

Abstract
An improved understanding of how a parasite species exploits its genetic repertoire to colonize novel hosts and environmental niches is crucial to establish the epidemiological risk associated with emergent pathogenic genotypes. Trypanosoma cruzi, a genetically heterogeneous, multi‐host zoonosis, provides an ideal system to examine the sylvatic diversification of parasitic protozoa. In Bolivia, T. cruzi I, the oldest and most widespread genetic lineage, is pervasive across a range of ecological clines. High‐resolution nuclear (26 loci) and mitochondrial (10 loci) genotyping of 199 contemporaneous sylvatic TcI clones was undertaken to provide insights into the biogeographical basis of T. cruzi evolution. Three distinct sylvatic parasite transmission cycles were identified: one highland population among terrestrial rodent and triatomine species, composed of genetically homogenous strains (A r = 2.95; PA/L = 0.61; DAS = 0.151), and two highly diverse, parasite assemblages circulating among predominantly arboreal mammals and vectors in the lowlands (A r = 3.40 and 3.93; PA/L = 1.12 and 0.60; DAS = 0.425 and 0.311, respectively). Very limited gene flow between neighbouring terrestrial highland and arboreal lowland areas (distance ~220 km; FST = 0.42 and 0.35) but strong connectivity between ecologically similar but geographically disparate terrestrial highland ecotopes (distance >465 km; FST = 0.016–0.084) strongly supports ecological host fitting as the predominant mechanism of parasite diversification. Dissimilar heterozygosity estimates (excess in highlands, deficit in lowlands) and mitochondrial introgression among lowland strains may indicate fundamental differences in mating strategies between populations. Finally, accelerated parasite dissemination between densely populated, highland areas, compared to uninhabited lowland foci, likely reflects passive, long‐range anthroponotic dispersal. The impact of humans on the risk of epizootic Chagas disease transmission in Bolivia is discussed.
Keywords: ecological fitting, microsatellites, mitochondria, population genetics, sylvatic transmission, Trypanosoma cruzi
Introduction
Host–parasite relationships are assumed to be ecologically specialized, tightly co‐evolved systems driven by either mutual modification (synchronous cospeciation) or exaptation into novel environmental niches, often accompanied by host switching (ecological fitting) (Janzen 1985; Brooks et al. 2006). Ecological fitting occurs when an organism co‐opts an existing suite of genetic traits to exploit an unfamiliar resource or colonize and persist in a new or modified environment (Agosta & Klemens 2008). Distinguishing between host–parasite relationships that result from ecological fitting or long‐term co‐evolution remains challenging. However, understanding how a species exploits their current genetic repertoire to form novel host associations is of primary interest to the study of emerging infectious diseases, with considerable implications for the design of disease control programmes (Brooks & Ferrao 2005; Agosta et al. 2010). In this regard, Trypanosoma cruzi (Kinetoplastida: Trypanosomatidae), the aetiological agent of Chagas disease, a pervasive zoonosis that is eclectic with respect to host species and tissues it can inhabit, provides a model system to examine the genetic diversification of parasitic protozoa.
Chagas disease is the most important vector‐borne infection in Latin America, affecting an estimated 7–8 million individuals (World Health Organization 2014). Following acute disease, which is often undiagnosed, the majority of patients are clinically asymptomatic for life. Without treatment, approximately 20–30% will develop irreversible, potentially fatal cardiomyopathy or, more rarely, dilatation of the gastrointestinal tract (megaesophagus or megacolon) (Rassi et al. 2010). The geographical distribution of T. cruzi extends from the southern United States to Argentinean Patagonia, where it is transmitted by more than 100 species of hematophagus triatomine bugs (Hemiptera: Reduviidae: Triatominae) (Lent & Wygodzinsky 1979; Galvão et al. 2003). Human disease is primarily confined to areas of Central and South America where individuals are exposed to infected faeces of domiciliated or invasive triatomines through contact with intact mucosae or abraded skin (Coura & Dias 2009) or by oral ingestion of contaminated food/drink (Shikanai‐Yasuda & Carvalho 2012). In addition, enzootic T. cruzi infection is naturally sustained by an extensive range of domestic, synanthropic and sylvatic mammalian hosts (Noireau et al. 2009).
Trypanosoma cruzi is an ancient parasite, estimated to have diverged from its most recent common ancestor 3–4 Ma (Lewis et al. 2011), and as such, it is characterized by considerable genetic diversity (Stevens et al. 1999). Current international consensus recognizes a minimum of six stable genetic lineages or discrete typing units (DTUs) (TcI‐TcVI) (Zingales et al. 2009), which have distributions loosely defined by geography, ecology and transmission cycle (Miles et al. 2009). The level of nuclear sequence divergence between major T. cruzi DTUs is equivalent to interspecies diversity among New World Leishmania species (Yeo et al. 2011; Boité et al. 2012). TcI is the most widely distributed DTU; it is the principal cause of human chagasic cardiomyopathy in Colombia and Venezuela (Ramírez et al. 2010; Carrasco et al. 2012) and is ubiquitous among sylvatic transmission cycles across the parasite's endemic range (Llewellyn et al. 2009a). Multiple molecular markers consistently identify high levels of genetic diversity within sylvatic TcI populations (Herrera et al. 2007, 2009; O'Connor et al. 2007; Falla et al. 2009; Llewellyn et al. 2009a; Ocaña‐Mayorga et al. 2010; Lima et al. 2014), and divergent, but genetically homogeneous, strains isolated from human infections (Llewellyn et al. 2009a; Cura et al. 2010; Ramírez et al. 2012; Zumaya‐Estrada et al. 2012). However, the genetic determinants that drive natural T. cruzi diversification are largely unknown. Some have proposed that T. cruzi lineages co‐evolved in close concert with discrete vertebrate hosts and insect vectors (Miles et al. 1981; Gaunt & Miles 2000; Yeo et al. 2005), while others favour ecological fitting as a more parsimonious explanation for contemporary host associations (Hamilton et al. 2007; Agosta & Klemens 2008; Llewellyn et al. 2009a). Evidence to support the latter is increasing; TcI transmission is now known to span multiple ecological niches (Lisboa et al. 2004; Herrera et al. 2005, 2008a,b; Llewellyn et al. 2009a; Rocha et al. 2013; Lima et al. 2014), and genetic diversity of terrestrial TcIII appears similarly independent of host species (Llewellyn et al. 2009b; Marcili et al. 2009).
Bolivia comprises diverse sylvatic ecotopes where TcI transmission persists unabated and thus provides a perfect platform to test ecological hypotheses. Colonies of Triatoma infestans, infected with TcI (Barnabé et al. 2011; Breniere et al. 2012), have been reported in highland Andean valleys (Cortez et al. 2006, 2007; Buitrago et al. 2010) and to the south in the arid, lowland Chaco region (Ceballos et al. 2011; Waleckx et al. 2012), where their potential for domestic re‐invasion threatens the success of the National Control Programme (Noireau et al. 2005; Noireau 2009). Sylvatic TcI transmission also extends northwards to sparsely populated Amazonian Beni, where disease ecology is poorly described (Matias et al. 2003; Justi et al. 2010). Bolivia suffers the greatest human burden of T. cruzi infection in Latin America, impacting approximately 6.75% of the population (Jannin & Salvatella 2006). Chagas disease is endemic across two‐thirds of the country and concentrated disproportionally among lower socio‐economic rural populations with seroprevalence reaching 72.7–97.1% among adults of some communities (Medrano‐Mercado et al. 2008; Samuels et al. 2013). In these areas, continuing domestic transmission has been attributed to a decrease in intensity of residual insecticide spraying (Samuels et al. 2013; Espinoza et al. 2014), the emergence of insecticide resistance (Germano et al. 2010; Lardeux et al. 2010) and decentralized vector control initiatives in areas of recurrent political, social and economic instability (Gürtler 2009).
To date, few studies have adopted rigorous sampling strategies and genetic markers with sufficient resolution to elucidate fully the biogeographical basis of T. cruzi evolution. Ideally, parasite samples should be minimally subdivided biologically, spatially and temporally, with multiple clones examined from each host (Prugnolle & De Meeus 2010). In practice, low circulating parasitaemia often prohibits parasite isolation, and thus, many studies are heavily reliant on historical collections of reference isolates. T. cruzi genetic analysis is further complicated by the presence of mixed DTU infections (Bosseno et al. 1996; Yeo et al. 2005; Burgos et al. 2008) and multiclonal parasite populations within individual hosts and vectors (Llewellyn et al. 2011), requiring strains to be biologically cloned prior to genotyping, a laborious caveat often overlooked by researchers.
In this study, we applied high‐resolution nuclear and mitochondrial genotyping to contemporaneous biologically cloned TcI strains, isolated from triatomines and mammalian hosts across Bolivia, to identify key determinants of sylvatic T. cruzi genetic diversification. We also explore genetic diversity and potential hybridization along two ecological clines, first between highland and lowland ecotopes and second within lowland Bolivia itself. Finally, we examine the spatial genetic structure of natural TcI populations and consider the implications of our data for human Chagas disease transmission in Bolivia.
