

Abstract
Background:
Toll-like receptor 4 (TLR4) is a crucial receptor in the innate immune system and noninfectious immune responses. It has been reported that TLR4 participates in the pathological course of ischemia/reperfusion (I/R) injury. However, the role of TLR4 in the process of I/R injury after cardiac arrest (CA) and cardiopulmonary resuscitation (CPR) is still unknown. In this study, we investigated the effects of TLR4 mutation on survival and neurological outcome in a mouse model of CA/CPR.
Methods:
A model of potassium-induced CA was performed on TLR4-mutant mice (C3H/HeJ) and wild-type mice (C3H/HeN). After 3 min of untreated CA, resuscitation was attempted with chest compression, ventilation, and intravenous epinephrine. Behavioral tests were performed on mice on day 3 after CPR. The morphological changes in hippocampal neurons were assessed by light and electron microscopy. Expressions of TLR4 and intercellular adhesion molecule-1 (ICAM-1) were detected by Western blot. Levels of tumor necrosis factor-α (TNF-α) and myeloperoxidase (MPO) were measured with enzyme-linked immunosorbent assay (ELISA).
Results:
On day 3 after resuscitation the overall mortality was 33.33% in C3H/HeJ group compared with 53.33% in C3H/HeN group (P < 0.05). And there was much higher central tendency in C3H/HeJ group than C3H/HeN group during open field test (P < 0.05). Meanwhile, the percentage of nonviable neurons was 21.16% in C3H/HeJ group compared with 53.11% in C3H/HeN group (P < 0.05). And there were significantly lower levels of hippocampal TNF-α and MPO in C3H/HeJ mice (TNF-α: 6.85±1.19 ng/mL, MPO: 0.33±0.11 U/g) than C3H/HeN mice (TNF-α: 11.36±2.12 ng/mL, MPO: 0.54±0.17 U/g) (all P < 0.01). CPR also significantly increased the expressions of TLR4 and ICAM-1 in C3H/HeN group. However, the expression of ICAM-1 was much lower in C3H/HeJ group than in C3H/HeN group after CPR (P < 0.01).
Conclusion:
TLR4 signaling is involved in brain damage and in inflammation triggered by CA/CPR.
Keywords: Cardiac Arrest, Cardiopulmonary Resuscitation, Cerebral Injury, Toll-like Receptor 4
INTRODUCTION
Although cardiopulmonary resuscitation (CPR) has been studied for half a century, survival and neurological outcome after cardiac arrest (CA) remain unsatisfactory.[1,2] It has been demonstrated that the high mortality rate of patients who initially achieve resumption of spontaneous circulation (ROSC) after CA can be attributed to whole-body ischemia and reperfusion (I/R) injury.[3] In particular, neurological injury following CA/CPR primarily results from the global I/R.[4]
It is well-known that brain injury caused by cerebral I/R is a complicated pathological course, in which inflammatory response plays a crucial role.[5] Toll-like receptors (TLRs) are a family of signal transduction molecules and are implicated in the induction of innate and adaptive immunity.[6,7] Recently, accumulating evidence have shown that TLR4 is activated by endogenous proteins released from damaged brain and participates in mediating the cerebral injury following I/R.[8,9,10] However, these previous studies mainly focus on the regional cerebral I/R models induced by the occlusion of the middle cerebral artery, which are obviously different from those global I/R models induced by CA/CPR. A current study has proved that the TLR4 mRNA expression is upregulated in a rat model of CPR following asphyxial CA.[11] In the present study, we investigated the role of TLR4 for survival and neurological function after CA/CPR using TLR4 genetically mutant mice.
METHODS
Experimental animals
TLR4 mutant male mice (C3H/HeJ) of 10–12 weeks of age were purchased from Shanghai SLAC Animal Center. Their intracellular region of TLR4 amino acids had a mutation at 712 site from proline to histidine which resulted in no response of TLR4 to its ligand lipopolysaccharide.[12] TLR4 wild-type male mice (C3H/HeN) of 10–12 weeks of age were purchased from Beijing Vitalriver Experimental Animal Center. All mice were kept and bred in-house under pathogen-free condition, and they were divided into four groups at random which were C3H/HeN sham group (n = 15), C3H/HeJ sham group (n = 15), C3H/HeN CPR group (n = 30) and C3H/HeJ CPR group (n = 30). Animal protocols were approved by the Animal Care and Use Committee of the Huazhong University of Science and Technology.
