

Abstract
Fetal hemoglobin (HbF) is a recognized modulator of sickle cell disease (SCD) severity. HbF levels are strongly influenced by genetic variants at three major genetic loci, Xmn1‐HBG2, HMIP‐2, and BCL11A, but the effect of these loci on the hematological phenotype in SCD, has so far not been investigated. In a cohort of individuals with SCD in Tanzania (HbSS and HbS/β° thalassemia, n = 726, aged 5 or older), HbF levels were positively correlated with hemoglobin, red blood cell (RBC) indices, mean corpuscular volume (MCV), and mean corpuscular hemoglobin (MCH), and negatively with white blood cell (WBC) and platelet counts (all P < 0.0001). We subsequently assessed the contribution of the three HbF modifier loci and detected diverse effects, including a reduction in anemia, leukocytosis, and thrombocytosis associated with certain HbF‐promoting alleles. The presence of the ‘T’ allele at Xmn1‐HBG2 led to a significant increase in hemoglobin (P = 9.8 × 10−3) but no changes in cellular hemoglobin content. Xmn1‐HBG2 ‘T’ also has a weak effect decreasing WBC (P = 0.06) and platelet (P = 0.06) counts. The BCL11A variant (rs11886868‐‘C’) increases hemoglobin (P = 2 × 10−3) and one of the HBS1L‐MYB variants decreases WBC values selectively (P = 2.3 × 10−4). The distinct pattern of effects of each variant suggests that both, disease alleviation through increased HbF production, and ‘pleiotropic’ effects on blood cells, are involved, affecting a variety of pathways. Am. J. Hematol. 90:E1–E4, 2015. © 2014 Wiley Periodicals, Inc.
Introduction
Sickle cell disease (SCD) is an inherited hemoglobin disorder caused by the Glu6Val mutation in the β globin chain. It has a devastating impact in Sub‐Saharan Africa, where it is highly prevalent and a significant cause of childhood mortality 1. The severity of the disease presentation is variable, and is usually reduced in patients retaining high levels of fetal hemoglobin (HbF) into adulthood 2, a condition that is strongly influenced by secondary genetic factors. Common genetic variants promoting HbF persistence have been identified at three loci: a promoter variant of the gene encoding the Gγ globin chain of HbF (termed Xmn1‐HBG2) and clusters of variants in regulative elements for two hematopoietic transcription factors, BCL11A and MYB. BCL11A acts as a repressor of γ globin gene expression, whereas genetic variation near MYB (termed HMIP‐2, HBS1L‐MYB intergenic polymorphism, block 2) affects the levels of HbF indirectly by altering the kinetics of erythropoiesis 3. In healthy, nonanemic individuals, most HbF‐associated variants have small, but significant effects on general hematological parameters (“pleiotropic effects”). The HMIP‐2 locus influences average volume (MCV), hemoglobin content (MCH), and number (RBC) of erythrocytes 4. To a lesser degree, HMIP‐2 affects hemoglobin (Hb), hematocrit (HCT), mean corpuscular hemoglobin concentration (MCHC) 5, and also numbers of platelets (PLT), monocytes, and total white blood cells (WBC) 4, 5, 6, 7, 8, 9, 10. BCL11A variants have generally weaker pleiotropic effects, on RBC, MCV, and MCH 5, 8. Subtle effects such as these might be hard to discern in SCD, where blood parameters are expected to primarily reflect disease‐related processes, such as variable degrees of anemia, leukocytosis, and thrombocytosis 11, 12, 13, 14. HbF levels in SCD are also affected by the disease itself. They are generally increased, due to a combination of stress erythropoiesis, which releases more F cells 15, 16, 17, immature erythrocytes that contain relatively significant amounts of HbF 18, and also due to the selective survival of such cells 19, 20, 21. Nonetheless, a wide variation of HbF levels has been observed in patients with HbSS, which is mainly ascribed to the underlying genetic background of other coinherited factors.
We wanted to know how the natural variability in HbF levels and the known genetic HbF modifiers influence the hematological phenotype of SCD patients. For this purpose, we studied general blood cell parameters, peripheral HbF levels, and genotype at the three main HbF modifier loci, BCL11A, HMIP‐2, and Xmn1‐HBG2, in 726 Tanzanian SCD patients, who have minimal disease intervention such as regular blood transfusion or hydroxyurea therapy.