Materials and methods
Study area and parasite sampling
Parasite strains were isolated from sylvatic terrestrial and arboreal transmission cycles in five localities across three departments in Bolivia (Cochabamba, Potosí and Beni) (Fig. 1). Study sites were situated at altitudes that ranged from ~143 to 3200 m and selected to span five major ecoregions: savannah grassland and Madeira‐Tapajós moist forests (Beni), dry Andean puna and Yungas (Cochabamba) and wet Andean puna (Potosí). Triatomine vectors were sampled using a combination of manual microhabitat dissection and live‐baited Noireau traps (Noireau et al. 2002). Wild mammalian reservoir hosts were captured using Sherman and Tomahawk box and cage traps. Parasite sampling was undertaken from 2004 to 2010 and is described for each study site individually.
Figure 1.
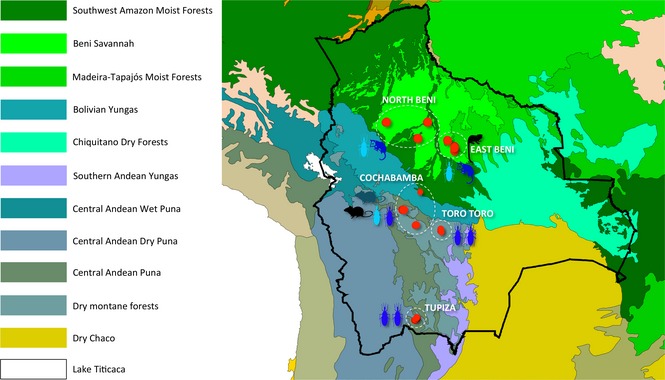
Map of Bolivia showing distribution of sylvatic TcI isolates among different ecotopes. Parasite strains were isolated from terrestrial and arboreal transmission cycles in five localities across three departments: Cochabamba, Potosí and Beni. Study sites were situated at altitudes that ranged from ~143 to 3200 m and spanned five different ecoregions: savannah grassland and Madeira‐Tapajós moist forests (Beni), dry Andean puna and Yungas (Cochabamba) and dry montane forests (Potosí). Geographical origins of individual strains are shown by closed red circles. Circle areas are proportionate to sampling density. Images indicate sample host/vector origin (rodent, marsupial, primate or triatomine). Open white circles designate five a priori populations: Cochabamba, Tupiza, Toro Toro, North Beni and East Beni used for population genetic analyses. Population and department names are indicated in uppercase and lowercase, respectively.
The Cotopachi site in Cochabamba department is located in a densely populated area of open dry Andean puna (thorny scrub vegetation interspersed with rocky outcrops and large, spiny cacti), ~20 km southwest of Cochabamba city at an elevation of ~2600 m. Here, TcI parasites were sampled from wild Triatoma infestans and terrestrial rodents (Akodon boliviensis and Phyllotis osilae). Triatomine sampling (T. infestans and T. guasayana) was also undertaken in neighbouring Toro Toro, an area of similar ecology to Cotopachi, situated at ~2700 m. North of Cotopachi, sylvatic Rhodnius robustus were collected from Chapare, a dense temperate montane forest (Yungas) in the westernmost foothills of the Andes.
South of Cochabamba, TcI parasites were isolated from wild T. infestans in Tupiza, a region of high‐altitude (~3200 m) montane grasslands in Potosí department.
Sampling was undertaken in two regions of Beni department, a sparsely populated province in eastern lowland Bolivia. Ecologically, Beni is a patchwork of two principal vegetation types. The majority of the department is covered by lush savannah grassland (Llanos de Moxos). Along riverine alluvial plains and to the northern and western borders of the area, this ecotope is supplanted by dense Amazonian moist forests. To the east, Beni borders another moist forest (Madeira‐Tapajós), which extends into Brazil and Santa Cruz department. In East Beni (Nueva Alianza, San Juan de Aguas Dulces and San Juan de Mocovi), TcI parasites were isolated from triatomines (Rhodnius pictipes) and mammals (Didelphis marsupialis, Philander opossum and Sciureus species) in areas of savannah grassland, interspersed with large stands of evergreen palm trees, on the boundary between Llanos de Moxos and the moist forests of northwestern Santa Cruz. The study sites in North Beni (Mercedes, San Cristobal and Santa Maria de Apere) were remote, largely uninhabited, open savannah grasslands with occasional lone standing trees, bordered by riverine forests. Here TcI parasites were isolated from R. robustus, P. opossum and D. marsupialis. Both study sites in North and East Beni were situated at low‐lying altitudes (~143 and ~160 m, respectively), and parasite sampling was undertaken using similar methods described for other departments.
All parasite strains were isolated by direct inoculation of triatomine faeces or heparinized venous animal blood into biphasic hemoculture media (Miles 1993).
Parasite strains and DTU confirmation
A panel of 199 biological clones derived from 68 Trypanosoma cruzi TcI isolates was assembled for analysis (Table S1, Supporting information). Biological clones were obtained from primary cultures by plate cloning according to Yeo et al. (2007) to minimize any loss of genetic diversity incurred by long‐term maintenance in culture. Parasites (epimastigotes) were expanded to logarithmic phase at 28 °C in RPMI‐1640 liquid media supplemented with 0.5% (w/v) tryptone, 20 mm HEPES buffer (pH 7.2), 30 mm haemin, 10% (v/v) heat‐inactivated foetal calf serum, 2 mm sodium glutamate, 2 mm sodium pyruvate and 25 μg/mL gentamycin (all Sigma‐Aldrich, UK). Genomic DNA was extracted using the Gentra PureGene Tissue Kit (Qiagen, UK), according to the manufacturer's protocol. Clone genotypes were confirmed as TcI using a triple‐marker assay (Lewis et al. 2009), to exclude the potential presence of mixed infections with several DTUs, and classified a priori into five populations according to geographical origin: Cochabamba (n = 28), Tupiza (n = 15), Toro Toro (n = 43), North Beni (n = 26) and East Beni (n = 87).
High‐resolution genotyping: multilocus microsatellite typing
Twenty‐six microsatellite loci were amplified for all 199 clones, as previously described by Llewellyn et al. (2009a). These markers were distributed across 10 putative chromosomes, including six groups of physically linked loci (Weatherly et al. 2009). A full list of microsatellite targets and primers are given in Table S2 (Supporting information). Allele sizes were determined using an automated capillary sequencer (AB3730, Applied Biosystems, UK), in conjunction with a fluorescently tagged size standard, and were manually checked for errors. All isolates were typed ‘blind’ to control for user bias.
High‐resolution genotyping: mitochondrial multilocus sequence typing
Ten maxicircle gene fragments were sequenced for a subset of 78 clones, chosen to be representative of total nuclear genetic diversity (indicated in Table S1, Supporting information) (Messenger et al. 2012). For NADH dehydrogenase subunit 4 (ND4), an alternate set of primers was designed to improve amplification efficiency: ND4 forward (5′‐TTYTTCCCAATATGTATBGTMAG‐3′) and ND4 reverse (5′‐TGTATTAYCGAYCAATTYGC‐3′), and reactions were performed using the same conditions as previously (Messenger et al. 2012).
Microsatellite analysis
Individual‐level sample clustering was initially defined using a neighbour‐joining (NJ) tree based on pairwise distances (D AS: 1 − proportion of shared alleles at all loci/n) between microsatellite genotypes calculated in microsat v1.5d (Minch et al. 1997) under the infinite‐alleles model. To accommodate multi‐allelic genotypes (≥3 alleles per locus), a script was written in Microsoft Visual Basic to generate random multiple diploid resamplings of each multilocus profile (software available upon request). A final pairwise distance matrix was derived from the mean across multiple resampled data sets and used to construct a NJ phylogenetic tree in phylip v3.67 (Felsenstein 1989). Majority rule consensus analysis of 10 000 bootstrap trees was performed in phylip v3.67 by combining 100 bootstraps generated in microsat v1.5d, each drawn from 100 respective randomly resampled data sets.
A second analysis to define the number of putative populations in the data set was performed using a nonparametric approach (free from Hardy–Weinberg assumptions). A K‐means clustering algorithm, implemented in adegenet (Jombart 2008), was used to determine the optimal number of ‘true’ populations, with reference to the Bayesian information criterion (BIC), which reaches a minimum when approaching the best supported assignment of individuals to the appropriate number of clusters. The relationship between these clusters and the individuals within them was then evaluated via a discriminant analysis of principal components (DAPC) according to Jombart et al. (2010).
A single randomly sampled diploid data set was used for all subsequent analyses (Appendix S1, Supporting information; available from Dryad doi: 10.5061/dryad.b8465). Population‐level genetic diversity was evaluated using sample‐size‐corrected allelic richness (A r) in fstat 2.9.3.2 (Goudet 1995). In addition, mean F IS, which measures the distribution of heterozygosity within and between individuals, was calculated per population in fstat 2.9.3.2. F IS can vary between −1 (all loci are heterozygous for the same alleles) and +1 (all loci are homozygous for different alleles). F IS = 0 indicates Hardy–Weinberg allele frequencies. Sample‐size‐corrected private (population‐specific) allele frequency per locus (PA/L) was calculated in hp‐rare (Kalinowski 2005).