Cardiac arrest/cardiopulmonary resuscitation model
The mice were anesthetized with 40 mg/kg bodyweight 1% pentobarbital sodium delivered by intraperitoneal injection. Rectal temperature was maintained at near 37°C during surgery with a heating lamp. The mice were orally intubated with a 22-gauge catheter, connected to a mouse ventilator (ALC-V8S, Shanghai Alcbio Company, China) set to a respiratory rate of 130 breaths/min. Needle electrodes were placed subcutaneously on the chest for electrocardiogram monitoring throughout the experimental procedures. CA was induced by injection of 0.08 mg/g body weight potassium chloride via the jugular catheter and confirmed by the appearance of asystole on the electrocardiography monitor and no spontaneous breathing.[13,14] At this time, mechanical ventilation was interrupted for the duration of CA. CPR was begun 3 min after induction of CA by injection of 0.4 μg epinephrine, chest compressions at a rate of approximately 300 beats/min, and ventilation with 100% oxygen at a respiratory rate of 160 breaths/min. As soon as ROSC was achieved, defined as electrocardiographic activity with visible cardiac contractions, chest compressions were stopped.[13,14] If ROSC could not be achieved within 10 min of CPR, resuscitation was stopped, and the animal would be excluded from the study as a result. Sham animals underwent anesthesia, oral intubation, mechanical ventilation, surgical preparation, and insertion of vascular catheters. An equivalent volume isotonic saline was given as a placebo control of potassium chloride and epinephrine in the sham groups.
Open field test
General activity and anxiety-like behavior were assessed during a 5-min session in an open field apparatus (25 cm × 25 cm × 38 cm) on day 3 after CPR using Ugo Basile instruments (Mouse Open Field, Ugo Basile, Italia). Central tendency is the percent time spent (or distance traveled) in a 12 cm × 12 cm zone in the center of the apparatus. Data were analyzed to determine general locomotor activity and the relative amount of activity occurring in the periphery versus the center of the apparatus (anxiety-like behavior).
Histological evaluation
Upon cardiac puncture and exsanguination on day 3 after CPR, 4 mice in each group underwent transcardial perfusion and whole body fixation with 10% phosphate-buffered formalin. Brains were removed, postfixed with paraformaldehyde and embedded in paraffin. Brain samples were sectioned at 5 μm intervals and stained with hematoxylin and eosin. Nonviable neurons were determined by the presence of hypereosinophilic cytoplasm and pyknotic nuclei.[14] Viable as well as nonviable neurons were counted in three microscopic fields per section, and the percentage of nonviable neurons was averaged for three sections per animal (high-power fields, ×400).
Observation of ultrastructural organization
Fresh hippocampi were obtained from 4 mice in each group on day 3 after CPR and were prefixed with 4% glutaraldehyde, stored at 4°C until processed and then postfixed with 1% osmiun tetroxide. Then the specimens were immersed in propylene oxide after dehydration with gradient ethanol, embedded with epoxy resin and made into ultrathin sections (0.1 μm). The sections were stained subsequently with lead-uranium and the changes in ultrastructure organization were then observed by a transmission electron microscope.
Determination of tumor necrosis factor-α and myeloperoxidase
Six mice in each group were used to quantify tumor necrosis factor-α (TNF-α) and myeloperoxidase (MPO) levels on day 3 after CPR. Mice were euthanized and then brains were removed using aseptic techniques. Hippocampi were dissected out, homogenized and centrifuged. Then, the supernatant was harvested and stored at −70°C. The TNF-α level and MPO activity in the supernatant were detected using an enzyme-linked immunosorbent assay (ELISA) kit respectively (Wuhan Jingmei Biotechnology Co., China).
Western blotting analysis
Murine brain tissue from the hippocampi was lysed and homogenized in cold lysis buffer. The homogenate was centrifuged at 15,000 r/min (Radius=9cm) for 15 min at 4°C. Supernatants were collected, and then the protein concentration was determined with an assay kit based on the Bradford method (Bio-Rad). Proteins were then transferred to nitrocellulose membranes, and these membranes were blocked for 2 h at room temperature with 5% nonfat milk. The blots were then incubated with primary antibody against TLR4 and intercellular adhesion molecule-1 (ICAM-1), β-actin, respectively (diluted in 1:1000, Santa Cruz, USA) overnight at 4°C, followed by incubation for 1 h with horseradish peroxidase-conjugated goat anti-murine IgG (diluted in 1:5000). Fresh DAB was used for staining and the image scanning was done using UVP computerized image analysis system (UVP Co., USA) to measure the staining intensity of each protein bands.