Methods
The Muhimbili Sickle Cell Cohort has been described previously 1. Ethics approval is in place from the Muhimbili University Research and Publications Committee (MU/RP/AEC/VOLX1/33).
Confirmation of diagnosis (Hb SS or HbS/β0 thalassemia genotype) and HbF quantification were carried out by High Performance Liquid Chromatography (Variant I, Biorad, Hercules, CA). Hematological parameters (Hb, RBC, MCV, MCH, MCHC, WBC, PLT, and platelet volume‐MPV) were measured with an ABX Pentra 60 Analyzer (Horiba, Kyoto, Japan). Mean/median values for our cohort are shown in the Supporting Information Table SI.
Patients were excluded if they were on hydroxyurea therapy, younger than 60 months of age, tested malaria‐positive, had pain, fever, or had been hospitalized 30 days before or after study, or were lacking alpha thalassemia (3.7 deletion) data. Only patients with Hb SS or HbS/β0 thalassemia genotype were included. This resulted in a study population of 726 patients (52% females), aged 5–43 years (median 11 years, interquartile range 8–15 years).
Genetic variants were selected from ten SNPs genotyped at the three main HbF modifier loci, resulting a set of four, one effectively tagging HbF‐associated genetic variability at each locus 22, including the two sub‐loci (A and B) present at HMIP‐2 in individuals of African descent (Table 1). They were genotyped by TaqMan procedure (Applied Biosystems, Foster City, CA): rs11886868 (for BCL11A), rs9389269 (for HMIP‐2B), and rs7482144 (Xmn1‐HBG2, after PCR), or by PCR fragment sizing (rs66650371, for HMIP‐2A) 22, 23, 24. Alpha thalassemia (3.7 deletion) was genotyped using a PCR based method 26.
Table 1.
SNP Markers Used to Tag the Main HbF Modifier Loci
| Chromosome locus | Chr. 2 BCL11A | Chr. 6 HMIP‐2A | HMIP‐2B | Chr. 11 Xmn1‐HBG2 |
|---|---|---|---|---|
| SNP | rs11886868 | rs66650371 | rs9389269 | rs7482144 |
| Position | 60,720,496 | 135,418,633 | 135,427,159 | 5,276,419 |
| Allele change | T→C | In→Del | T→C | C→T |
| N/MAF | 764/0.29 | 727/0.03 | 764/0.03 | 723/0.01 |
| H/W_(p (1DF) | 0.29 | 0.85 | 0.80 | 0.96 |
| G. success (%) | 99 | 94.29 | 99 | 95.71 |
Chromosomal position is given in hg19 co‐ordinates.
The HMIP locus is divided into HMIP‐2A and HMIP‐2B, as recently proposed (34).
rs66650371 is characterized by presence/absence of a ‘TAY’ trinucleotide.
MAF: Minor allele frequency within the patient cohort.
H/W_(p (1DF): Hardy‐Weinberg P‐value, i.e., all four markers are in equilibrium.
G. success (Genotyping success): percent of individuals with genotype among those tested.
Multiple linear regression (STATA v.12, Stata Corp, College Station, TX) was used to test for association of genetic markers with hematological parameters and HbF, as well as for the influence of HbF on blood cell parameters. Alpha‐thalassemia status (3.7 deletion), age (fitted as square or native value) and sex were included a priori in all regression models. WBC, MPV, and HbF were log‐transformed. The genetic association analysis of the four tag markers with hematological parameters was repeated with ln[HbF] as a covariate, to test for the dependence of genetic effects on HbF levels.
Results
We detected a significant influence of HbF levels and of variants at three major HbF modifier loci, BCL11A, HMIP, and Xmn1‐HBG2, on the hematological phenotype of Tanzanian SCD patients.