Population subdivision was estimated using pairwise F ST, linearized with Slatkin's correction, in arlequin v3.11 (Excoffier et al. 2005). Statistical significance was assessed via 10 000 random permutations of alleles between populations. Within‐population subdivision was evaluated in arlequin v3.11 using a hierarchal analysis of molecular variance (amova). Population‐level heterozygosity indices were also calculated in arlequin v3.11 and associated significance levels for P‐values derived after performing a sequential Bonferroni correction to minimize the likelihood of type I errors (Rice 1989). Multilocus linkage disequilibrium, estimated by the index of association (I A), was calculated in multilocus 1.3b (Agapow & Burt 2001), and statistical significance was evaluated by comparison with a null distribution of 1000 randomizations. Mantel's tests for the effect of isolation by distance (IBD) within populations (pairwise genetic vs. geographical distance) were implemented in genaiex 6.5 using 10 000 random permutations (Peakall & Smouse 2012).
Mitochondrial analysis
Sequence data from 10 maxicircle gene fragments were concatenated for each isolate according to Messenger et al. (2012) and are available from Dryad (doi: 10.5061/dryad.b8465). Additional mitochondrial multilocus sequence typing (mtMLST) data from 24 previously published TcI strains were included in selected analyses, as indicated (Messenger et al. 2012). The most appropriate nucleotide substitution model was selected from 1624 candidates, based on the Akaike information criterion (AIC), in jmodeltest 2.1.4 (Darriba et al. 2012). Alternate maximum‐likelihood (ML) phylogenies were constructed using the TrN+G model (six substitution rate categories) in mega 5.10 (Tamura et al. 2011). Bootstrap support for clade topologies was estimated following the generation of 1000 pseudoreplicate data sets. Bayesian phylogenetic analysis was performed with mrbayes, implemented through topali v2.5, using the best‐fit model based on the BIC (GTR+G) (Milne et al. 2009). Five independent analyses were run for one million generations, with sampling every 100 simulations (30% burn‐in). Statistically supported topological incongruence between alternate mitochondrial and nuclear phylogenies was evaluated using Kishino–Hasegawa (KH) (Kishino & Hasegawa 1989) and Shimodaira–Hasegawa (SH) (Shimodaira & Hasegawa 1999) likelihood tests in paml v.4 (Yang 2007). Haplotype diversity (H d) was calculated using dnasp v5.10.1 (Librado & Rozas 2009).
Results
Strain characteristics
One hundred and ninety‐nine biological clones were genotyped across 26 polymorphic microsatellite loci (Appendix S1, Supporting information). In total, 10 122 alleles were identified, corresponding to 178 unique multilocus genotypes (MLGs). Multiple (≥3) alleles were observed at 0.83% of loci. Levels of intrastrain genetic diversity were high; multiclonality, that is the presence of multiple, different genetic clones, was observed in 65/68 strains. Identical intraclonal genotypes were sampled in five isolates (1/18 Toro Toro, 1/11 Cochabamba and 3/26 East Beni). Clones were initially categorized into five populations based on geographical origin, consisting of three high‐altitude (Cochabamba, Tupiza and Toro Toro) and two low‐altitude groups (North and East Beni). All populations demonstrated uniformly high numbers of unique MLGs and low frequencies of repeated MLGs (Table 1).
Table 1.
Population genetic parameters for sylvatic populations of Trypanosoma cruzi TcI in Bolivia
| Populationa | G/N | Max. Freq. MLG | H d (H/N) | PL | PA/L ±SE | A r ±SE | H o | H e | % H e | % H d | F IS ±SE | I A | I A P‐value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All highlands | 75/86 | 3 | 0.54 (9/46) | 21 | 0.61 ± 0.15 | 2.95 ± 0.37 | 0.26 | 0.23 | 33.3 | 19 | −0.158 ± 0.02 | 2.06 | <0.001 |
| Cochabamba (highlands) | 25/28 | 2 | 0.40 (4/14) | 20 | 0.42 ± 0.12 | 2.22 ± 0.20 | 0.29 | 0.24 | 30 | 5 | −0.206 ± 0.10 | 2.56 | <0.001 |
| Tupiza (highlands) | 14/15 | 2 | 0.73 (3/6) | 15 | 0.21 ± 0.07 | 2.21 ± 0.29 | 0.28 | 0.28 | 6.7 | 13.3 | 0.026 ± 0.08 | 3.54 | <0.001 |
| Toro Toro (highlands) | 39/43 | 2 | 0.46 (4/26) | 18 | 0.19 ± 0.06 | 1.92 ± 0.21 | 0.25 | 0.20 | 22.2 | 11.1 | −0.241 ± 0.09 | 1.48 | <0.001 |
| North Beni (lowlands) | 22/26 | 2 | 0.81 (4/7) | 19 | 0.60 ± 0.16 | 3.93 ± 0.39 | 0.37 | 0.45 | 10.5 | 63.2 | 0.176 ± 0.06 | 2.70 | <0.001 |
| East Beni (lowlands) | 78/87 | 3 | 0.84 (9/25) | 21 | 1.12 ± 0.29 | 3.40 ± 0.46 | 0.39 | 0.48 | 9.5 | 52.3 | 0.203 ± 0.05 | 2.23 | <0.001 |
N, number of isolates in population; G, number of multilocus genotypes (MLGs) per population based on microsatellite data of 26 loci analysed; Max. Freq. of MLG, frequency of the most common MLG within the population; H, number of haplotypes in population; H d, haplotype diversity measures the uniqueness of a particular haplotype in a given population, calculated using available mitochondrial sequence data in dnasp v5.10.1 (Librado & Rozas 2009); PL, number of polymorphic loci out of 26 loci analysed; A r, allelic richness as a mean over loci ±SE, calculated in fstat 2.9.3.2 (Goudet 1995); PA/L, mean number of private alleles per locus ±SE, calculated in HP‐Rare (Kalinowski 2005); H o, mean observed heterozygosity across all loci; H e, mean expected heterozygosity across all loci; %H E, proportion of loci showing a significant excess in heterozygosity after a sequential Bonferroni correction (Rice 1989); %H d, proportion of loci showing a significant deficit in heterozygosity after a sequential Bonferroni correction (Rice 1989); F IS, mean fixation index ±SE, calculated in fstat 2.9.3.2 (Goudet 1995); I A, index of association calculated in multilocus 1.3b; P‐value estimated by comparison with a null distribution of 1000 randomizations (Agapow & Burt 2001); DAPC, discriminant analysis of principal components.
Population designation based on a priori geographical populations and DAPC/D AS strain assignments.
Nuclear genetic clustering among isolates
Patterns of isolate clustering were evaluated using two different methodologies: nonparametric population assignment (DAPC) and a NJ analysis based on pairwise genetic distances (D AS). Ten genetic clusters were defined among the 199 clones submitted to DAPC, once three principal components (PCs) were retained and analysed (representing 80% of the total variation). A full list of isolate assignments to DAPC populations is included in Table S1 (Supporting information), and a multidimensional scaling plot of the DAPC results is shown in Fig. 2. We observed a slight ‘elbow’ in the distribution of the BIC values across optimal cluster numbers at K = 10 (Fig. 2). DAPC‐derived clusters were largely congruent with a priori allocations of strains to geographical populations. The 10 DAPC clusters separated into three genetically distinct groups: highlands (clusters 1, 8 and 10), lowlands 1 (clusters 2, 3 and 6) and lowlands 2 (clusters 4, 5, 7 and 9). The highlands group corresponded exclusively to samples from Cochabamba, Tupiza and Toro Toro, with the exception of a single clone from Rhodnius robustus in Chapare (CV‐05 cl1), which was instead assigned to cluster 2 in the lowlands 1 group. Within the highlands group, isolates from different sampling areas and sources (hosts and vectors) were distributed across clusters 8 and 10, while cluster 1 comprised only a subset of clones from Triatoma infestans found in Tupiza and Toro Toro. The lowlands 1 group encompassed all strains from North Beni (only cluster 2) and approximately half of the isolates from Rhodnius spp. and Didelphis marsupialis in East Beni (interspersed among clusters 2, 3 and 6). Lastly, the lowlands 2 group contained all remaining East Beni clones, including those isolated from Rhodnius spp., D. marsupialis, Philander opossum and Sciureus spp.
Figure 2.
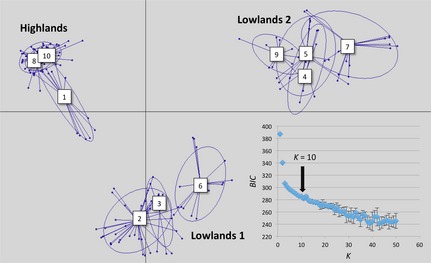
Nuclear genetic clustering among 199 sylvatic Bolivian TcI clones. Multidimensional scaling plot based on discriminant analysis of principal component (DAPC) analysis for 10 clusters defined via K‐means clustering algorithm (109 iterations, three principal components representing 80% of total variation in the data set). Bayesian information criterion (BIC) curve is inserted with error bars representing the standard deviation about the mean of five independent runs. Inertia ellipses correspond to the optimal (as defined by the BIC minimum) number of population clusters among the genotypes analysed. Individual clones are indicated by dots. The 10 DAPC clusters are separated into three genetically distinct groups: highlands (clusters 1, 8 and 10), lowlands 1 (clusters 2, 3 and 6) and lowlands 2 (clusters 4, 5, 7 and 9).
