

Abstract
Background:
Pyroptosis is the term for caspase-1-dependent cell death associated with pro-inflammatory cytokines. The role of alveolar macrophage (AM) pyroptosis in the pathogenesis of the acute lung injury and acute respiratory distress syndrome (ALI/ARDS) remains unclear.
Methods:
C57BL/6 wild-type mice were assigned to sham, lipopolysaccharide (LPS) + vehicle, LPS + acetyl-tyrosyl-valyl- alanyl-aspartyl-chloromethylketone (Ac-YVAD-CMK) and LPS + Z-Asp-Glu-Val-Asp-fluoromethylketone groups. Mice were given intraperitoneal (IP) injections of LPS. Drugs were IP injected 1 h before LPS administration. Mice were sacrificed 16 h after LPS administration, and AMs were isolated. Western blot analysis for active caspase-1 and cleaved caspase-3, evaluation of lung injury and a cytokine release analysis were performed. AMs were treated with LPS and adenosine triphosphate (ATP); caspase-1-dependent cell death was evaluated using flow cytometry; the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) pyroptosomes were examined by immunofluorescence.
Results:
The expression of activated caspase-1 in AMs was enhanced following LPS challenge compared with the sham group. In the ex vivo study, the caspase-1/propidium iodide-positive cells, caspase-1 specks and ASC pyroptosomes were up-regulated in AMs following LPS/ATP stimulation. The specific caspase-1 inhibitor Ac-YVAD-CMK inhibited the activation of caspase-1 and pyroptotic cell death. Ac-YVAD-CMK also reduced the lung injury, pulmonary edema and total protein in bronchoalveolar lavage fluid (BALF). In addition, Ac-YVAD-CMK significantly inhibited interleukin-β (IL-1β) release both in serum and BALF and reduced the levels of IL-18, tumor necrosis factor-α (TNF-α), High Mobility Group Box 1 (HMGB1) in BALF during LPS-induced ALI/ARDS.
Conclusions:
This study reported AM pyroptosis during LPS-induced ALI/ARDS in mice and has demonstrated that Ac-YVAD-CMK can prevent AM-induced pyroptosis and lung injury. These preliminary findings may form the basis for further studies to evaluate this pathway as a target for prevention or reduction of ALI/ARDS.
Keywords: Acute Lung Injury/Acute Respiratory Distress Syndrome, Alveolar Macrophage, Caspase-1, Pyroptosis
INTRODUCTION
Acute lung injury and acute respiratory distress syndrome (ALI/ARDS) is a complex and severe disorder of the lungs and remains a leading cause of death in the Intensive Care Unit.[1] In a recent meta-analysis of 72 studies from 1994 to 2006, the overall pooled morality rate of 11,426 patients with ALI/ARDS was 43%.[2] Despite the use of supportive pulmonary mechanical ventilation, effective prevention, reduction and treatment of ALI/ARDS remain clinically elusive.[3]
Pathophysiologically, ARDS is characterized by a severe acute inflammatory process that induces increased alveolar permeability and pulmonary edema, accumulation of leukocytes, diffuse alveolar damage and epithelial injury.[4] Apoptotic cell death has been considered to be an underlying mechanism in ALI.[5] However, apoptosis is a form of cell death that lacks an associated inflammatory response, and so apoptosis alone does not explain the severe inflammatory response in the lung that is so characteristic of both ALI/ARDS.
Recently, a new form of programmed cell death has been described - pyroptosis. Pyroptosis is a caspase-1-dependent cell death with morphological and biochemical features of necrosis and apoptosis. Pyroptosis is found in monocytes, macrophages, and dendritic cells when associated with infection.[6,7] During pyroptosis, activated macrophages undergo caspase-1-dependent, nuclease-mediated cleavage of DNA, without the characteristic oligonucleosomal fragmentation pattern associated with apoptosis.[6] In contrast to apoptosis, pyroptosis is associated with potassium efflux, water influx, cell swelling, release of pro-inflammatory intracellular contents and formation of pores (1–2 nm) in the plasma membrane, which are positive for propidium iodide (PI) staining.[8,9] Following inflammasome pathway activation, pro-caspase-1 is processed into two cleaved subunits called p10 and p20, leading to extracellular release of pro-inflammatory cytokines, including interleukin-β (IL-1β) and IL-18.[10,11] IL-1β, a key inflammatory cytokine involved in host defense against pathogens, acts as a “gate keeper” of inflammation.[12,13]
Lipopolysaccharide (LPS) is a major component of the outer membrane of Gram-negative bacteria. LPS and adenosine triphosphate (ATP) can work together to induce the formation of an NLRP3 inflammasome, a Nod-like receptor forming a complex composed of apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and caspase-1.[14,15] This leads to oligomerization and assembly of a high-molecular-weight multimeric inflammasome complex, which results in the conversion of pro-caspase-1 into catalytically active form.