Statistical analysis
All the data were showed as mean ± standard deviation (SD). One-way ANOVA was employed for result analysis. Survival data for Kaplan–Meier curves were tested with the log-rank test. A P < 0.05 was considered as statistically significant.
RESULTS
In the present study, 35 C3H/HeN mice and 34 C3H/HeJ mice underwent CA/CPR, and 5 C3H/HeN mice and 4 C3H/HeJ mice were excluded because of unsuccessful resuscitation. There was no significant difference in these unresuscitated mice between C3H/HeN and C3H/HeJ group.
Mortality
A significantly better 72 h survival was observed in the C3H/HeJ CPR group when compared with the C3H/HeN CPR group (P < 0.05). The overall mortality in the latter group was 53.33% (16/30 mice died). By comparison, the overall mortality in the former group was significantly lower, which was 33.33% (10/30 mice died). No animal in the both sham groups died [Figure 1].
Figure 1.
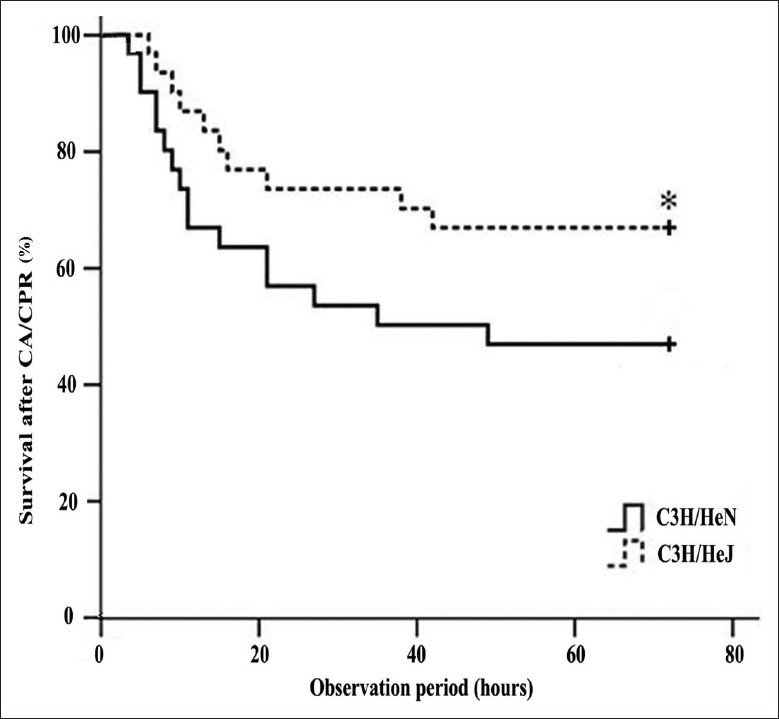
Survival after successful resuscitation in an observation period of 72 h. The overall mortality was significantly lower in the C3H/HeJ cardiopulmonary resuscitation group. Data were analyzed by Kaplan–Meier log-rank survival analysis. *P < 0.05 versus C3H/HeN cardiopulmonary resuscitation group.
Cell death response to cardiopulmonary resuscitation
CPR animals displayed more severe cell damage within the hippocampus on day 3 after CPR. The hippocampal region in CA/CPR-treated mice was especially affected by vacuolization of cells, and the above changes of neurons were more obvious in C3H/HeN mice than C3H/HeJ mice after CPR [Figure 2a–2d]. In addition, electron microscopic results showed that neurons presented the reduced ribosome, the swollen endoplasmic reticulum and mitochondria with broken cristae after CPR, which were more obvious in C3H/HeN mice than C3H/HeJ mice [Figure 3a–3d].
Figure 2.