A NJ tree based on the same microsatellite data was constructed and further corroborated the DAPC strain assignments. A clear division between highland and lowland populations was observed, with isolates segregating into two well‐supported clades (64% BS) (Fig. 3). Similar to the DAPC results, the D AS topology supported the delineation of isolates from Beni into two groups (71% BS), one composed of all North Beni clones and the same portion of East Beni clones (D AS lowlands 1), the other containing the remaining East Beni strains (D AS lowlands 2). As previously, CV‐05 cl1 from Chapare clustered as an outlier among North and East Beni isolates. Comparison of branch lengths in Fig. 3 between the two lowland populations indicated high and consistent levels of genetic variation across strains. By contrast, highland isolates were less diverse overall (mean pairwise D AS = 0.151 and 0.431 for highlands and lowlands, respectively). Within this clade, there was strong evidence for the existence of local geographical clusters in Tupiza (100% BS) and Toro Toro (73% BS), which clustered basally to the remaining highland strains.
Figure 3.
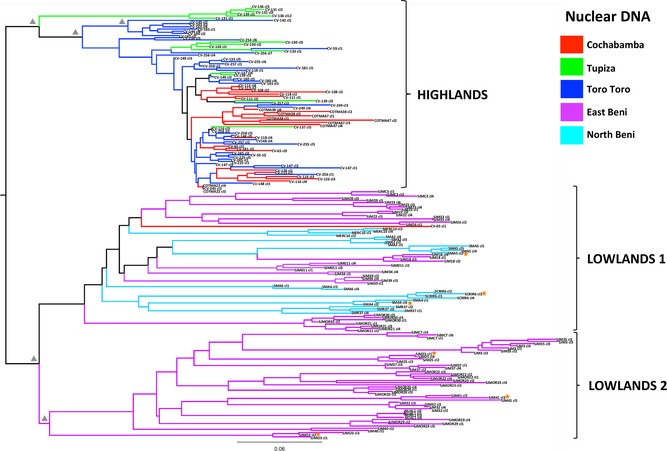
Unrooted neighbour‐joining tree based on DAS values between multilocus genotypes generated for 199 sylvatic Bolivian TcI clones. DAS values were calculated as the mean across 1000 random diploid resamplings of the data set. Branch colours indicate isolate a priori population (Cochabamba, Tupiza, Toro Toro, East Beni and North Beni; see legend). Closed grey triangles are adjacent to nodes that receive >60% bootstrap support. Isolates are grouped into three statistically supported clades (highlands, lowlands 1 and lowlands 2). Orange stars denote clones which have phylogenetically incongruent positions between nuclear and mitochondrial topologies.
Population characteristics
Population genetic indices were calculated using both a priori geographical and DAPC/D AS‐supported strain assignments (Table 1). Overall, a clear division in genetic diversity and heterozygosity was apparent between highland and lowland areas. The three highland populations were characterized by lower levels of genetic diversity, as evidenced by smaller estimates of allelic richness (A r = 1.92–2.22) and numbers of private alleles per locus (PA/L = 0.19–0.42), compared to the lowlands (A r = 3.40 and 3.93 and PA/L = 1.12 and 0.60, respectively) (Table 1 and Fig. 4A). All highland groups had moderately excess heterozygosity (F IS = −0.241 to 0.026, 5–13.3% polymorphic loci with significant deficit in heterozygosity), whereas both lowland populations demonstrated more pronounced deviations from H–W allele frequencies (F IS = 0.176 and 0.203, 63.2% and 52.3% polymorphic loci with significant deficit in heterozygosity, respectively) (Table 1). Strongly significant multilocus linkage disequilibrium was observed among all study areas (I A = P < 0.0001 for all populations).
Figure 4.
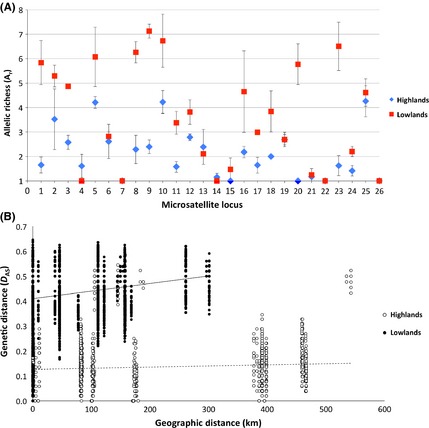
(A) Allelic richness (A r) per microsatellite locus for grouped a priori geographical highland (diamonds) and lowland (squares) populations. Highland populations were characterized by smaller estimates of allelic richness (A r), compared to the lowlands (average of A r = 1.92–2.22 and 3.40 and 3.93, respectively). Error bars represent ±SE about the mean. Values without error bars correspond to markers containing only a single variable locus. (B) Nuclear spatial genetic analysis among Trypanosoma cruzi isolates from highland (open circles) and lowland (closed circles) populations. Nuclear genetic isolation by distance (IBD) was observed among lowland populations (RXY = 0.209, P < 0.001; slope = 0.0003 ± 0.0000179), while no spatial structure was evident among highland populations spanning a much greater geographical area (RXY = 0.109, P = 0.085; slope = 0.0002 ± 0.0000307).
Interpopulation gene flow and intrapopulation subdivision
Estimates of subdivision (F ST) between a priori populations support a genetic demarcation between highland and lowland areas (Table 2). Little evidence for subdivision existed among the three highland study sites (F ST = 0.084, 0.016 and 0.079 and P = 0.00089, 0.0032 and 0.0001 for Cochabamba–Tupiza, Cochabamba–Toro Toro and Tupiza–Toro Toro, respectively) or between the two lowland populations (F ST = 0.087 and P < 0.0001 for North and East Beni). However, elevated F ST values between closest highland and lowland study sites (Cochabamba–Beni distance = ~220 km; F ST = 0.42 and 0.35 and P < 0.0001 for Cochabamba–North and East Beni, respectively) indicate very limited gene flow, suggesting a powerful role for altitude and/or ecotope in structuring parasite populations. Interestingly, the extent of genetic subdivision between the most geographically distant highland populations (Cochabamba–Tupiza; distance = ~465 km) and adjacent areas of Beni (distance = ~155 km) was equivalent (F ST = 0.084 and 0.087, respectively).
Table 2.
F ST values in a five way comparison between populations (P‐value indicated in brackets)
| Cochabamba (highlands) | Tupiza (highlands) | Toro Toro (highlands) | North Beni (lowlands) | East Beni (lowlands) | |
|---|---|---|---|---|---|
| Cochabamba (highlands) | * | ||||
| Tupiza (highlands) | 0.084 (0.00089 ± 0.0003) | * | |||
| Toro Toro (highlands) | 0.016 (0.00317 ± 0.0006) | 0.079 (0.00010 ± 0.0001) | * | ||
| North Beni (lowlands) | 0.42 (0.000 ± 0.000) | 0.25 (0.000 ± 0.000) | 0.50 (0.000 ± 0.000) | * | |
| East Beni (lowlands) | 0.35 (0.000 ± 0.000) | 0.26 (0.000 ± 0.000) | 0.40 (0.000 ± 0.000) | 0.087 (0.000 ± 0.000) | * |
Finally, a hierarchical amova was conducted, to evaluate the distribution of genetic diversity between groups of populations (highlands vs. lowlands), among populations within groups (Cochabamba, Tupiza, Toro Toro, North Beni and East Beni) and among individuals within populations. Strikingly, 23% of total genetic variation was attributed to differences between highlands and lowlands, while 4.5% and 7% were present at the population and the individuals within population levels, respectively.
Mitochondrial introgression across ecological clines
For a subset of 78 clones, 10 mitochondrial gene fragments (mtMLST) were sequenced and concatenated into a 3684‐bp alignment. Twenty‐four unique haplotypes were identified from a total of 48 variable sites (~1.3% sequence diversity). ML and Bayesian phylogenies constructed from concatenated data were not significantly different (KH test: ML tree L = −4845.23, Bayesian tree L = −4848.13, P = 0.12). A second ML tree was assembled using 24 additional outgroup sequences representing known TcI mitochondrial diversity, including a small population of domestic Bolivian isolates (ANDESBol/Chile, previously described in Llewellyn et al. 2009a and Messenger et al. 2012) (Fig. 5).
Figure 5.
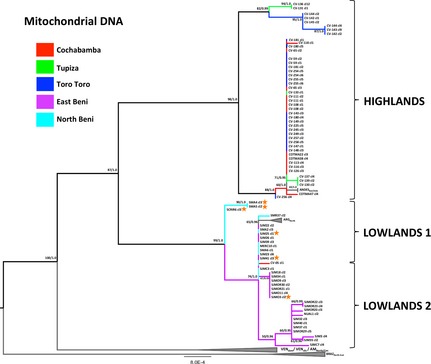
Maximum‐likelihood (ML) tree constructed from concatenated maxicircle sequences for 78 sylvatic Bolivian TcI clones and 24 additional TcI isolates from across the Americas. A ML topology was constructed from concatenated maxicircle sequences for 78 sylvatic Bolivian TcI clones and rooted using 24 additional TcI strains belonging to six previously characterized populations (AMN orth/Cen, ANDESB ol/Chile, ARGN orth, BRAZN orth‐East, VEN dom and VEN silv from Messenger et al. 2012). The most appropriate nucleotide substitution model was TrN+G (six substitution rate categories) based on the Akaike information criterion. Branch colours indicate sample a priori population (Cochabamba, Tupiza, Toro Toro, East Beni and North Beni; see legend). Statistical support for major clades are given as equivalent bootstraps and posterior probabilities from consensus ML (1000 pseudoreplicates) and Bayesian trees (based on the GTR+G model), respectively. Orange stars denote clones which have statistically supported phylogenetically incongruent positions between nuclear and mitochondrial topologies.