Influence of HbF levels on hematological parameters
HbF levels associated positively with hemoglobin (Hb Beta = 0.05, P = 5.49 × 10−6), and negatively with WBC (lnWBC Beta = −0.01, P = 4.23 × 10−5) and platelet counts (Beta = 7.62, P = 6.3 × 10−6). Hb gains with higher HbF were accompanied by increases in MCV (Beta = 0.43, P = 9.9 × 10−9) and MCH (Beta = 0.16, P = 2.95 × 10−9), while RBC was unchanged (P = 0.97).
Influence of genetic HbF modifier variants on HbF and on hematological parameters
HbF levels were strongly influenced by all four variants tested, confirming previous findings 24 (Table 2). The number of HbF‐promoting alleles across all genotyped markers (‘Summary Score’) was positively associated with Hb, MCV, and MCH, similar to the pattern of effects exerted by HbF itself. Individual loci; however, had diverse effects (Table 2). The rare HbF‐promoting allele at Xmn1‐HBG2 (rs7482144‐‘T’) had by far the largest allelic effect on Hb (Beta = 0.79, P = 9.8 × 10−3), with a tendency towards increased RBC (Beta = 0.28, P = 0.06), but no change in MCV and MCH values (P > 0.1). When adjusting for the influence of HbF levels, the Hb‐increasing effect of rs7482144‐‘T’ remained large and significant (Beta = 0.69, P = 0.03) suggesting independence of this relationship from HbF levels. BCL11A (rs11886868‐‘C’) also had a significant effect on Hb (Beta = 0.19, P = 2 × 10−3), but this ceased to be significant after adjusting for HbF levels (P > 0.1). The two HbF‐increasing variants at HMIP‐2 (rs66650371‐‘del’, representing sub‐locus HMIP‐2A and rs9389269‐‘C’, representing HMIP‐2B) are uncommon in our population and no significant effect on Hb was detected. However, the HMIP‐2B variant had a significant positive effect on MCV and MCH and the HMIP‐2A variant had a negative effect on WBC (Beta = −0.19, P = 2.3 × 10−4), which was not diminished when adjusting for HbF.
Table 2.
Regression Analysis Testing the Influence of HbF and HbF Modifier Loci on Hematological Parameters in Tanzanian Patients with Sickle Cell Disease
| Outcome variables | HbF (lnHbF%) | Genetic HbF modifiers | ||||
|---|---|---|---|---|---|---|
| rs11886868 (BCL11A) | rs66650371 (HMIP‐2A) | rs9389269 (HMIP‐2B) | rs7482144 (Xmn1‐HBG2) | Summary scorea | ||
| lnHbF% | – | 0.37 (5.23 × 10−21) | 0.51 (2.88 × 10−5) | 0.44 (3.86 × 10−5) | 0.55 (4.35 × 10−3) | 0.40 (8.57 × 10−31) |
| Hb | 0.05 (5.5 × 10−6) | 0.19 (2 × 10−3) | 0.29 (0.14) | 0.10 (0.54) | 0.79 (9.8 × 10−3) | 0.21 (1.51 × 10−4) |
| RBC | −0.0002 (0.97) | 0.03 (0.36) | 0.004 (0.96) | −0.05 (0.49) | 0.28 (0.06) | 0.03 (0.35) |
| MCV | 0.43 (9.9 × 10−9) | 0.72 (0.10) | 1.82 (0.18) | 2.74 (0.02) | 2.15 (0.32) | 1.05 (5.5 × 10−3) |
| MCH | 0.16 (3.0 × 10−9) | 0.26 (0.11) | 0.74 (0.14) | 0.96 (0.02) | 0.12 (0.88) | 0.36 (0.01) |
| MCHC | 0.02 (0.14) | 0.07 (0.44) | 0.13 (0.61) | 0.06 (0.78) | −0.29 (0.48) | 0.05 (0.49) |
| lnWBC | −0.01 (4.2 × 10−5) | 5.7 × 10−5 (1) | −0.19 (2.3 ×10−4) | 0.02 (0.67) | −0.149 (0.06) | −0.02 (0.21) |
| PLT | 7.62 (6.3 × 10−6) | −9.82 (0.31) | 33.29 (0.27) | 22.36 (0.39) | −92.19 (0.06) | −6.43 (0.45) |
| lnMPV | 0.001 (0.57) | 0.0002 (0.98) | −0.018 (0.28) | −0.0007 (0.97) | −0.009 (0.73) | −0.002 (0.73) |
Shown are regression coefficient (Beta) estimates and significance (in brackets). Age, sex, and alpha globin status were included as covariates. For the genetic data, Beta serves as a measure of the effect of an allele change from low‐HbF to high‐HbF allele. Nominally significant effects are in bold font. N = 664–721.