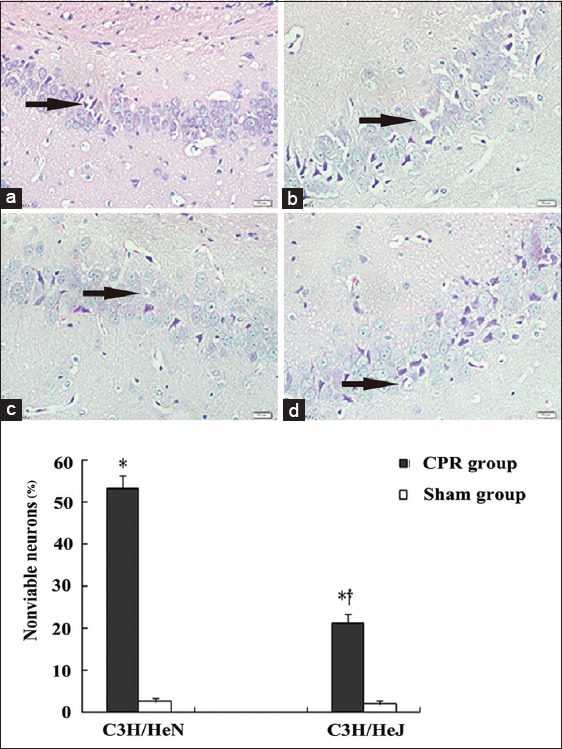
Representative photomicrographs of hippocampal neurons in sham-operated C3H/HeN mice (a) and cardiopulmonary resuscitation treated C3H/HeN mice (b) and sham operated C3H/HeJ mice (c) and cardiopulmonary resuscitation treated C3H/HeJ mice (d) (n = 4, per group). The black arrow indicates neuronal vacuolar change and dark pyknotic nuclei. *P < 0.01 versus sham group; †P < 0.05 versus C3H/HeN cardiopulmonary resuscitation group; H and E, original magnification: ×400.
Figure 3.
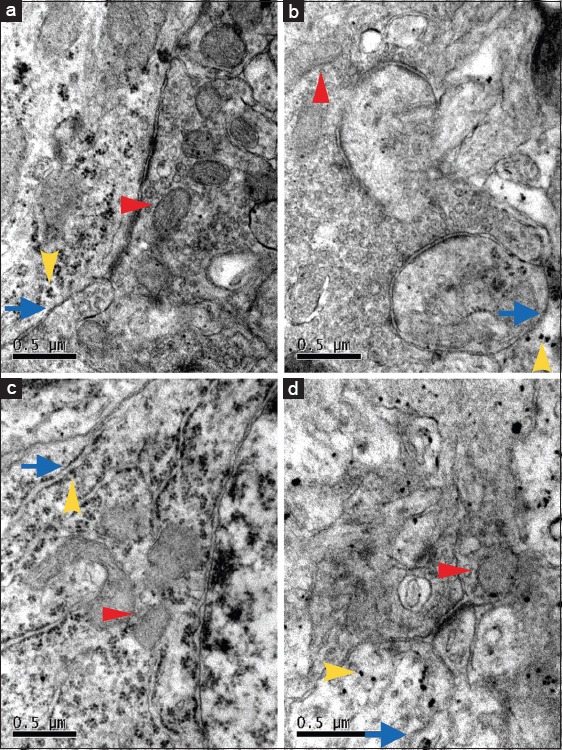
The ultrastructure of neurons in hippocampal CA1 region under electron microscope in sham operated C3H/HeN mice (a) and cardiopulmonary resuscitation treated C3H/HeN mice (b) and sham operated C3H/HeJ mice (c) and cardiopulmonary resuscitation treated C3H/HeJ mice (d). The red arrow indicates mitochondria, the yellow arrow indicates ribosome and the blue arrow indicates endoplasmic reticulum. The mitochondria and endoplasmic reticulum appeared more swollen with the reduction of ribosome in b and d (n = 4, per group). Scale bar = 0.5 μm.
Anxiety-like behaviors after cardiopulmonary resuscitation
Central tendency, a measure of anxiety-like behavior, as shown by the ratio of the time and distance in the central area was decreased in all mice exposed to CA/CPR (P < 0.01). In addition, there was a significant difference between the C3H/HeN and C3H/HeJ CPR group, indicating that the anxiety-like behavior to some extent was alleviated in TLR4-mutant mice after CA/CPR [Figure 4].
Figure 4.