The mitochondrial topology demonstrated the presence of considerable genetic variation among Bolivian TcI clones. The deepest and most robust internal branch (87/1.0) separated highland and lowland populations into two major clades, each with strongly supported internal structuring. The highland group was largely homogeneous, with a number of geographically dispersed strains sharing identical mitochondrial haplotypes. The mitochondrial topology also confirmed the existence of Tupiza‐ and Toro Toro‐specific populations (98/1.0), in agreement with the nuclear tree.
Human isolates from Cochabamba (ANDESBol/Chile), while genetically distinct from sylvatic strains circulating in the same area (63/1.0), were grouped within the main highlands clade. As previously, lowland strains were subdivided into two well‐supported clades (74/1.0) with higher overall levels of genetic diversity, compared to highland isolates (H d = 0.81 and 0.84 vs. 0.54, respectively; Table 1).
While the gross topology of the mitochondrial tree was broadly concordant with that of the nuclear phylogeny, internal branch patterns were significantly incongruent (SH test: ML tree L = −4845.86, Bayesian tree L = −4849.55 and D AS tree L = −5006.48, P = 0.001). No evidence of recombination between highland and lowland strains was observed, even in Chapare, a zone of ecological transition. Across the more ‘gentle’ ecological cline of East–North Beni, several instances of genetic hybridization were apparent. Three clones from the mixed East–North Beni group (D AS lowlands 1) and three isolates from the East Beni‐specific population (D AS lowlands 2) received unambiguously different phylogenetic positions in the maxicircle topology and are likely the progeny of multiple, independent mitochondrial introgression events (Fig. 5).
Geographical dispersal within populations
To determine the extent of spatial genetic structure (or IBD) among highland and lowland isolates, Mantel's tests were conducted using alternate nuclear and mitochondrial data sets. Nuclear IBD was detected within both highland and lowland populations (highland R XY = 0.307, P < 0.001, and lowland R XY = 0.209, P < 0.001). However, the strength of the effect was significantly larger among lowland isolates (highland slope = 0.0002 ± 0.00000873; lowland slope = 0.0003 ± 0.0000179). Furthermore, when focusing on highland clones from approximately the same spatial scale as their lowlands counterparts [i.e. omitting the local subpopulation of Tupiza isolates identified in the D AS tree (n = 6)], little evidence for spatial structuring remained (R XY = 0.109, P = 0.085). Concordant with estimates of F ST between populations, the differing extent of spatial genetic structuring suggests accelerated parasite dispersal among geographically disparate highland areas by comparison with adjacent lowland foci (Fig. 4B).
Interestingly, no IBD was detected in either highland (R XY = 0.068, P = 0.161; slope = 0.000001 ± 0.000000345) or lowland (R XY = 0.119, P = 0.0654; slope = 0.000001 ± 0.000000349) populations using mitochondrial sequence data, potentially the result of lower population genetic resolution at these loci, but also consistent with the occurrence of mitochondrial introgression among lowland isolates.
Discussion
This study exploited rigorous population genetic analyses of contemporaneous parasite clones. Herein, we provide several insights into the biogeographical basis of Trypanosoma cruzi genetic diversification in Bolivia. Additionally, our study undertook an in‐depth dissection of TcI spatial genetic diversity and hybridization across two ecological clines.
Lowland arboreal and highland terrestrial sylvatic populations show different genetic structures
A clear dichotomy in population structure emerged between highland and lowland areas. Lowland parasites from two adjacent arboreal transmission cycles were strongly subdivided within a restricted contact zone in East Beni (~15 km2). Deep internal nuclear branching patterns in both lowland groups were indicative of stable, undisturbed, long‐term genetic diversification, with correspondingly high levels of diversity. Mitochondrial introgression occurring among genetically distinct strains in Beni supports prolonged historical interactions between these two populations. Consistent with high intrahost and vector clonal diversity, these data support intense, local transmission and/or low rate of genotypic extinction (Criscione & Blouin 2006). MLGs were rarely repeated, indicating only a fraction of total population genetic diversity was sampled.
In contrast, highland populations were considerably less diverse compared to their lowland counterparts and widespread dispersal of genetically homogeneous strains was observed across geographically disparate terrestrial highland populations, supported by little evidence of genetic substructuring (low F ST). Dissimilar heterozygosity estimates between highlands (excess) and lowlands (deficit) suggest a recent hybrid origin for some highland strains or fundamental differences in mating systems between these two populations (Ramírez & Llewellyn 2014). Importantly, human isolates from Cochabamba were closely related to adjacent sylvatic highland strains.
Gross differences between highland and lowland population structures may be partially explained in the context of their respective ecological niches. Most lowland parasites were isolated from Didelphimorphia mammals, prominent disease reservoirs which are susceptible to high circulating parasitaemia (Legey et al. 2003) and have a propensity for nonvectoral routes of infection, including oral transmission via predation of infected vectors or mammals (Jansen & Roque 2010; Rocha et al. 2013) and exposure to contaminated anal scent gland secretions (Carreira et al. 2001). These biological features may predispose these hosts to multiplicity of infection which will be directly related to intensity and efficiency of parasite transmission and duration and course of disease (Roellig et al. 2010; Nouvellet et al. 2013). The high levels of genetic diversity among Bolivian lowland strains are consistent with this hypothesis.
While minimal parasite interaction was observed between neighbouring terrestrial and arboreal transmission cycles (high F ST values between Cochabamba and Beni), a single clone (CV‐05 cl1) isolated from Rhodnius robustus in the Andean foothills was more closely related to lowland Beni strains on the basis of both nuclear and mitochondrial markers, suggesting the existence of an additional, under‐sampled transmission cycle and potential hybridization zone in Chapare, northern Cochabamba.
The remaining lowland strains were isolated from Rhodnius vectors (R. robustus and Rhodnius pictipes). In general, sylvatic Rhodnius species are promiscuous feeders, which can actively migrate at night to colonize domestic environments (Feliciangeli et al. 2007; Fitzpatrick et al. 2008), thus promoting the accumulation of mixed DTU infections (Bosseno et al. 1996; Yeo et al. 2005), as well as infrahost multiclonality and co‐infections with other trypanosome species, such as Trypanosoma rangeli (Dias et al. 2014). The lower genetic diversity observed among highlands strains may reflect more restricted feeding preferences and limited independent dispersal of their host vector species Triatoma infestans (<500 m) (Rabinovich & Himschoot 1990; Richer et al. 2007). As a more recent host of TcI, vector competency of sylvatic T. infestans may also vary, particularly in terms of bottlenecks during transmission, which can further reduce genetic diversity, as demonstrated in tsetse fly vectors of other digenetic trypanosome species (Ruepp et al. 1997; Oberle et al. 2010).
Ecological fitting is a driver of contemporary T. cruzi genetic diversification
No clear association of genotype by host or vector was observed among any sylvatic Bolivian TcI population, with the exception of a small subset of coclustering T. infestans clones sampled in Tupiza and Toro Toro (DAPC cluster 1 and D AS highlands; n = 14). Previous T. cruzi studies that favoured constrained, extant co‐evolutionary scenarios were probably limited by sampling bias (O'Connor et al. 2007); Didelphimorphia mammals continue to be oversampled as sources of sylvatic TcI due to their aforementioned high circulating parasitaemia, which can facilitate greater hemoculture positivity rates and thus parasite isolation, as well as their ease of capture.
With improved and more exhaustive sampling strategies, TcI has now been detected among a range of Mammalia (Lisboa et al. 2004; Herrera et al. 2005, 2008a,b; Yeo et al. 2005; Rocha et al. 2013; Lima et al. 2014), cautioning the interpretation of putative host associations. Here we demonstrate that parasite genetic diversity was principally partitioned by ecotope: arboreal lowland or terrestrial highland. Limited gene flow between neighbouring arboreal and terrestrial transmission cycles and low levels of subdivision among similar ecotopes, spanning much larger geographical distances (F ST), strongly suggest ecological host fitting is the predominant mechanism of sylvatic T. cruzi diversification (Llewellyn et al. 2009a,b). Our observations support a current model for wider trypanosome evolution where ecological host fitting has been proposed to define major parasite clades (Hamilton et al. 2007; Lukes et al. 2014).
Mitochondrial introgression is a common phenomenon among natural T. cruzi populations
The majority of field evidence indicates T. cruzi does not conform to strict clonality or panmixia and that recombination is common, nonobligatory and idiosyncratic, potentially involving independent exchange of kinetoplastid and nuclear genetic material and both canonical meiotic and parasexual mechanisms (Carrasco et al. 1996; Machado & Ayala 2001; Ocaña‐Mayorga et al. 2010; Lewis et al. 2011; Messenger et al. 2012; Ramírez et al. 2012; Roellig et al. 2013; Baptista Rde et al. 2014). The relative contributions of alternate mating strategies to T. cruzi population structures are as yet unclear and strongly debated (Tibayrenc & Ayala 2012, 2013; Ramírez & Llewellyn 2014).
One aim of our study was to evaluate the extent of genetic recombination within two putative hybrid zones. Due to limited sample size (only a single isolate could be recovered from the politically unstable Chapare region), we were unable to detect hybridization across the highland–lowland cline. However, mitochondrial introgression was observed among a subset of lowland strains between East and North Beni. Evidence of intra‐TcI genetic exchange in a primary Amazonian forest (Carrasco et al. 1996), between domestic/peri‐domestic populations in Ecuador (Ocaña‐Mayorga et al. 2010) and within an endemic focus in Colombia (Ramírez et al. 2012) suggests that intensive local sampling of transmission cycles is an effective strategy to detect recombination.