Activation of the NLRP3 inflammasome in alveolar macrophages (AMs) leads to caspase-1 activation and IL-1β production, which aggravates lung injury during mechanical ventilation in patients with ALI.[16] In patients with ARDS, caspase-1, IL-18, and IL-1β expression increase in the peripheral blood and serve as novel biomarkers of morbidity and mortality. Genetic deletion of caspase-1 reduces lung injury in mice and results in resistance to LPS-induced endotoxic shock.[17,18] Inhibitors of the cysteine protease caspase-1 have been considered as potential therapeutic approaches in ALI/ARDS.[18] The caspase-1 inhibitor acetyl-tyrosyl-valyl-alanyl-aspartyl- chloromethylketone (Ac-YVAD-CMK) can reduce LPS-induced tissue damage in rats.[19] These findings indicate that the activation of caspase-1 in AMs has a role in the development of ARDS. Therefore, the inhibition of pyroptosis in AMs may be a future approach to the clinical prevention or treatment of ALI/ARDS in patients.
AMs account for 90% of the cells in bronchoalveolar lavage fluid (BALF), forming the first line of defense against airborne particles and microbes. They are the main immune cells to recognize pathogen-associated molecular patterns and trigger innate immunity and host defense.[20,21] Recent studies support the importance of macrophages in the initiation of the inflammatory response during ALI/ARDS, as well as the resolution of lung inflammation and repair.[20] Pyroptosis has been widely observed in infected macrophages. Because the role of pyroptosis and the AM in ALI/ARDS remains to be elucidated, we chose a murine study model of LPS-induced ALI.
Caspase-1 activation in AMs has been observed during the development of ALI/ARDS, and NLRP3/ASC inflammasome activation participates in this process; treatment with Ac-YVAD-CMK has been shown to reduce LPS-induced lung injury in mice. These findings supported the approach of this in vivo mouse study to investigate a component of the complex mechanism of ALI/ARDS, namely AM pyroptosis.
METHODS
Materials
LPS (Escherichia coli 0111:B4) and the specific caspase-1 inhibitor
Ac-YVAD-CMK were obtained from Sigma-Aldrich (St. Louis, MO, USA). The specific caspase-3 inhibitor Z-Asp-Glu-Val-Asp-fluoromethylketone (Z-DEVD-FMK) was purchased from BioVision (Milpitas, CA, USA). Rabbit polyclonal caspase-1 P10 (M-20) antibody was from Santa Cruz (Santa Cruz, CA, USA). Rabbit cleaved caspase-3 and GAPDH antibody were all obtained from Cell Signaling (Boston, MA, USA). Rabbit NLRP3 antibody was obtained from NOVUS (Littleton, CO, USA). Rabbit polyclonal ASC antibody was obtained from Immunoway (Newark, NJ, USA). FLICA® 660 Caspase-1 Assay Kit, far-red fluorescence was obtained from ImmunoChemistry (Blooming, MN, USA), and PI from Life Technologies (Carlsbad, CA, USA).