The total number of high‐HbF alleles present in a patient.
Additive effect of Xmn1‐HBG2 and BCL11A alleles
When the impact of both loci was analyzed in a joint regression model, estimates of allelic effects were similar to those obtained in separate analysis (rs11886868: Beta = 0.19, P = 0.002, rs7482144: Beta = 0.79, P = 9.8 × 10−3), suggesting that they contribute independently and therefore beneficial effects might add their effects when occurring in the same individual. Accordingly, patients with HbF‐promoting alleles at both loci (one at Xmn1‐HBG2 and either one or two at BCL11A) had Hb levels of up to 8.5 g/dl on average, compared with 7.3 g/dl for patients lacking any such allele (Fig. 1).
Figure 1.
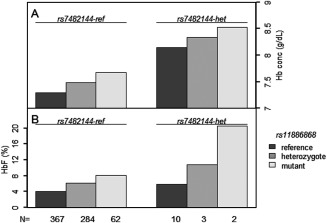
Coinheritance of rs11886868 and rs7482144 alleles and hemoglobin concentration. Panel (A) effect of coinheritance of rs7482144 reference genotype (left) or heterozygous (right) with either reference, heterozygous or mutant genotypes for rs11886868 on Hb levels. Panel B: effect of coinheritance of rs7482144 reference genotype (left) or heterozygous (right) with either reference, heterozygous or mutant genotypes for rs11886868 on HbF levels. There were no homozygotes for rs7482144 high‐HbF allele. The number of individuals in each combination is presented at the bottom.
Discussion
We report that both, increased HbF levels and HbF‐promoting alleles at HbF modifier loci significantly reduce anemia, leukocytosis, and thrombocytosis in Tanzanian patients with SCD.
The beneficial effects of higher HbF on hematological parameters, such as a higher Hb, lower WBC, and platelet counts, have previously been described in Jamaica 27, 28, 29, 30, but might be less evident in a setting where the most‐severely anemic patients are transfused regularly. The reduction in anemia we saw in patients with higher HbF levels was paralleled by an increase in two RBC indices (MCV and MCH), while RBC numbers were unchanged. Possibly, this indicates the presence of a larger F‐cell fraction in such patients, a hypothesis that will be investigated in further studies. It should be noted that iron status, an independent cause of variability in red blood cell indices particularly MCV, has only been assessed in a sub‐set of the patients.
Effects of the known genetic HbF modifiers (HbF‐increasing variants at Xmn1‐HBG2, BCL11A, and HMIP‐2) were, in general, similar to that of HbF itself: Hb, MCV, and MCH were increased when the loci were analyzed jointly, using a summary score. However, individual analysis of the four variants revealed distinct patterns of effects, suggesting that diverse biological mechanisms are involved. The ‘T’ allele at Xmn1‐HBG2 is a component of the ‘Arab‐Indian’ β globin gene cluster haplotype 31, 32, 33, known to be associated with higher HbF values and milder sickle disease. In the seven patients carrying a Xmn1‐HBG2‐T allele, Hb was raised, but MCV and MCH were not, probably due to the direct [34], ‘pancellular’ effect of β globin cluster variants on HbF production.
BCL11A (rs11886868‐C) had a significant effect on Hb, which was HbF‐dependent. The HbF increase due to this allele (Beta) was small, but as it is highly prevalent in this population (29% allele frequency), it created an overall significant impact. Five patients had HbF increasing alleles at both BCL11A and Xmn1‐HBG2 loci, resulting in maximum Hb levels (Fig. 1). Joint regression analysis of both loci supports a model of independence of their effects on Hb levels, and an additive contribution to overall hemoglobin variability. However, this will have to be confirmed in a larger population. HbF‐promoting alleles at HMIP are infrequent in Tanzanian patients (frequency of <0.20) and we detected no effect on Hb. HMIP‐2B (rs9389269) does influence MCV and MCH, a finding that resembles pleiotropic HMIP‐2 effects observed in nonanemic individuals 4, 5, 7, 8, 10. HMIP‐2A, but not HMIP‐2B, has a striking effect on WBC, independent of HbF. HMIP‐2 variants have been reported to influence the WBC count in healthy populations 9, but a possible influence of population stratification, given the ethnic diversity of Tanzania, will be evaluated in further studies.