Arboreal lowland populations in Beni provide an example of an undisturbed epidemiological situation where genetic exchange might be expected (Carrasco et al. 1996). Two divergent TcI populations overlap in this region, one sharing affinities to TcI populations from the Chaco region to the South (East Beni), the other more related to Amazonian TcI to the North (North Beni) (Llewellyn et al. 2009a; Lima et al. 2014). Experimental recombination in T. cruzi was shown to arise in mammalian cell cultures (Gaunt et al. 2003). The aforementioned Didelphimorphia maintain high levels of multiclonal parasite populations, providing ample opportunities for hybridization to occur. Multiple mitochondrial introgression events were detected in East Beni, which appeared independent of parasite nuclear genotype, mammalian host species and study site. Consistent with previous studies, no evidence of reciprocal nuclear hybridization was detected among recombinant strains (Messenger et al. 2012; Ramírez et al. 2012). While the biological cues that initiate genetic exchange remain unresolved (Gaunt et al. 2003; Lewis et al. 2010), in these populations we speculate that asymmetric introgression may act as a mechanism to facilitate ecological fitting (e.g. host range extension or resource tracking), considering the crucial role that mitochondria play in parasite metabolism, growth and development and their elevated need to escape Muller's ratchet compared to the nuclear genome (Neiman & Taylor 2009; Ramírez & Llewellyn 2014).
Dispersal of Chagas disease in highland Bolivia
Multiple lines of evidence suggest that there is no ‘bona fide’ sylvatic transmission cycle in the Bolivian highlands. Little spatial differentiation was detected among geographically disparate highland populations (~465 km), and this level was comparable to that observed between neighbouring lowland areas (~155 km). Terrestrial clones also displayed limited genetic IBD, a lack of private alleles and excess heterozygosity, all potentially attributable to a recent population bottleneck and/or founder event followed by clonal propagation.
The putative accelerated parasite dispersal between highland sites in comparison with lowland areas does not accord with the ecology expected for local established sylvatic transmission. Indeed, didelphid marsupials and Rhodnius vectors have a far greater capacity for auto‐dissemination than T. infestans and smaller rodents (Richer et al. 2007). Instead, parasite dispersal across the highlands may be recent and anthroponotic. Substantial population genetic evidence indicates that T. infestans has a precedent for passive dissemination by human populations throughout history, initially during pre‐Incan times throughout the Western Andes (Schofield 1988; Bargues et al. 2006; Cortez et al. 2010) and subsequently, post‐Colombian, eastwards into Argentina, Paraguay, Uruguay and Brazil (Panzera et al. 2004; Piccinali et al. 2009). Trafficking of genetically homogeneous, human‐infective (at least in Cochabamba), highland TcI clones is reminiscent of the epidemic propagation of hybrid T. cruzi lineages TcV and TcVI by domestic T. infestans across the Southern Cone (Lewis et al. 2011). All highland study sites coincided with major, densely populated, transport routes transecting the departments of Cochabamba and Potosí and the distribution of highland strains closely reflected human migratory movements.
Genetic continuity between human and sylvatic strains in the highlands adjacent to Cochabamba by mitochondrial MLST confirms the existence of gene flow from local sylvatic to domestic transmission cycles. More widespread highland domestic infestation with T. infestans might be expected if its sylvatic distribution was the result of anthropogenic propagation. Thus, the extent to which humans are responsible for long‐range parasite distribution throughout highland Bolivia remains to be resolved. Importantly, the widespread dispersal of limited diversity genotypes in Bolivia has significant biological and medical implications with respect to virulence, transmissibility and drug susceptibility, and the potential risk for emergent epizootic Chagas disease.
L.A.M. designed and performed the experiments, analysed the data and drafted the manuscript. L.G. participated in fieldwork, contributed materials and analysed the data. M.V. contributed materials and analysed the data. C.H., M.B. and M.T. participated in fieldwork. F.T. contributed materials. M.A.M. drafted the manuscript. M.S.L. designed the study, participated in fieldwork, analysed the data and drafted the manuscript.
Data accessibility
Strain panel (Table S1, Supporting information), microsatellite primers (Table S2, Supporting information), microsatellite genotypes (Appendix S1, Supporting information), concatenated maxicircle sequence alignment, mitochondrial ML phylogeny and microsatellite NJ phylogeny are all available from Dryad: doi: 10.5061/dryad.b8465.
Supporting information
Appendix S1 Dataset
Table S1 Panel of Bolivian T. cruzi TcI biological clones assembled for analysis.
Table S2 Panel of microsatellite loci and primers employed in this study.
Acknowledgements
This research was supported by the Wellcome Trust and the European Commission Framework Programme Project ‘Comparative epidemiology of genetic lineages of Trypanosoma cruzi’ ChagasEpiNet (contract #223034). L.A.M. was supported by a BBSRC Doctoral Training Grant and a travelling fellowship from the Chadwick Trust, University College London. M.S.L. would like to thank Kendra Shanley and Anna Warren for their assistance in the field. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
The copyright line for this article was changed on 03 August, 2015 after original online publication.
References
- Agapow PW, Burt A (2001) Indices of multilocus linkage disequilibrium. Molecular Ecology Notes, 1, 101–102. [Google Scholar]
- Agosta SJ, Klemens JA (2008) Ecological fitting by phenotypically flexible genotypes: implications for species associations, community assembly and evolution. Ecology Letters, 11, 1123–1134. [DOI] [PubMed] [Google Scholar]
- Agosta SJ, Janz N, Brooks DR (2010) How specialists can be generalists: resolving the “parasite paradox” and implications for emerging infectious disease. Zoologia, 27, 151–162. [Google Scholar]
- Baptista Rde P, D'Avila DA, Segatto M et al (2014) Evidence of substantial recombination among Trypanosoma cruzi II strains from Minas Gerais. Infection, Genetics and Evolution, 22, 183–191. [DOI] [PubMed] [Google Scholar]
- Bargues MD, Klisiowicz DR, Panzera F et al (2006) Origin and phylogeography of the Chagas disease main vector Triatoma infestans based on nuclear rDNA sequences and genome size. Infection, Genetics and Evolution, 6, 46–62. [DOI] [PubMed] [Google Scholar]
- Barnabé C, De Meeus T, Noireau F et al (2011) Trypanosoma cruzi discrete typing units (DTUs): microsatellite loci and population genetics of DTUs TcV and TcI in Bolivia and Peru. Infection, Genetics and Evolution, 11, 1752–1760. [DOI] [PubMed] [Google Scholar]
- Boité MC, Mauricio IL, Miles MA et al (2012) New insights on taxonomy, phylogeny and population genetics of Leishmania (Viannia) parasites based on multilocus sequence analysis. PLoS Neglected Tropical Diseases, 6, e1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosseno MF, Telleria J, Vargas F et al (1996) Trypanosoma cruzi: study of the distribution of two widespread clonal genotypes in Bolivian Triatoma infestans vectors shows a high frequency of mixed infections. Experimental Parasitology, 83, 275–282. [DOI] [PubMed] [Google Scholar]
- Breniere SF, Aliaga C, Waleckx E et al (2012) Genetic characterization of Trypanosoma cruzi DTUs in wild Triatoma infestans from Bolivia: predominance of TcI. PLoS Neglected Tropical Diseases, 6, e1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DR, Ferrao AL (2005) The historical biogeography of co‐evolution: emerging infectious diseases are evolutionary accidents waiting to happen. Journal of Biogeography, 32, 1291–1299. [Google Scholar]