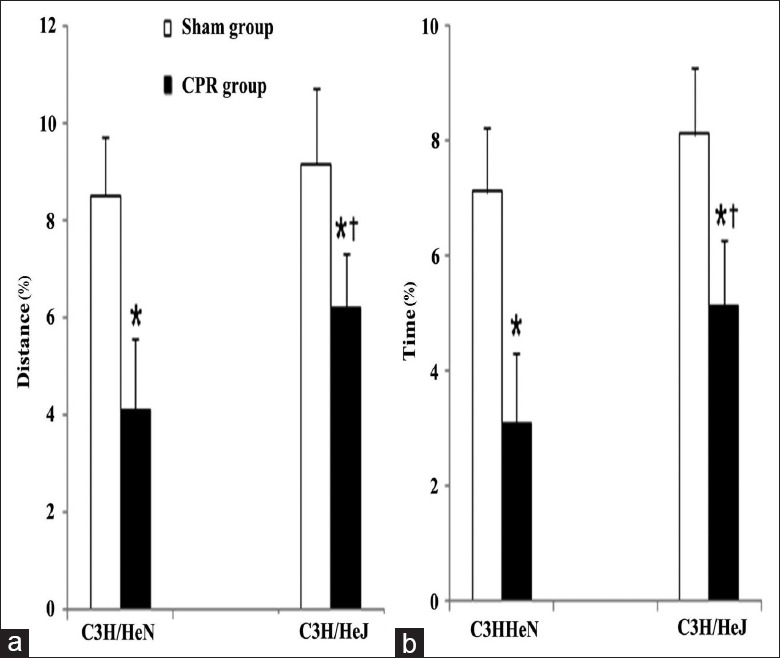
Central tendency, a measure of anxiety-like behavior, was evaluated by the percentage of distance (distance traveled in central area/total distance traveled in whole area×100%) (a) and the percentage of time (time spent in central area/total time spent in whole area×100%) (b) during open field test. Both sham groups n = 15, respectively; C3H/HeN cardiopulmonary resuscitation group n = 14; C3H/HeJ cardiopulmonary resuscitation group n = 20; *P < 0.01 versus sham group; †P < 0.05 versus C3H/HeN cardiopulmonary resuscitation group.
Expression of TNF-α and myeloperoxidase after cardiopulmonary resuscitation
To explore the implication of TNF-α in TLR4 signaling, we measured alteration of hippocampal TNF-α contents in mice with ELISA. After CPR, there were significant increases of TNF-α in the hippocampi of the C3H/HeN and C3H/HeJ CPR groups (11.36 ± 2.12 ng/ml and 6.85 ± 1.19 ng/ml, respectively) compared with those of the C3H/HeN and C3H/HeJ sham groups (3.07 ± 0.25 ng/ml and 2.26 ± 0.23 ng/ml, respectively) (all P < 0.01). However, as compared with the mice in C3H/HeN CPR group, the mice in C3H/HeJ CPR group displayed significantly lower levels of TNF-α (P < 0.01). And no significant difference was found between C3H/HeN and C3H/HeJ sham group.
Similar to TNF-α, MPO activity, a biochemical marker of neutrophil infiltration rose to 0.54 ± 0.17 U/g in the hippocampi in C3H/HeN mice after CPR (P < 0.01). CPR also resulted in a significant increase in the hippocampal MPO activity in C3H/HeJ mice (0.33 ± 0.11 U/g) (P < 0.01). However, MPO activity in C3H/HeJ CPR group was significantly lower than that in C3H/HeN CPR group (P < 0.01). And there was no significant difference in MPO activity between C3H/HeN and C3H/HeJ sham group (0.16 ± 0.09 U/g and 0.13 ± 0.07 U/g, respectively).
Expression of TLR4 and intercellular adhesion molecule-1 after cardiopulmonary resuscitation
To clarify whether TLR4 participate in the pathological course of cerebral injury after CA/CPR, we detected the TLR4 expression in C3H/HeN mice that underwent CA/CPR. As shown in Figure 5a, CA/CPR treatment caused an obvious increase in the level of TLR4 expression in C3H/HeN mice (n = 6) (P < 0.01).
Figure 5.
