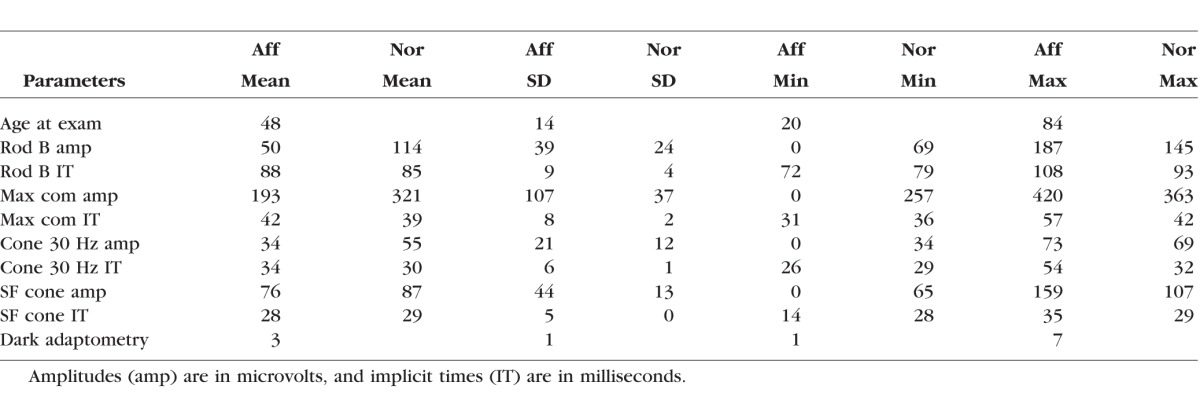
Figure 3D shows rod amplitudes as a function of age. All individuals with rod amplitudes significantly below normal (P < 0.05), with the exception of one individual with PD, had RP, CRD, or CACD (red symbols). Individuals of various subgroups of PD did not show any characteristic pattern of photoreceptor loss. Two individuals who had normal and near-normal amplitudes had vitelliform-like PD (Fig. 1A, #5790) and butterfly-like PD (Fig. 1B, #214). These findings suggested that, although the distribution of flecks results in a varied fundus appearance, the photoreceptor loss does not correspond to fundus appearance.
Long-term follow-up of some individuals over several years show differences in severity of progression. Patients with PD showed milder progressive vision loss (Fig. 4) than patients with CACD/RP-like phenotype (Fig. 5).
Figure 4.

Pattern dystrophy in a female aged 36 years (Fam8-6226). Milder vision loss and slow progression over a 9-year period, with relatively preserved rod and cone function at age 45. Lower limits of normal (P < 0.05) are 85 μV for rod ERG, 200 μV for standard combined ERG, 50 μV for 30-Hz flicker ERG, and 75 μV for single flash cone ERG.
Figure 5.

Retinitis pigmentosa in a male aged 48 years (Fam1-365). Rapid progression over a 20-year period from relatively preserved photoreceptors and RPE to a complete loss of rod and greatly reduced cone function. Lower limits of normal (P < 0.05) are 85 μV for rod ERG, 200 μV for standard combined ERG, 50 μV for 30-Hz flicker ERG, and 75 μV for single flash cone ERG.
Modifier Gene Screening
Peripherin/PRPH2.