- Brooks DR, Léon‐Règagnon V, McLennan DA et al (2006) Ecological fitting as a determinant of the community structure of platyhelminth parasites of anurans. Ecology, 87(Suppl), S76–S85. [DOI] [PubMed] [Google Scholar]
- Buitrago R, Waleckx E, Bosseno MF et al (2010) First report of widespread wild populations of Triatoma infestans (Reduviidae, Triatominae) in the valleys of La Paz, Bolivia. The American Journal of Tropical Medicine and Hygiene, 82, 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos JM, Begher S, Silva HM et al (2008) Molecular identification of Trypanosoma cruzi I tropism for central nervous system in Chagas reactivation due to AIDS. The American Journal of Tropical Medicine and Hygiene, 78, 294–297. [PubMed] [Google Scholar]
- Carrasco HJ, Frame IA, Valente SA et al (1996) Genetic exchange as a possible source of genomic diversity in sylvatic populations of Trypanosoma cruzi . The American Journal of Tropical Medicine and Hygiene, 54, 418–424. [DOI] [PubMed] [Google Scholar]
- Carrasco HJ, Segovia M, Llewellyn MS et al (2012) Geographical distribution of Trypanosoma cruzi genotypes in Venezuela. PLoS Neglected Tropical Diseases, 6, e1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira JC, Jansen AM, de Nazareth Meirelles M et al (2001) Trypanosoma cruzi in the scent glands of Didelphis marsupialis: the kinetics of colonization. Experimental Parasitology, 97, 129–140. [DOI] [PubMed] [Google Scholar]
- Ceballos LA, Piccinali RV, Marcet PL et al (2011) Hidden sylvatic foci of the main vector of Chagas disease Triatoma infestans: threats to vector elimination campaign? PLoS Neglected Tropical Diseases, 5, e1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MR, Pinho AP, Cuervo P et al (2006) Trypanosoma cruzi (Kinetoplastida Trypanosomatidae): ecology of the transmission cycle in the wild environment of the Andean valley of Cochabamba, Bolivia. Experimental Parasitology, 114, 305–313. [DOI] [PubMed] [Google Scholar]
- Cortez MR, Emperaire L, Piccinali RV et al (2007) Sylvatic Triatoma infestans (Reduviidae, Triatominae) in the Andean valleys of Bolivia. Acta Tropica, 102, 47–54. [DOI] [PubMed] [Google Scholar]
- Cortez MR, Monteiro FA, Noireau F (2010) New insights on the spread of Triatoma infestans from Bolivia – implications for Chagas disease emergence in the Southern Cone. Infection, Genetics and Evolution, 10, 350–353. [DOI] [PubMed] [Google Scholar]
- Coura JR, Dias JC (2009) Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Memórias do Instituto Oswaldo Cruz, 104(Suppl 1), 31–40. [DOI] [PubMed] [Google Scholar]
- Criscione CD, Blouin MS (2006) Minimal selfing, few clones, and no among‐host genetic structure in a hermaphroditic parasite with asexual larval propagation. Evolution, 60, 553–562. [PubMed] [Google Scholar]
- Cura CI, Mejía‐Jaramillo AM, Duffy T et al (2010) Trypanosoma cruzi I genotypes in different geographical regions and transmission cycles based on a microsatellite motif of the intergenic spacer of spliced‐leader genes. International Journal for Parasitology, 40, 1599–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R et al (2012) jmodeltest 2: more models, new heuristics and parallel computing. Nature Methods, 9, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias FB, Quartier M, Diotaiuti L et al (2014) Ecology of Rhodnius robustus Larrousse, 1927 (Hemiptera, Reduviidae, Triatominae) in Attalea palm trees of the Tapajós River Region (Pará State, Brazilian Amazon). Parasites and Vectors, 7, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza N, Borrás R, Abad‐Franch F (2014) Chagas disease vector control in a hyperendemic setting: the first 11 years of intervention in Cochabamba, Bolivia. PLoS Neglected Tropical Diseases, 8, e2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S (2005) arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1, 47–50. [PMC free article] [PubMed] [Google Scholar]
- Falla A, Herrera C, Fajardo A et al (2009) Haplotype identification within Trypanosoma cruzi I in Colombian isolates from several reservoirs, vectors and humans. Acta Tropica, 110, 15–21. [DOI] [PubMed] [Google Scholar]
- Feliciangeli MD, Sanchez‐Martin M, Marrero R et al (2007) Morphometric evidence for a possible role of Rhodnius prolixus from palm trees in house re‐infestation in the State of Barinas (Venezuela). Acta Tropica, 101, 167–177. [DOI] [PubMed] [Google Scholar]
- Felsenstein J (1989) phylip – phylogeny inference package (version 3.2). Cladistics, 5, 164–166. [Google Scholar]
- Fitzpatrick S, Feliciangeli MD, Sanchez‐Martin MJ et al (2008) Molecular genetics reveal that sylvatic Rhodnius prolixus do colonise rural houses. PLoS Neglected Tropical Diseases, 2, e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvão C, Carcavallo R, Rocha DS et al (2003) A checklist of the current valid species of the subfamily Triatominae Jeannel, 1919 (Hemiptera, Reduviidae) and their geographical distribution, with nomenclatural and taxonomic notes. Zootaxa, 202, 1–36. [Google Scholar]
- Gaunt M, Miles M (2000) The ecotopes and evolution of triatomine bugs (triatominae) and their associated trypanosomes. Memórias do Instituto Oswaldo Cruz, 95, 557–565. [DOI] [PubMed] [Google Scholar]
- Gaunt MW, Yeo M, Frame IA et al (2003) Mechanisms of genetic exchange in American trypanosomes. Nature, 421, 936–939. [DOI] [PubMed] [Google Scholar]
- Germano MD, Roca Acevedo G, Mougabure Cueto GA et al (2010) New findings of insecticide resistance in Triatoma infestans (Heteroptera: Reduviidae) from the Gran Chaco. Journal of Medical Entomology, 47, 1077–1081. [DOI] [PubMed] [Google Scholar]
- Goudet J (1995) fstat (version 1.2): a computer program to calculate F‐statistics. Journal of Heredity, 86, 485–486. [Google Scholar]
- Gürtler R (2009) Sustainability of vector control strategies in the Gran Chaco Region: current challenges and possible approaches. Memórias do Instituto Oswaldo Cruz, 104(Suppl 1), 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton PB, Gibson WC, Stevens JR (2007) Patterns of co‐evolution between trypanosomes and their hosts deduced from ribosomal RNA and protein‐coding gene phylogenies. Molecular Phylogenetics and Evolution, 44, 15–25. [DOI] [PubMed] [Google Scholar]
- Herrera L, D'Andrea PS, Xavier SC et al (2005) Trypanosoma cruzi infection in wild mammals of the National Park ‘Serra da Capivara’ and its surroundings (Piaui, Brazil), an area endemic for Chagas disease. Transactions of the Royal Society of Tropical Medicine and Hygiene, 99, 379–388. [DOI] [PubMed] [Google Scholar]
- Herrera C, Bargues MD, Fajardo A et al (2007) Identifying four Trypanosoma cruzi I isolate haplotypes from different geographic regions in Colombia. Infection, Genetics and Evolution, 7, 535–539. [DOI] [PubMed] [Google Scholar]
- Herrera HM, Lisboa CV, Pinho AP et al (2008a) The coati (Nasua nasua, Carnivora, Procyonidae) as a reservoir host for the main lineages of Trypanosoma cruzi in the Pantanal region, Brazil. Transactions of the Royal Society of Tropical Medicine and Hygiene, 102, 1133–1139. [DOI] [PubMed] [Google Scholar]
- Herrera HM, Abreu UG, Keuroghlian A et al (2008b) The role played by sympatric collared peccary (Tayassu tajacu), white‐lipped peccary (Tayassu pecari), and feral pig (Sus scrofa) as maintenance hosts for Trypanosoma evansi and Trypanosoma cruzi in a sylvatic area of Brazil. Parasitology Research, 103, 619–624. [DOI] [PubMed] [Google Scholar]
- Herrera C, Guhl F, Falla A et al (2009) Genetic variability and phylogenetic relationships within Trypanosoma cruzi I isolated in Colombia based on miniexon gene sequences. Journal of Parasitology Research, 2009, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannin J, Salvatella R (2006) Estimacion cuantitiva de la enfermedad de Chagas en las Americas. Organizacion Panamericana de la Salud, p. 7.