Animals
C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mice used in this study were male, 8–10 weeks of age and weighed between 25 g and 30 g. Animals were maintained in a specific pathogen-free, laminar-flow housing apparatus under controlled temperature, humidity and 12-h light/dark regimen. All animal protocols were approved by the Animal Care and Use Committee of the Central South University. All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
In vivo experimental design
Eighty mice were randomly assigned to four groups: Sham (n = 20); LPS + vehicle (n = 20); LPS + AC-YVAD-CMK (n = 20) and LPS + Z-DEVD-FMK (n = 20). The ALI/ARDS model was induced by intraperitoneal (IP) injection with a lethal dose of LPS (20 mg/kg) in mice. Sham mice received injections of sterilized phosphate-buffered saline (PBS). Caspase-1 inhibitor Ac-YVAD-CMK (in PBS containing 1% DMSO) or caspase-3 inhibitor Z-DEVD-FMK (in PBS containing 1% DMSO) was injected at a dose of 6.5 mg/kg 1 h before administration of LPS. Sixteen hours following PBS or LPS administration, the animals were sacrificed after blood sample had been collected. Following euthanasia, the lungs (n = 6 per group) were excised from the mice by opening the chest via a median sternotomy. The wet weight (W) of the left lung was measured using an electronic scale and then desiccated in an oven at 65°C for 48 h to determine the dry weight (D). The water content was obtained by calculating the W/D weight ratio. The right lung was removed and fixed in 4% paraformaldehyde (PFA) for 24 h. Lungs were also lavaged with 0.5 ml of sterile saline each time through an intratracheal catheter as descried previously,[22] and a total of 2 ml of bronchoalveolar lavage was instilled and withdrawn from each mouse to detect the total protein level in the BALF. The BALF and serum were collected for the assays of IL-1β, IL-18, High Mobility Group Box 1 (HMGB1) and tumor necrosis factor-α (TNF-α).
Isolation of alveolar macrophages and cell culture
The mouse lungs were lavaged with 1 ml of sterile saline through an intratracheal catheter, and a total of 10 ml of bronchoalveolar lavage was instilled and withdrawn from each mouse. The BALF was centrifuged at 300 ×g for 10 min at 4°C to pellet the AMs. The pelleted cells were resuspended and cultured in 60-mm sterilized polystyrene Petri dishes for 3 h at 37°C in an atmosphere of 5% CO2. Primary AMs were isolated from the lavage for further study in vitro. The mouse AMs were cultured in RPMI1640 supplemented with 10% fetal bovine serum, 1 mmol/L glutamine, 10 mmol/L 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid, 50 U/ml penicillin and 50 μg/ml streptomycin. After 3 h of adherence, the cells adhering to the bottom of dish were washed twice, and the total number of AMs in BALF was calculated. Then whole cell lysates were harvested and subjected to western blotting. In vitro AMs were also stimulated for 5 h with or without LPS (500 ng/ml) and ATP (5 mmol) added during the last hour of culture, in the absence or presence of Ac-YVAD-CMK (50 μmol).
Cellular protein extraction and western blotting
Collected cells were centrifuged at 5000 rpm for 5 min. These pellets were then resuspended on ice with RIPA lysis buffer containing proteinase inhibitors for 30 min, then centrifuged at 12,000 rpm for 10 min at 4°C. Protein concentration was quantitated with bicinchoninic acid protein assay reagent (Pierce Chemical Co., Rockford, IL, USA), and 25 μg of protein per sample was mixed with sample loading buffer and boiled for 8 min. Protein samples were electrophoresed in 12% sodium dodecyl sulfate-polyacrylamide gel and transferred onto polyvinylidene fluoride membranes (Bio-Rad Laboratories, Berkeley, CA, USA). After blocking with 5% nonfat milk in TBS-T for 1 h at room temperature, membranes were incubated overnight at 4°C with primary antibodies against caspase-1 P10 (1:1000), cleaved caspase-3 (1:1000), GAPDH (1:2000) and NLRP3 (1:200). After three washes, the membranes were incubated with the secondary antibody conjugated with horseradish peroxidase at room temperature for 1 h. The blots were developed with the Super Signal chemiluminescent substrate (Pierce Chemical Co.) and exposed to film.
Confocal immunofluorescence imaging
AMs were placed onto glass cover slides, fixed with 4% PFA and permeabilized with 0.1% Triton X-100 for 10 min and blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. Slides were incubated with rabbit polyclonal ASC antibody (1:200 in 1% BSA in PBS) overnight at 4°C. After being washed three times with PBS, the cells were stained with Alexa555-conjugated secondary antibody (molecular probes, diluted 1:1000) for 90 min at room temperature, followed by nuclear counterstaining with 4’,6-diamidino-2-phenylindole (DAPI). Slides were visualized with an Olympus Fluoroview 500 confocal microscope.