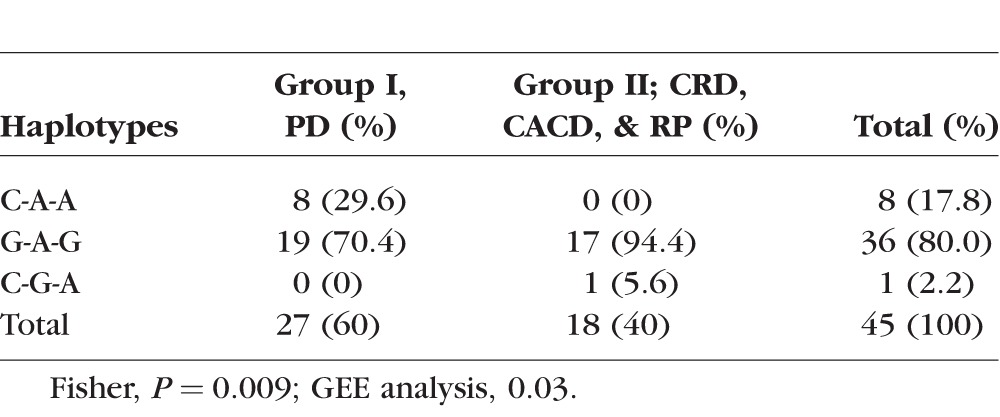
The distribution of PRPH2 (NM_000322.4) haplotypes in trans for PD, CRD, CACD, and RP phenotypes are shown in Tables 3 and 4. For individuals over 43 years, in Group I, representing milder disease with different types of PDs, Gln304-Lys310-Asp338 (C910-A929-A1014) was identified in eight individuals (29.6% vs. 0%) and 19 had Glu304-Lys310-Gly338 (G910-A929-G1013). In Group II, with CRD, RP, and CACD, 17 individuals had Glu304-Lys310-Gly338 (G910-A929-G1013; 94.4% vs. 70.4%), and one had Gln304-Arg310-Asp338 (C910-G929-A1013). Gln304-Lys310-Asp338 (C910-A929-A1013) was absent (Table 4). A significant difference in the haplotype distribution between the two phenotypic groups among individuals older than 43 years was seen using Fisher's exact probability test (P = 0.0092). A GEE analysis that accounted for differences in age and familial correlation of genotypes confirmed that the increased frequency of the Glu304-Lys310-Gly338 haplotype among individuals with CRD, RP, and CACD was significant with a relevant P value of <2e-16. However, because the asymptotic P value may not be robust to the “zero” cell counts in two of the haplotype-phenotype categories (see Tables 3, 4), we reran the GEE model taking the conservative approach of collapsing the Gln304-Lys310-Asp338 (C-A-A) and Gln304-Arg310-Asp338 (C-G-A) haplotypes into a single category (P = 0.03, N = 45 due to removal of individuals with age <43). The estimated odds ratio for this comparison is 7.16 (95% CI = [2.8–18.4]), suggesting that the odds of having the CACD/RP-like phenotype (versus the PD phenotype) are 7.16 times greater for affected individuals with a Glu304-Lys310-Gly338 haplotype (G-A-G) haplotype in trans than for affected individuals with one of the other two haplotypes in trans (Gln304-Lys310-Asp338 [C-A-A] and Gln304-Arg310-Asp338 [C-G-A]).
Table 3.
Frequency of Haplotypes in trans in the Two Groups
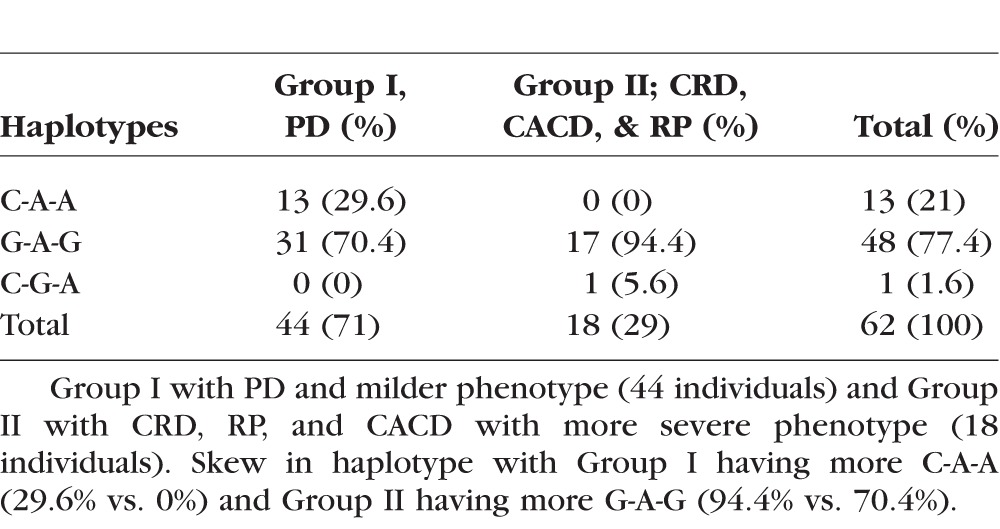
Table 4.