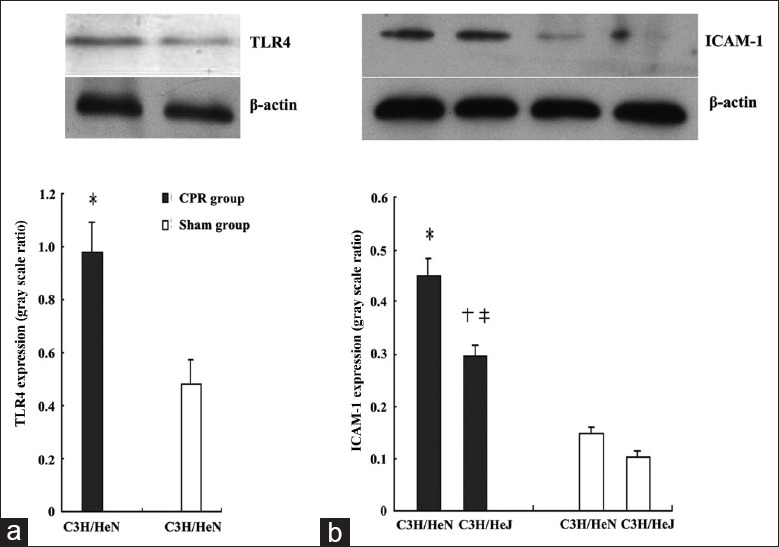
Hippocampal toll-like receptor 4 (a) and intercellular adhesion molecule-1 (b) expression on day 3 after cardiopulmonary resuscitation. Cardiac arrest/cardiopulmonary resuscitation treatment resulted in an obvious increase of toll-like receptor 4 and intercellular adhesion molecule-1 expressions in C3H/HeN mice. *P < 0.01 versus C3H/HeN sham group, n = 6; †P < 0.01 versus C3H/HeN cardiopulmonary resuscitation group, n = 6, ‡P < 0.01 versus C3H/HeJ sham group, n = 6.
Since the increased neutrophils adhesion has been demonstrated to be one of the most important factors in the pathogenesis of inflammatory injury during I/R injury of the brain,[15] we also examined the change of ICAM-1 expression in mice brain after CA/CPR. As shown in Figure 5b, ICAM-1 expressions were strongly upregulated in C3H/HeN mice exposed to CA/CPR (n = 6) (P < 0.01). Also, ICAM-1 expression also increased in C3H/HeJ mice but to a much lesser extent (P < 0.01). There was no significant ICAM-1 expression difference between C3H/HeN and C3H/HeJ sham group.
DISCUSSION
The present study demonstrated that TLR4-mutant mice had lower mortality, minor cerebral injury, and less inflammatory response after CA/CPR. Our results indicated that TLR4 signaling might be involved in brain damage and in inflammation triggered by CA/CPR.
To establish a stable and reliable model of CA is the most important work for CPR research. It has been proved that the injection of potassium chloride is a feasible method to induce immediate CA and result in successful resuscitation in a high fraction of animals. A 3-min period of CA can produce significant organ and neurological damage though this time period seems to be short.[16] We demonstrated that mice that express TLR4 normally presented a mortality with 53.33% on day 3 after CPR, which was quantitatively similar to the previously reported data.[16] Meanwhile, we found that mice that lack expression of TLR4 presented a significantly better CPR outcome, as shown by a remarkable reduction of the mortality by 33.33%. The major proportion of deaths occurred during the first 12 h after ROSC, which was closely related to the cardiac dysfunction and the proinflammatory cytokine response after resuscitation.[17] Also, both microscopic and electron microscopic results revealed that cerebral injury is less severe in TLR4-mutant mice than wild-type mice, indicating that TLR4 might play an important role in brain injury after CPR.
We also showed that both C3H/HeN and C3H/HeJ mice that underwent CA/CPR presented a significantly reduced central tendency, suggesting that an increased anxiety-like behavior might be induced by CA/CPR. These data confirm the previous studies that have shown that global ischemia-induced in rodents by CA/CPR resulted in neuronal cell death and increased anxiety-like behavior.[18,19] In addition, our data support that CA/CPR-induced anxiety-like behavior could be attenuated in TLR4-mutant mice indicating that TLR4 signaling might have an effect on anxiety induced by CA/CPR. A current study has reported that the type 1 diabetes model mice exhibit anxiety and cognitive impairment which is less severe in TLR4-deficient mice compared to wild-type mice, showing that TLR4 might contribute to the negative impact of the type 1 diabetes model mice on anxiety and cognition.[20] However, this improved neurological behavior in TLR4-deficient animals as a neuroprotective mechanism will need to be further addressed.