Hematoxylin and eosin staining
A portion of the lung from the right lobe was fixed in 4% PFA and embedded in paraffin wax for histopathological examination. Sections of 5 μm thickness were placed onto glass slides and stained with hematoxylin and eosin. The stained lung tissue sections were viewed with a light photomicroscope and were evaluated for pathological changes using a double-blind method. The severity of lung injury was evaluated using a semi-quantitative histological index, as has been described previously.[23] The histological index of lung damage included alveolar edema, hemorrhage, alveolar septal thickening and infiltration of polymorphonuclear leukocytes. Each item was divided into four grades from 0 to 3 (0 = normal; 1 = mild; 2 = moderate; 3 = severe) and then calculated for a total ALI score.[23]
Quantification of inflammatory indicators
IL-1, IL-18, TNF-α and HMGB1 levels in the serum and BALF were determined using a commercially available mouse IL-1β, IL-18 and TNF-α enzyme-linked immunosorbent assay (ELISA) kit (R and D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. HMGB1 levels in the samples were determined by a HMGB1 Detection Kit (Chondrex Inc., Redmond, WA, USA) according to the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Group comparisons were assessed with the one-way ANOVA and Student's t-test. A value of P <0.05 (two-tailed) was considered as statistically significant.
RESULTS
Lipopolysaccharide-induced alveolar macrophage pyroptosis in acute lung injury
In the caspase-1-activated AMs, p10 was markedly increased in the LPS + vehicle group compared with the sham group (P < 0.05; Figure 1) and was inhibited by the caspase-1 inhibitor Ac-YVAD-CMK but not by the caspase-3 inhibitor Z-DEVD-FMK. This date demonstrated that caspase-1 was activated in AMs in vivo during LPS-induced ALI/ARDS. The NLRP3 inflammasome appeared following LPS stimulation and could not be inhibited by Ac-YVAD-CMK and Z-DEVD-FMK. In the ex vivo study, LPS + ATP-stimuli were strong inducers of caspase-1 cleavage. We measured caspase-1 activation by utilizing a cell-permeable fluorescent detection probe called FLICA660-YVAD, which specifically binds to activated caspase-1[24] with PI available through the cell membrane to stain nucleic acid. The rate of pyroptotic cells was calculated by the formula: (pyroptotic cells/total cells) ×100%. The representative histograms [Figure 2a] showed a great increase in activated caspase-1- and PI-positive cells in response to LPS and ATP stimulation and could be inhibited by Ac-YVAD-CMK, but not by Z-DEVD-FMK (P < 0.05) which is similar to what was observed by the western blot method. We found a distinct population of AMs stimulated by LPS + ATP that were expressing active caspase-1. The PI-positive and caspase-1-negative cells may arise through other types of cell death, such as apoptosis and necrosis.
Figure 1.

Alveolar macrophage pyroptosis occurs in vivo after LPS stimulation. Mice were treated with YVAD (6.5 mg/kg, in 1% DMSO in PBS) or DEVD (6.5 mg/kg, in 1% DMSO in PBS) by IP injection 1 h before LPS (20 kg/mg) administration. Alveolar macrophages were isolated from mice after 16 h of LPS administration. Proteins were obtained and caspase-1 P10 and cleaved caspase-3, NLRP3 inflammation activation were analyzed by Western blotting. *P < 0.05 vs. the sham group; †P < 0.05 vs. the LPS + vehicle group; ‡P < 0.05 vs. the LPS + YVAD group. Results are representative of three separate independent experiments.
Figure 2.
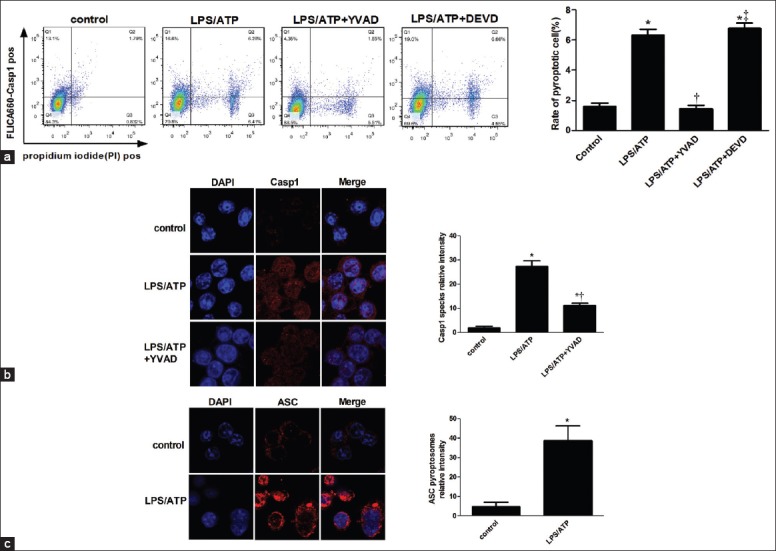
Alveolar macrophage pyroptosis occurs ex vivo after LPS stimulation. Alveolar macrophages isolated from mice were stimulated for 5 h with or without LPS (500 ng/ml) and ATP (5 mmol) added during the last hour of culture, in the absence or presence of 50 μmol of YVAD or DEVD. (a) Cells were stained with FLICA, and pyroptotic cells were detected by flow cytometry. Caspase-1 speck (b) and ASC (c) cells were analyzed by immunofluorescence. *P < 0.05 vs. the control group; †P < 0.05 vs. the LPS/ATP group. ‡P < 0.05 vs. the lipopolysaccharide/ATP + YVAD group. Results are representative of three separate independent experiments.