Individuals Older than Age 43 Were Used for Statistical Analysis and Continue to Exhibit the Skew in Haplotype
ROM1 Gene.
No significant sequence variants were identified by bidirectional sequencing of ROM1 (NM_000327.3), in 10 affected individuals representing different phenotypes.
GUCA1A Gene.
A GUCA1A (NM_000409.3), c.149C>T (p.Pro50Leu), sequence variant was identified in 4 individuals (3 affected—1 with macular atrophy and 2 with PD; 1 individual had normal retina) by bidirectional sequencing of GUCA1A in 62 individuals.
NXNL1 Gene.
Several variants including c.46G>A (p.Asp16Asn), c.83G>C (p.Arg28Pro), c.172_174delTTC, (p.delPhe58), c.190G>A (p.Glu64Lys), and c.461A>T (p.Glu154Val), were identified by bidirectional screening of NXNL1 (NM_138454.1) in 62 affected individuals and 103 CEPH normal controls. However, none showed any significant association.
Discussion
This study demonstrated that the c.828+3A>T splice site mutation in the PRPH2 gene is the primary cause of retinal disease in all 62 affected individuals in these 19 families with complete penetrance but highly variable expressivity. It results in clinically diverse, autosomal dominant retinal dystrophies, including PDs, CRD, CACD, and RP (Figs. 1, 2), in 19 families derived from a common ancestor.2 The phenotypes fall into two broad clinical categories: Individuals in Group I (N = 44; AVMD, BMD, and AOFMD types of PD) have milder disease than individuals in Group II (CRD, CACD, and RP). There is limited progression from Groups I to II. In our cohort, almost all individuals had reduced photoreceptor responses relative to age-matched controls, regardless of the phenotype (Fig. 3).
We previously had established a founder effect for the PRPH2 c.828+3A>T mutation by haplotype analysis which excludes sequence variants in cis within 5 megabases of the PRPH2 locus as modifiers.2 In this study, we analyzed the haplotypes in trans and showed that Gln304-Lys310-Asp338 (C910-A929-A1014) is significantly associated with Group I pattern dystrophies and Glu304-Lys310-Gly338 (G910-A929-G1013) with Group II (P value = 0.03 taking family structure, progression with age and restricting our analysis to only individuals (N = 45) older than 43 years of age. This suggested the possible modifier role of the polymorphic protein haplotypes in trans, with an estimated 7.16-fold increase in the odds of the more severe phenotype for affected individuals harboring a Glu304-Lys310-Gly338 (G-A-G) haplotype in trans compared to other affected individuals, and a possible protective role for having a second Gln304-Lys310-Asp338 (C-A-A) haplotype in trans. Given that the two alleles are similar in their temporal and spatial expression and interact directly, the protein haplotype in trans is likely to contribute to the phenotypic variation seen in the PRPH2 splice site mutation families.
Several sequence variants were found in the other potential modifier genes that were screened; however, none of them had a significant association with any one phenotype. Progressive photoreceptor loss with age explains continued vision loss in some individuals. Another possible modifier may be variations in expression of normal versus aberrant transcripts due to leaky aberrant splicing.2 Functional studies with more accurate quantification of the truncated protein and its interaction with different RDS protein mini-haplotypes may help determine the effect of variable splicing and protein mini-haplotypes in modifying the retinal phenotype. Future studies with larger number of patients and whole exome sequencing of additional retinal disease-causing genes and/or whole exome and genome sequencing may identify additional modifiers.
This study demonstrated the variability of autosomal dominant retinal dystrophies resulting from PRPH2 mutations and the possibility of common polymorphic protein variants functioning as modifiers of disease-causing mutations in Mendelian disorders. Determining the protein haplotype in trans in these families may help predict the severity of the retinal phenotype and may aid in counseling of the families. The potential for splice site modification also is of therapeutic interest in preserving vision in the affected individuals and needs further evaluation.