We believe that the significant effects of the three modifier loci on general blood traits we have shown represent a combination of both, disease amelioration through HbF modification and pleiotropic effects, and that the mechanisms underlying both phenomena are diverse and gene‐specific. To explore this further, we will increase the power provided by our cohort by recruiting more patients and by broadening the scope of biological systems tested. Inclusion of additional hematological data in future analysis is expected to account for part of the background variability, thus improving our ability to detect more subtle genetic effects.
Acknowledgments
The authors thank the patients and staff of Muhimbili National Hospital, Muhimbili University of Health and Allied Sciences (MUHAS), Tanzania, Hematology Outpatient Unit and staff of King's College Hospital, London, and members of Professor Thein's Molecular Hematology group, King's College London. We extend our special thanks to J. Mgaya, H. Mariki of the Muhimbili Wellcome Programme, MUHAS and H. Rooks from Professor Thein's Molecular Hematology group. The sponsors of this study are nonprofit organizations that support science in general. They had no role in gathering, analyzing, or interpreting the data. Siana Nkya Mtatiro is a PhD candidate at Muhimbili University of Health and Allied Sciences and this work is submitted in partial fulfillment of the requirement for the PhD.
Author Contributions
S.L.T., S.M., S.N.M., S.E.C., and J.M. designed the study. S. M and S.N.M. designed and performed the genotyping assays. B.M. performed the initial analysis. S.N.M., S.M., S.L.T., and S.E.C. wrote the manuscript and all authors commented on the drafts of the manuscript.
Supporting information
Supporting Information
Conflict of interest: The authors declare no competing financial or other interests
†S.M. and S.E.C. contributed equally to this work.
References
- 1. Makani J, Cox SE, Soka D, et al. Mortality in sickle cell anemia in Africa: A prospective cohort study in Tanzania. PLoS One 2011;6:e14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–1644. [DOI] [PubMed] [Google Scholar]
- 3. Thein S. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet 2009;18:R216–R223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Menzel S, Jiang J, Silver N, et al. The HBS1L‐MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood 2007;110:3624–3626. [DOI] [PubMed] [Google Scholar]
- 5. van der Harst P, Zhang W, Mateo Leach I, et al. Seventy‐five genetic loci influencing the human red blood cell. Nature 2012;492:369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okada Y, Kamatani Y. Common genetic factors for hematological traits in humans. J Hum Genet 2012;57:161–169. [DOI] [PubMed] [Google Scholar]
- 7. Ganesh SK, Zakai NA, van Rooij FJ, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet 2009;41:1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamatani Y, Matsuda K, Okada Y, et al. Genome‐wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 2010;42:210–215. [DOI] [PubMed] [Google Scholar]
- 9. Ferreira MA, Hottenga JJ, Warrington NM, et al. Sequence variants in three loci influence monocyte counts and erythrocyte volume. Am J Hum Genet 2009;85:745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. N Soranzo, TD Spector, M Mangino, et al. A genome‐wide meta‐analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 2009;41:1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Francis RB Jr. Platelets, coagulation, and fibrinolysis in sickle cell disease: Their possible role in vascular occlusion. Blood coagulation & fibrinolysis: an international journal in haemostasis and thrombosis 1991;2:341–353. [DOI] [PubMed] [Google Scholar]
- 12. Ataga KI, Key NS. Hypercoagulability in sickle cell disease: New approaches to an old problem. Hematol Am Soc Hematol Educ Program 2007:91–96. [DOI] [PubMed] [Google Scholar]