To confirm that the pyroptosis occurred in AMs, caspase-1 expression and ASC oligomerization in AMs in response to LPS/ATP stimulation were detected by immunofluorescence. Both caspase-1 specks [Figure 2b] and ASC pyroptosomes [Figure 2c] were low in the unstimulated control group, but were induced over time by LPS/ATP stimulation. In contrast to the NLRP3 inflammasome, ASC was constitutively expressed in unstimulated cells,[25] and the accumulation of ASC specks were triggered to redistribute and form a paranuclear protein speck in activated cells. In summary, these data indicate that caspase-1 activation occurs in AMs following LPS challenge both in vivo and ex vivo and that NLRP3/ASC inflammasomes participate in the process.
Inhibition of alveolar macrophage pyroptosis reduces lipopolysaccharide-induced lung injury
Histopathological light microscopic findings of the mouse lung tissue are shown in Figure 3a. The pulmonary organizational structure of the sham group was normal. The LPS + vehicle group developed marked lung inflammation after 16 h, which induced hemorrhage, interstitial edema and inflammatory cells, observed in most of the alveolar spaces. The amount of inflammatory cells infiltration and alveolar edema were reduced in the LPS + Ac-YVAD-CMK group when compared with the LPS + Z-DEVD-FMK group. The lung injury score [Figure 3b] and the total numbers of AM in BALF [Figure 3c] were also significantly higher in the LPS + vehicle group when compared with sham group (P < 0.05; P < 0.05). Ac-YVAD-CMK treatment reduced lung injury score and the total numbers of AM in BALF more significantly than in the LPS + Z-DEVD-FMK group (P < 0.05; P < 0.05). To further evaluate the lung injury, the pulmonary W/D weight [Figure 3d], and the total proteins in the BALF [Figure 3e] were assessed. Consistent with histological changes, pulmonary W/D weight and levels of total proteins in BALF in the LPS + vehicle group were much higher than those in sham group (P < 0.05) and can be inhibited more effectively by the caspase-1 inhibitor than caspase-3 inhibitor (P < 0.05; P < 0.05).
Figure 3.
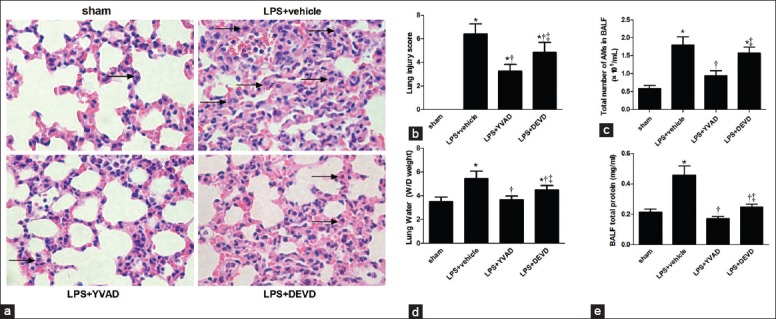
Mice were treated with YVAD (6.5 mg/kg, in 1% DMSO in PBS) or DEVD (6.5 mg/kg, in 1% DMSO in PBS) by IP injection 1 h before LPS (20 kg/mg) administration. (a) Changes in the histology of lung injury after 16 h of LPS administration (H and E, ×400). The arrowheads show alveolar macrophages; (b) The lung injury score; (c)The total number of alveolar macrophages in bronchoalveolar lavage fluid;(d) Water content of lung; (e)The total protein concentration in bronchoalveolar lavage fluid. *P < 0.05 vs. the sham group; †P < 0.05 vs. the LPS + vehicle group; ‡P < 0.05 vs. the LPS + YVAD group. n = 6 mice/group. Results are representative of three separate independent experiments.