Supplementary Material
Acknowledgments
The authors thank Jean Andorf for excellent technical assistance and critical review of the manuscript; Petra Kozma, MD, and Kirsten Locke for their help coordinating and obtaining patient information; Richard Ruiz, MD, the Ruiz Department of Ophthalmology and Visual Sciences, University of Texas Health Science Center Houston, Gerald Fishman, MD, Chicago Lighthouse for the Blind and Visually Impaired; Jonathon Sears, MD, Cole Eye Institute, and Richard Weleber, MD, Casey Eye Institute, for providing access to patients and clinical information. The authors also thank the patients and their families for participating in our study.
Supported in part by an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology, Emory University. Supported by grants from the Foundation Fighting Blindness, William Stamps Farish Fund, the Hermann Eye Fund, Howard Hughes Medical Institute, and by National Institutes of Health (NIH; Bethesda, MD, USA) Grants EY007142 (SPD) and EY09076 (DGB). Research to Prevent Blindness provides an unrestricted grant to the Department of Ophthalmology, Emory University (P30EY006360, SPS).
Disclosure: S.P. Shankar, Emory (E); D.K. Hughbanks-Wheaton, None; D.G. Birch, Shire (C), Acucela (C), NightStarX (C), AGTC (C), Thrombogenics (C), Allergan (C), QLT (C); L.S. Sullivan, None; K.N. Conneely, None; S.J. Bowne, None; E.M. Stone, None; S.P. Daiger, None
References
- 1. Sohocki MM,, Daiger SP,, Bowne SJ,, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001; 17: 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shankar SP,, Birch DG,, Ruiz RS,, et al. Founder effect of a c.828+3A>T splice site mutation in peripherin 2 (PRPH2) causing autosomal dominant retinal dystrophies. JAMA Ophthalmol. 2015; 133: 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gorin MB,, Jackson KE,, Ferrell RE,, et al. A peripherin/retinal degeneration slow mutation (Pro-210-Arg) associated with macular and peripheral retinal degeneration. Ophthalmology. 1995; 102: 246–255. [DOI] [PubMed] [Google Scholar]
- 4. Weleber RG,, Carr RE,, Murphey WH,, Sheffield VC,, Stone EM. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993; 111: 1531–1542. [DOI] [PubMed] [Google Scholar]
- 5. Payne AM,, Downes SM,, Bessant DA,, Bird AC,, Bhattacharya SS. Founder effect seen in the British population, of the 172 peripherin/RDS mutation-and further refinement of genetic positioning of the peripherin/RDS gene. Am J Hum Genet. 1998; 62: 192–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Downes SM,, Fitzke FW,, Holder GE,, et al. Clinical features of codon 172 RDS macular dystrophy: similar phenotype in 12 families. Arch Ophthalmol. 1999; 117: 1373–1383. [DOI] [PubMed] [Google Scholar]
- 7. Michaelides M,, Holder GE,, Bradshaw K,, Hunt DM,, Moore AT. Cone-rod dystrophy intrafamilial variability, and incomplete penetrance associated with the R172W mutation in the peripherin/RDS gene. Ophthalmology. 2005; 112: 1592–1598. [DOI] [PubMed] [Google Scholar]
- 8. Poloschek CM,, Bach M,, Lagreze WA,, et al. ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2010; 51: 4253–4265. [DOI] [PubMed] [Google Scholar]
- 9. Connell GJ,, Molday RS. Molecular cloning, primary structure, and orientation of the vertebrate photoreceptor cell protein peripherin in the rod outer segment disk membrane. Biochemistry. 1990; 29: 4691–4698. [DOI] [PubMed] [Google Scholar]
- 10. Travis GH,, Sutcliffe JG,, Bok D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron. 1991; 6: 61–70. [DOI] [PubMed] [Google Scholar]