Our data demonstrated that CA/CPR increased the expression of TLR4 in C3H/HeN mice. We have mentioned that TLR4 can evoke the inflammatory chain reaction and play a crucial role in innate immune response. It has been known that TLR4 activates nuclear factor κ-B signaling pathways that produce the transcription of many proinflammatory genes and cytokines such as TNF-α.[6,7] Indeed, we found that wild-type mice have significantly higher TNF-α expression than TLR4-mutant mice, suggesting that TNF-α expression may mediate brain damage through TLR4-dependent pathway. Because TNF-α mediates cytotoxicity in many cell systems such as the ischemic brain,[8,9] the reduction of the cerebral injury found in TLR4-mutant mice might be explained in part by the reduced expression of TNF-α. Our data strongly support that TLR4 signaling is implicated in brain damage after CA/CPR through the expression of TNF-α.
Neutrophil infiltration reflects persisting injury to the brain under the condition of systemic inflammatory signals.[21] MPO activity in brain tissue is evaluated as an index of neutrophil accumulation.[22] Our data showed that CA/CPR increased hippocampal MPO activity in both C3H/HeN and C3H/HeJ mice. More importantly, the hippocampal MPO activity was significantly lower in C3H/HeJ mice than in C3H/HeN mice, indicating that TLR4 mutation might reduce neutrophil infiltration into the brain. Our results also showed that C3H/HeN mice expressed much higher ICAM-1 expression in hippocampus than C3H/HeJ mice. Because the inhibition of ICAM-1 expression reduces polymorphonuclear leukocytes (PMN) recruitment and organ damage after resuscitation in mice,[23] the reduction of ICAM-1 expression found in TLR4-mutant mice after CA/CPR is likely to be one of the main mechanisms responsible for the protective effect of the deletion of TLR4-mediated signaling.
TLR4 is found to present on various innate immune cells. Within brain, TLR4 is mainly located on glial cells including microglia, astrocytes, and oligodendrocytes. TLR4 is also expressed on circulating inflammatory cells and vascular endothelial cells in a variety of other tissues.[24] It has been reported that neuronal damage can be exacerbated by peripheral inflammatory.[25] Therefore, it is possible that the actions of TLR4 mutation may result from its ability to inhibit peripheral inflammatory responses to cerebral injury after CPR. Given this limitation of the present study, further studies are needed to identify the role of central versus peripheral TLR4 following CA/CPR.
Our study also had other limitations. First, it is better to use the mechanical compressor instead of the manual chest compression after CA. However, as far as we know, mechanical compressor for small animals is not commercial available at the time we carried out our experiments. Secondly, we did not measure the inflammatory cytokines in the first 12 h after ROSC. The immediate reperfusion period after ROSC is characterized by an abrupt increase in plasma TNF-α concentration.[17] And it is better to observe the dynamic changes in inflammatory cytokines after resuscitation. Thirdly, there are different rheological and metabolic properties between the human brain and the rodent animal brain. Therefore, the outcome of this study in a rat model of CPR remains to be demonstrated in large-animal and clinical studies.
In summary, our results documented that TLR4 signaling might play the dominant role in mediating inflammatory responses to CPR. These data suggested that TLR4 might represent a potential target for the pharmacological treatment of postresuscitation reperfusion injury following CPR.