Cytokines (IL-18, IL-1β, TNF-α and HMGB1) were measured in serum and BALF in all four groups [Figure 4]. The Ac-YVAD-CMK reduced IL-1β levels both in serum (P < 0.05; Figure 4b) and BALF (P < 0.05; Figure 4f) significantly during LPS-induced ALI/ARDS. Since the processing of the pro-inflammatory cytokine-IL-1β depends on caspase-1 and the production of mature IL-1β is tightly regulated, the caspase-1 inhibitor may be the cause of a dramatic reduction of IL-1β secretion. Also, Ac-YVAD-CMK reduced IL-18 (P < 0.05; Figure 4e), TNF-α (P < 0.05; Figure 4g), HMGB1 (P < 0.05; Figure 4h) in BALF, but without affecting these in serum during LPS-induced ALI/ARDS. These findings may have occurred because the AMs account for the large number of cells in the BALF, and the drug is able to access the target sites. Surprisingly, the caspase-1 function is also independent of processing and releasing IL-18, but the IL-18 level in serum was not significantly reduced (P > 0.05; Figure 4a), suggesting that IL-18 may have less susceptibility than IL-1β imposed by Ac-YVAD-CMK. The Z-DEVD-FMK administration had no effects on LPS-induced circulating cytokine responses in mice. These results indicate that the caspase-1 specific inhibitor Ac-YVAD-CMK decreases cytokine responses more effectively than the caspase-3 inhibitor Z-DEVD-FMK, especially for IL-1β. These findings indicate that blocking caspase-1 activation by Ac-YVAD-CMK treatment decreased inflammation and organ damage in LPS-induced ALI/ARDS in mice.
Figure 4.
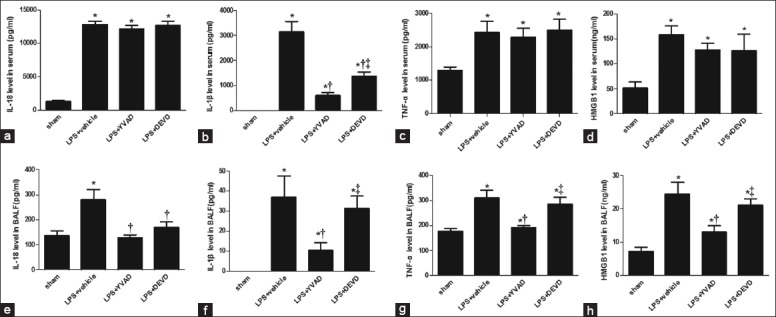
The influences of YVAD and DEVD treatment of LPS-induced cytokine levels in plasma and bronchoalveolar lavage fluid. After YVAD and DEVD treatment and LPS injection at predetermined time points, serum and bronchoalveolar lavage fluid samples were prepared and tested for levels for IL-18, IL-1β, TNF-α and HMGB1 in serum (a–d) and bronchoalveolar lavage fluid (e–h). *P < 0.05 vs. the sham group; †P < 0.05 vs. the LPS + vehicle group. ‡P < 0.05 vs. the LPS + YVAD group. n = 6 mice/group. All data shown are representative of at least three separate independent experiments.
DISCUSSION
ALI/ARDS has a complex mechanism, which is mediated by a complex, but common array of immunological cascades. Caspase-1 plays an important role in both the innate and acquired immune responses. Recent studies have shown that caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria.[26] Evidence of extensive apoptosis has been shown in the analysis of organs and tissues of patients during ALI/ARDS.[27] Apoptosis has a noninflammatory outcome, whereas a severe inflammatory response is observed in the lung during sepsis. In contrast to apoptosis, pyroptotic cell death elicits inflammation due to the release of cytosolic contents. Previous studies have shown that mice lacking capase-1 are highly resistant to LPS-induced endotoxemia.[28] For these reasons, we hypothesized that pyroptosis in AMs participate in the pathogenesis of LPS-induced ALI/ARDS.