- 13. Robert B. Francis JaRPH. Hemostasis In: Stephen H. Embury RPH, Mohandas N, Steinberg MH, editors. Sickle Cell Disease: Basic Principles and Clinical Practise. New York: Raven Press; 1994. [Google Scholar]
- 14. Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br J Haematol 2013;162:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blau CA, Constantoulakis P, al‐Khatti A, et al. Fetal hemoglobin in acute and chronic states of erythroid expansion. Blood 1993;81:227–233. [PubMed] [Google Scholar]
- 16. Swank RA, Stamatoyannopoulos G. Fetal gene reactivation. Curr Opin Genet Dev 1998;8:366–370. [DOI] [PubMed] [Google Scholar]
- 17. Rees DC, Porter JB, Clegg JB, et al. Why are hemoglobin F levels increased in HbE/beta thalassemia? Blood 1999;94:3199–3204. [PubMed] [Google Scholar]
- 18. Dover GJ, Boyer SH. Fetal hemoglobin‐containing cells have the same mean corpuscular hemoglobin as cells without fetal hemoglobin: A reciprocal relationship between gamma‐ and beta‐globin gene expression in normal subjects and in those with high fetal hemoglobin production. Blood 1987;69:1109–1113. [PubMed] [Google Scholar]
- 19. Dover GJ, Boyer SH, Charache S, et al. Individual variation in the production and survival of F cells in sickle‐cell disease. N Engl J Med 1978;299:1428–1435. [DOI] [PubMed] [Google Scholar]
- 20. Franco RS, Yasin Z, Palascak MB, et al. The effect of fetal hemoglobin on the survival characteristics of sickle cells. Blood 2006;108:1073–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagel RL, Platt OS. General pathophysiology of sickle cell anemia In: Steinberg MH, Forget BG, Higgs DR, et al. editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge, UK: Cambridge University Press; 2001. pp 494–526. [Google Scholar]
- 22. Menzel S, Rooks H, Zelenika D, et al. Global genetic architecture of an erythroid quantitative trait locus, HMIP‐2. Ann Hum Genet 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Creary LE, Ulug P, Menzel S, et al. Genetic variation on chromosome 6 influences F cell levels in healthy individuals of African descent and HbF levels in sickle cell patients. PLoS One 2009;4:e4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makani J, Menzel S, Nkya S, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood 2011;117:1390–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox SE, Makani J, Soka D, et al. Haptoglobin, alpha‐thalassaemia and glucose‐6‐phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br J Haematol 2014;165:699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Donaldson A, Thomas P, Serjeant BE, et al. Foetal haemoglobin in homozygous sickle cell disease: A study of patients with low HBF levels. Clin Lab Haematol 2001;23:285–289. [DOI] [PubMed] [Google Scholar]
- 27. Maude GH, Hayes RJ, Serjeant GR. The haematology of steady state homozygous sickle cell disease: Interrelationships between haematological indices. Br J Haematol 1987;66:549–558. [DOI] [PubMed] [Google Scholar]
- 28. Olatunji PO, Davies SC. The predictive value of white cell count in assessing clinical severity of sickle cell anaemia in Afro‐Caribbeans patients. Afr J Med Med Sci 2000;29:27–30. [PubMed] [Google Scholar]
- 29. Ikuta T, Adekile AD, Gutsaeva DR, et al. The proinflammatory cytokine GM‐CSF downregulates fetal hemoglobin expression by attenuating the cAMP‐dependent pathway in sickle cell disease. Blood Cells Mol Dis 2011;47:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol 2009;145:455–467. [DOI] [PubMed] [Google Scholar]
- 31. Rahimi Z, Karimi M, Haghshenass M, et al. Beta‐globin gene cluster haplotypes in sickle cell patients from southwest Iran. Am J Hematol 2003;74:156–160. [DOI] [PubMed] [Google Scholar]
- 32. Padmos MA, Roberts GT, Sackey K, et al. Two different forms of homozygous sickle cell disease occur in Saudi Arabia. Br J Haematol 1991;79:93–98. [DOI] [PubMed] [Google Scholar]
- 33. Gilman JG, Huisman TH. DNA sequence variation associated with elevated fetal G gamma globin production. Blood 1985;66:783–787. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information