Financial support and sponsorship
This study was funded by the grants from the National Nature Science Foundation of China (No. 81201444, No. 81101401) and should be attributed to Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Xiu-Yuan Hao and Peng Lyu
REFERENCES
- 1.Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, et al. Heart disease and stroke statistics – 2008 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 2.Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med. 2004;30:2126–8. doi: 10.1007/s00134-004-2425-z. [DOI] [PubMed] [Google Scholar]
- 3.Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Böttiger BW, et al. Post cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation. Circulation. 2008;118:2452–83. doi: 10.1161/CIRCULATIONAHA.108.190652. [DOI] [PubMed] [Google Scholar]
- 4.Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1–21. doi: 10.1016/j.ncl.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke. 2003;34:2518–32. doi: 10.1161/01.STR.0000089015.51603.CC. [DOI] [PubMed] [Google Scholar]
- 6.Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–63. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- 7.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 8.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 9.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–14. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 10.Sun P, Xu L, Zhang Q, Li Q. Impact of Toll-like receptor 4 deficiency on cerebrocardiac syndrome. J Huazhong Univ Sci Technolog Med Sci. 2014;34:161–4. doi: 10.1007/s11596-014-1251-y. [DOI] [PubMed] [Google Scholar]
- 11.Sui B, Li Y, Ma L. Postconditioning improvement effects of ulinastatin on brain injury following cardiopulmonary resuscitation. Exp Ther Med. 2014;8:1301–7. doi: 10.3892/etm.2014.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 13.Wang QY, Sun P, Zhang Q, Yao SL. Minocycline attenuates microglial response and reduces neuronal death after cardiac arrest and cardiopulmonary resuscitation in mice. J Huazhong Univ Sci Technolog Med Sci. 2015;35:225–9. doi: 10.1007/s11596-015-1415-4. [DOI] [PubMed] [Google Scholar]
- 14.Deng G, Yonchek JC, Quillinan N, Strnad FA, Exo J, Herson PS, et al. A novel mouse model of pediatric cardiac arrest and cardiopulmonary resuscitation reveals age-dependent neuronal sensitivities to ischemic injury. J Neurosci Methods. 2014;222:34–41. doi: 10.1016/j.jneumeth.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J Leukoc Biol. 2010;87:779–89. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menzebach A, Bergt S, von Waldthausen P, Dinu C, Nöldge-Schomburg G, Vollmar B. A comprehensive study of survival, tissue damage, and neurological dysfunction in a murine model of cardiopulmonary resuscitation after potassium-induced cardiac arrest. Shock. 2010;33:189–96. doi: 10.1097/SHK.0b013e3181ad59a3. [DOI] [PubMed] [Google Scholar]
- 17.Niemann JT, Rosborough JP, Youngquist S, Shah AP, Lewis RJ, Phan QT, et al. Cardiac function and the proinflammatory cytokine response after recovery from cardiac arrest in swine. J Interferon Cytokine Res. 2009;29:749–58. doi: 10.1089/jir.2009.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neigh GN, Kofler J, Meyers JL, Bergdall V, La Perle KM, Traystman RJ, et al. Cardiac arrest/cardiopulmonary resuscitation increases anxiety-like behavior and decreases social interaction. J Cereb Blood Flow Metab. 2004;24:372–82. doi: 10.1097/01.WCB.0000112323.75217.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norman GJ, Zhang N, Morris JS, Karelina K, Berntson GG, DeVries AC. Social interaction modulates autonomic, inflammatory, and depressive-like responses to cardiac arrest and cardiopulmonary resuscitation. Proc Natl Acad Sci U S A. 2010;107:16342–7. doi: 10.1073/pnas.1007583107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawamoto EM, Cutler RG, Rothman SM, Mattson MP, Camandola S. TLR4-dependent metabolic changes are associated with cognitive impairment in an animal model of type 1 diabetes. Biochem Biophys Res Commun. 2014;443:731–7. doi: 10.1016/j.bbrc.2013.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sladojevic N, Stamatovic SM, Keep RF, Grailer JJ, Sarma JV, Ward PA, et al. Inhibition of junctional adhesion molecule-A/LFA interaction attenuates leukocyte trafficking and inflammation in brain ischemia/reperfusion injury. Neurobiol Dis. 2014;67:57–70. doi: 10.1016/j.nbd.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rymaszewski AL, Tate E, Yimbesalu JP, Gelman AE, Jarzembowski JA, Zhang H, et al. The role of neutrophil myeloperoxidase in models of lung tumor development. Cancers (Basel) 2014;6:1111–27. doi: 10.3390/cancers6021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larmann J, Schmidt C, Gammelin H, Van Aken HK, Frenzel T, Lanckohr C, et al. Intercellular adhesion molecule-1 inhibition attenuates neurologic and hepatic damage after resuscitation in mice. Anesthesiology. 2005;103:1149–55. doi: 10.1097/00000542-200512000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Ge P, Zhu Y. TLR2 and TLR4 in the brain injury caused by cerebral ischemia and reperfusion. Mediators Inflamm 2013. 2013 doi: 10.1155/2013/124614. 124614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spencer SJ, Mouihate A, Pittman QJ. Peripheral inflammation exacerbates damage after global ischemia independently of temperature and acute brain inflammation. Stroke. 2007;38:1570–7. doi: 10.1161/STROKEAHA.106.476507. [DOI] [PubMed] [Google Scholar]