The findings of this study have shown for the first time that the activation of caspase-1 occurs in AM during LPS-induced ALI/ARDS in an in vivo mouse model. This study also included an ex vivo analysis to confirm the findings observed in vivo. In contrast to apoptosis, pyroptosis is an established inflammasome-dependent form of cell death.[29] The NLRP3 inflammasome is a protein complex composed of NLRP3, caspase-1, and ASC, which results in the activation of caspase-1 and the secretion of IL-1β.[30,31] NLRP3 is involved in sensing microbes and a variety of endogenous danger signals, including silica, asbestos, bacterial toxins, hyaluronan, fibrillary amyloid-β peptide, as well as DAMP including ATP.[32] It has previously been shown that ATP stimulates the nucleotide receptor P2X7 gated ion channel resulting in intracellular K + efflux,[14] leading to activation of NLRP3 inflammasome.[33] Oligomerization of NLRP3/ASC inflammasome can be visualized as discrete intracellular specks in macrophages and dendritic cells.[34] In the present study, we confirmed that the NLRP3 inflammasome are generated in AMs during LPS-induced ALI/ARDS, and caspase-1 specks and ASC pyroptosome are formed in activated AMs stimulated by LPS combined and ATP ex vivo. These findings suggest that NLRP3/ASC inflammasome participates in the pathway. In ex vivo study, cells showed positive staining for both caspase-1 and PI when stimulated by LPS and ATP, indicating induction of pyroptosis in 6% of AM population. When compared with the in vivo experiment [Figures 1 and 3], this finding may be due to the following: (1) Suboptimal does or time for LPS and ATP stimulation; (2) limited sensitivity of the flow cytometry pyroptosis method or (3) LPS and ATP may not directly acting on the AMs.
The findings of this study also showed that caspase-1 inhibition can reduce pulmonary tissue damage, reduce edema and decrease the total number of AMs in the BALF. As shown in Figure 3a, the caspase-1 inhibitor may also modulate macrophages recruitment to the lung following LPS treatment. In this study, we tested for changes in plasma and BALF cytokines in sepsis when compared to Ac-YVAD-CMK- and Z-DEVD-FMK-treated mice. The study showed that Ac-YVAD-CMK treatment significantly inhibited LPS-induced IL-1β [Figure 4b and 4f]. IL-1β is a potent pro-inflammatory cytokine, which can stimulate the release of other cytokines and initiate and amplify lung inflammation in patients.[35] HMGB1 was identified originally as a DNA-binding protein and has also been proposed as a mediator of ALI.[36] The decrease in IL-1β and HMGB1 in BALF reduced pulmonary inflammatory responses. However, IL-18 levels in serum were not reduced significantly, unlike those in BALF. The basis for this phenomenon has yet to be determined. In contrast, TNF-α levels in serum were not affected in the presence of Ac-YVAD-CMK, which may indicate that TNF-α is not required for pyroptosis. The lack of effect of Ac-YVAD-CMK on other circulating cytokines might be attributable to inability of the drug to access target sites, such as monocytes and macrophages. In general, cytokine responses were decreased in the presence of Ac-YVAD-CMK which showed a more potent anti-pyroptotic and anti-inflammatory effects than Z-DEVD-FMK, by inhibiting the release of IL-1β and reducing the caspase-1 pathway in ALI/ARDS-induced by LPS.
In summary, this study has demonstrated that pyroptosis of AMs occurs in the pathogenisis of ALI/ARDS and indicates that caspase-1 could be an effective target for the clinical treatment of these important conditions. The findings of this study also shed some light on the regulatory role of caspase-1 activation. Future studies are recommended to define the pathways and factors that induce pyroptosis during ALI/ARDS in vivo.