- 11. Bascom RA,, Manara S,, Collins L,, Molday RS,, Kalnins VI,, McInnes RR. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron. 1992; 8: 1171–1184. [DOI] [PubMed] [Google Scholar]
- 12. Kajiwara K,, Berson EL,, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994; 264: 1604–1608. [DOI] [PubMed] [Google Scholar]
- 13. Subbaraya I,, Ruiz CC,, Helekar BS,, et al. Molecular characterization of human and mouse photoreceptor guanylate cyclase-activating protein (GCAP) and chromosomal localization of the human gene. J Biol Chem. 1994; 269: 31080–31089. [PubMed] [Google Scholar]
- 14. Payne AM,, Downes SM,, Bessant DA,, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998; 7: 273–277. [DOI] [PubMed] [Google Scholar]
- 15. Sokal I,, Dupps WJ,, Grassi MA,, et al. A novel GCAP1 missense mutation (L151F) in a large family with autosomal dominant cone-rod dystrophy (adCORD). Invest Ophthalmol Vis Sci. 2005; 46: 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leveillard T,, Mohand-Said S,, Lorentz O,, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004; 36: 755–759. [DOI] [PubMed] [Google Scholar]
- 17. Birch DG,, Fish GE. Rod ERGs in retinitis pigmentosa and cone-rod degeneration. Invest Ophthalmol Vis Sci. 1987; 28: 140–150. [PubMed] [Google Scholar]
- 18. Birch DG,, Peters AY,, Locke KL,, Spencer R,, Megarity CF,, Travis GH. Visual function in patients with cone-rod dystrophy (CRD) associated with mutations in the ABCA4(ABCR) gene. Exp Eye Res. 2001; 73: 877–886. [DOI] [PubMed] [Google Scholar]
- 19. Fishman GA,, Farbman JS,, Alexander KR. Delayed rod dark adaptation in patients with Stargardt's disease. Ophthalmology. 1991; 98: 957–962. [DOI] [PubMed] [Google Scholar]
- 20. Glenn AM,, Fishman GA,, Gilbert LD,, Derlacki DJ. Effect of vitamin A treatment on the prolongation of dark adaptation in Stargardt's dystrophy. Retina. 1994; 14: 27–30. [DOI] [PubMed] [Google Scholar]
- 21. Aaberg TM,, Han DP. Evaluation of phenotypic similarities between Stargardt flavimaculatus and retinal pigment epithelial pattern dystrophies. Trans Am Ophthalmol Soc. 1987; 85: 101–119. [PMC free article] [PubMed] [Google Scholar]
- 22. Deutman AF,, van Blommestein JD,, Henkes HE,, Waardenburg PJ,, Solleveld-van Driest E. Butterfly-shaped pigment dystrophy of the fovea. Arch Ophthalmol. 1970; 83: 558–569. [DOI] [PubMed] [Google Scholar]
- 23. Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974; 72: 139–156. [PMC free article] [PubMed] [Google Scholar]
- 24. Krill AE,, Deutman AF,, Fishman M. The cone degenerations. Doc Ophthalmol. 1973; 35: 1–80. [DOI] [PubMed] [Google Scholar]
- 25. Heckenlively J. Retinitis pigmentosa. Philadelphia PA: J.B. Lippincott; 1988: 68–89. [Google Scholar]
- 26. Gass JDM. Stereoscopic Atlas of Macular Diseases Diagnosis and Treatment. Maryland Heights, MO: Mosby; 1997: 334–336. [Google Scholar]
- 27. Webster AR,, Heon E,, Lotery AJ,, et al. An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2001; 42: 1179–1189. [PubMed] [Google Scholar]
- 28. Hardin J,, Hilbe J. Generalized Estimating Equations. London: Chapman and Hall/CRc.; 2003. [Google Scholar]
- 29. Halekoh U,, Højsgaard S,, Yan J. The R package geepack for generalized estimating equations. J Stat Software. 2006; 15: 1–11. [Google Scholar]
- 30. Fish G;,, Grey R,, Sehmi KS. AC B. The dark choroid in posterior retinal dystrophies. Br J Ophthalmol. 1981; 65: 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.