Financial support and sponsorship
The author thanks the National Natural Science Foundation of China (No. 81470266).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Yi Cui
REFERENCES
- 1.Modrykamien AM, Gupta P. The acute respiratory distress syndrome. Proc (Bayl Univ Med Cent) 2015;28:163–71. doi: 10.1080/08998280.2015.11929219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008;133:1120–7. doi: 10.1378/chest.07-2134. [DOI] [PubMed] [Google Scholar]
- 3.Villar J, Blanco J, Añón JM, Santos-Bouza A, Blanch L, Ambrós A, et al. The ALIEN study: Incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. 2011;37:1932–41. doi: 10.1007/s00134-011-2380-4. [DOI] [PubMed] [Google Scholar]
- 4.Matuschak GM, Lechner AJ. Acute lung injury and the acute respiratory distress syndrome: Pathophysiology and treatment. Mo Med. 2010;107:252–8. [PMC free article] [PubMed] [Google Scholar]
- 5.Z’graggen BR, Tornic J, Müller-Edenborn B, Reyes L, Booy C, Beck-Schimmer B. Acute lung injury: Apoptosis in effector and target cells of the upper and lower airway compartment. Clin Exp Immunol. 2010;161:324–31. doi: 10.1111/j.1365-2249.2010.04175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–70. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 7.Sangiuliano B, Pérez NM, Moreira DF, Belizário JE. Cell death-associated molecular-pattern molecules: Inflammatory signaling and control. Mediators Inflamm 2014. 2014 doi: 10.1155/2014/821043. 821043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 10.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243:206–14. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang HH, Jun HK, Jung YJ, Choi BK. Enterococcus faecalis activates caspase-1 leading to increased interleukin-1 beta secretion in macrophages. J Endod. 2014;40:1587–92. doi: 10.1016/j.joen.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 13.Dinarello CA. A clinical perspective of IL-1ß as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–17. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 14.Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T, et al. Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS One. 2014;9:e85765. doi: 10.1371/journal.pone.0085765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones HD, Crother TR, Gonzalez-Villalobos RA, Jupelli M, Chen S, Dagvadorj J, et al. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol. 2014;50:270–80. doi: 10.1165/rcmb.2013-0087OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185:1225–34. doi: 10.1164/rccm.201201-0003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–92. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathiak G, Grass G, Herzmann T, Luebke T, Zetina CC, Boehm SA, et al. Caspase-1-inhibitor ac-YVAD-cmk reduces LPS-lethality in rats without affecting haematology or cytokine responses. Br J Pharmacol. 2000;131:383–6. doi: 10.1038/sj.bjp.0703629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol. 2014;306:L709–25. doi: 10.1152/ajplung.00341.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Cardinal JS, Bahar R, Evankovich J, Huang H, Nace G, et al. Interferon regulatory factor-1 regulates the autophagic response in LPS-stimulated macrophages through nitric oxide. Mol Med. 2012;18:201–8. doi: 10.2119/molmed.2011.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang C, Wang SH, Lasbury ME, Tschang D, Liao CP, Durant PJ, et al. Toll-like receptor 2 mediates alveolar macrophage response to Pneumocystis murina. Infect Immun. 2006;74:1857–64. doi: 10.1128/IAI.74.3.1857-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuigan RM, Mullenix P, Norlund LL, Ward D, Walts M, Azarow K. Acute lung injury using oleic acid in the laboratory rat: Establishment of a working model and evidence against free radicals in the acute phase. Curr Surg. 2003;60:412–7. doi: 10.1016/S0149-7944(02)00775-4. [DOI] [PubMed] [Google Scholar]
- 24.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–55. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, et al. Neutrophil IL-1ß processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J Immunol. 2015;194:1763–75. doi: 10.4049/jimmunol.1401624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–42. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Zeng M, He W, Huang X, Luo L, Zhang H, et al. Ghrelin protects alveolar macrophages against lipopolysaccharide-induced apoptosis through growth hormone secretagogue receptor 1a-dependent c-Jun N-terminal kinase and Wnt/ß-catenin signaling and suppresses lung inflammation. Endocrinology. 2015;156:203–17. doi: 10.1210/en.2014-1539. [DOI] [PubMed] [Google Scholar]
- 28.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 29.Aziz M, Jacob A, Wang P. Revisiting caspases in sepsis. Cell Death Dis. 2014;5:e1526. doi: 10.1038/cddis.2014.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1ß and IL-18 processing during infection. Trends Immunol. 2011;32:110–6. doi: 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Compan V, Martín-Sánchez F, Baroja-Mazo A, López-Castejón G, Gomez AI, Verkhratsky A, et al. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J Immunol. 2015;194:1261–73. doi: 10.4049/jimmunol.1301676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 33.Triantafilou K, Triantafilou M. Ion flux in the lung: Virus-induced inflammasome activation. Trends Microbiol. 2014;22:580–8. doi: 10.1016/j.tim.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodman RB, Pugin J, Lee JS, Matthay MA. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14:523–35. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 36.Kudo D, Toyama M, Aoyagi T, Akahori Y, Yamamoto H, Ishii K, et al. Involvement of high mobility group box 1 and the therapeutic effect of recombinant thrombomodulin in a mouse model of severe acute respiratory distress syndrome. Clin Exp Immunol. 2013;173:276–87. doi: 10.1111/cei.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]