- Jansen AM, Roque ALR (2010) Domestic and wild mammalian reservoirs In: American Trypanosomiasis Chagas Disease One Hundred Years of Research (eds Telleria J, Tibayrenc M.), pp. 249–276. Elsevier, London. [Google Scholar]
- Janzen DH (1985) On ecological fitting. Oikos, 45, 308–310. [Google Scholar]
- Jombart T (2008) adegenet: a r package for the multivariate analysis of genetic markers. Bioinformatics, 24, 1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics, 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justi SA, Noireau F, Cortez MR et al (2010) Infestation of peridomestic Attalea phalerata palms by Rhodnius stali, a vector of Trypanosoma cruzi in the Alto Beni, Bolivia. Tropical Medicine and International Health, 15, 727–732. [DOI] [PubMed] [Google Scholar]
- Kalinowski ST (2005) hp‐rare: a computer program for performing rarefaction on measures of allelic diversity. Molecular Ecology, 5, 187–189. [Google Scholar]
- Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. Journal of Molecular Evolution, 29, 170–179. [DOI] [PubMed] [Google Scholar]
- Lardeux F, Depickère S, Duchon S et al (2010) Insecticide resistance of Triatoma infestans (Hemiptera, Reduviidae) vector of Chagas disease in Bolivia. Tropical Medicine and International Health, 15, 1037–1048. [DOI] [PubMed] [Google Scholar]
- Legey AP, Pinho AP, Xavier SC et al (2003) Trypanosoma cruzi in marsupial didelphids (Philander frenata and Didelphis marsupialis): differences in the humoral immune response in natural and experimental infections. Revista da Sociedade Brasileira de Medicina Tropical, 36, 241–248. [DOI] [PubMed] [Google Scholar]
- Lent H, Wygodzinsky P (1979) Revision of the Triatominae (Hemiptera, Reduviidae) and their significance as vectors of Chagas disease. Bulletin of the American Museum of Natural History, 163, 1–520. [Google Scholar]
- Lewis MD, Ma J, Yeo M et al (2009) Genotyping of Trypanosoma cruzi: systematic selection of assays allowing rapid and accurate discrimination of all known lineages. The American Journal of Tropical Medicine and Hygiene, 81, 1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MD, Llewellyn MS, Yeo M et al (2010) Experimental and natural recombination in Trypanosoma cruzi In: American Trypanosomiasis Chagas Disease One Hundred Years of Research (eds Telleria J, Tibayrenc M.), pp. 459–474. Elsevier, London. [Google Scholar]
- Lewis MD, Llewellyn MS, Yeo M et al (2011) Recent, independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Neglected Tropical Diseases, 5, e1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J (2009) dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. [DOI] [PubMed] [Google Scholar]
- Lima V, Jansen AM, Messenger LA et al (2014) Wild Trypanosoma cruzi I genetic diversity in Brazil suggests admixture and disturbance in parasite populations from the Atlantic Forest region. Parasites and Vectors, 7, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisboa CV, Mangia RH, De Lima NR et al (2004) Distinct patterns of Trypanosoma cruzi infection in Leontopithecus rosalia in distinct Atlantic coastal rainforest fragments in Rio de Janeiro‐Brazil. Parasitology, 129, 703–711. [DOI] [PubMed] [Google Scholar]
- Llewellyn MS, Miles MA, Carrasco HJ et al (2009a) Genome‐scale multilocus microsatellite typing of Trypanosoma cruzi discrete typing unit I reveals phylogeographic structure and specific genotypes linked to human infection. PLoS Pathogens, 5, e1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn MS, Lewis MD, Acosta N et al (2009b) Trypanosoma cruzi IIc: phylogenetic and phylogeographic insights from sequence and microsatellite analysis and potential impact on emergent Chagas disease. PLoS Neglected Tropical Diseases, 3, e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn MS, Rivett‐Carnac JB, Fitzpatrick S et al (2011) Extraordinary Trypanosoma cruzi diversity within single mammalian reservoir hosts implies a mechanism of diversifying selection. International Journal for Parasitology, 41, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukes J, Skalicky T, Tyc J et al (2014) Evolution of parasitism in kinetoplastid flagellates. Molecular and Biochemical Parasitology, 195, 115–122. [DOI] [PubMed] [Google Scholar]
- Machado C, Ayala FJ (2001) Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi . Proceedings of the National Academy of Sciences, USA, 98, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcili A, Lima L, Valente VC et al (2009) Comparative phylogeography of Trypanosoma cruzi TCIIc: new hosts, association with terrestrial ecotopes, and spatial clustering. Infection, Genetics and Evolution, 9, 1265–1274. [DOI] [PubMed] [Google Scholar]
- Matias A, De La Riva J, Martinez E et al (2003) Domiciliation process of Rhodnius stali (Hemiptera: Reduviidae) in Alto Beni, La Paz, Bolivia. Tropical Medicine and International Health, 8, 264–268. [DOI] [PubMed] [Google Scholar]
- Medrano‐Mercado N, Ugarte‐Fernandez R, Butron V et al (2008) Urban transmission of Chagas disease in Cochabamba, Bolivia. Memórias do Instituto Oswaldo Cruz, 103, 423–430. [DOI] [PubMed] [Google Scholar]
- Messenger LA, Llewellyn MS, Bhattacharyya T et al (2012) Multiple mitochondrial introgression events and heteroplasmy in Trypanosoma cruzi revealed by maxicircle MLST and Next Generation Sequencing. PLoS Neglected Tropical Diseases, 6, e1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles MA (1993) Culturing and biological cloning of Trypanosoma cruzi . In: Protocols in Molecular Parasitology. Methods in Molecular Biology, 21, 15–28. [DOI] [PubMed] [Google Scholar]
- Miles MA, De Souza AA, Povoa M (1981) Chagas disease in the Amazon Basin III. Ecotopes of ten triatomine bug species (Hemiptera: Reduviidae) from the vicinity of Belém, Pará State, Brazil. Journal of Medical Entomology, 18, 266–278. [DOI] [PubMed] [Google Scholar]
- Miles MA, Llewellyn MS, Lewis MD et al (2009) The molecular epidemiology and phylogeography of Trypanosoma cruzi and parallel research on Leishmania: looking back and to the future. Parasitology, 136, 1509–1528. [DOI] [PubMed] [Google Scholar]
- Milne I, Lindner D, Bayer M et al (2009) topali v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC cluster and multi‐core desktops. Bioinformatics, 25, 126–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minch E, Ruiz‐Linares A, Goldstein D et al (1997) microsat v1.5d: A Computer Programme for Calculating Various Statistics on Microsatellite Allele Data. Department of Genetics, Stanford University, Stanford, California. [Google Scholar]
- Neiman M, Taylor DR (2009) The causes of mutation accumulation in mitochondrial genomes. Proceedings of the Royal Society Biological Sciences, 276, 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noireau F (2009) Wild Triatoma infestans, a potential threat that needs to be monitored. Memórias do Instituto Oswaldo Cruz, 104(Suppl 1), 60–64. [DOI] [PubMed] [Google Scholar]
- Noireau F, Abad‐Franch F, Valente SA et al (2002) Trapping triatominae in silvatic habitats. Memórias do Instituto Oswaldo Cruz, 97, 61–63. [DOI] [PubMed] [Google Scholar]
- Noireau F, Cortez MG, Monteiro FA et al (2005) Can wild Triatoma infestans foci in Bolivia jeopardize Chagas disease control efforts? Trends in Parasitology, 21, 7–10. [DOI] [PubMed] [Google Scholar]
- Noireau F, Diosque P, Jansen AM (2009) Trypanosoma cruzi: adaptation to its vectors and its hosts. Veterinary Research, 40, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouvellet P, Dumonteil E, Gourbiere S (2013) The improbable transmission of Trypanosoma cruzi to human: the missing link in the dynamics and control of Chagas disease. PLoS Neglected Tropical Diseases, 7, e2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle M, Balmer O, Brun R et al (2010) Bottlenecks and the maintenance of minor genotypes during the life cycle of Trypanosoma brucei . PLoS Pathogens, 6, e1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocaña‐Mayorga S, Llewellyn MS, Costales JA et al (2010) Sex, subdivision, and domestic dispersal of Trypanosoma cruzi lineage I in Southern Ecuador. PLoS Neglected Tropical Diseases, 4, e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor O, Bosseno MF, Barnabé C et al (2007) Genetic clustering of Trypanosoma cruzi I lineage evidenced by intergenic miniexon gene sequencing. Infection, Genetics and Evolution, 7, 587–593. [DOI] [PubMed] [Google Scholar]
- Panzera F, Dujardin JP, Nicolini P et al (2004) Genomic changes of Chagas disease vector, South America. Emerging Infectious Diseases, 10, 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse P (2012) genaiex 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics, 28, 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinali RV, Marcet PL, Noireau F (2009) Molecular population genetics and phylogeography of the Chagas disease vector Triatoma infestans in South America. Journal of Medical Entomology, 46, 796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, De Meeus T (2010) Apparent high recombination rates in clonal parasitic organisms due to inappropriate sampling design. Heredity, 104, 135–140. [DOI] [PubMed] [Google Scholar]
- Rabinovich JE, Himschoot P (1990) A population‐dynamics simulation model of the main vectors of Chagas' disease transmission, Rhodnius prolixus and Triatoma infestans . Ecological Modelling, 52, 249–266. [Google Scholar]
- Ramírez JD, Llewellyn MS (2014) Reproductive clonality in protozoan pathogens – truth or artefact? Molecular Ecology, 23, 4195–4202. [DOI] [PubMed] [Google Scholar]
- Ramírez JD, Guhl F, Rendón LM et al (2010) Chagas cardiomyopathy manifestations and Trypanosoma cruzi genotypes circulating in chronic Chagasic patients. PLoS Neglected Tropical Diseases, 4, e899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez JD, Guhl F, Messenger LA et al (2012) Contemporary cryptic sexuality in Trypanosoma cruzi . Molecular Ecology, 21, 4216–4226. [DOI] [PubMed] [Google Scholar]
- Rassi A Jr, Rassi A, Marin‐Neto JA (2010) Chagas disease. The Lancet, 375, 1388–1402. [DOI] [PubMed] [Google Scholar]
- Rice W (1989) Analyzing tables with statistical tests. Evolution, 43, 223–225. [DOI] [PubMed] [Google Scholar]
- Richer W, Kengne P, Cortez MR et al (2007) Active dispersal by wild Triatoma infestans in the Bolivian Andes. Tropical Medicine and International Health, 12, 759–764. [DOI] [PubMed] [Google Scholar]
- Rocha FL, Roque AL, De Lima JS et al (2013) Trypanosoma cruzi infection in neotropical wild carnivores (Mammalia: Carnivora): at the top of the T. cruzi transmission chain. PLoS One, 8, e67463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roellig DM, McMillan K, Ellis AE et al (2010) Experimental infection of two South American reservoirs with four distinct strains of Trypanosoma cruzi . Parasitology, 137, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roellig DM, Savage MY, Fujita AW et al (2013) Genetic variation and exchange in Trypanosoma cruzi isolates from the United States. PLoS One, 8, e56198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp S, Furger A, Kurath U et al (1997) Survival of Trypanosoma brucei in the tsetse fly is enhanced by the expression of specific forms of procyclin. The Journal Cell Biology, 137, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels AM, Clark EH, Galdos‐Cardenas G et al (2013) Epidemiology of and impact of insecticide spraying on Chagas disease in communities in the Bolivian Chaco. PLoS Neglected Tropical Diseases, 7, e2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ (1988) Biosystematics of the Triatominae In: Biosystematics of Haematophagous Insects (ed. Service MW.), vol. 37, pp. 284–312. Clarendon Press, Oxford. [Google Scholar]
- Shikanai‐Yasuda MA, Carvalho NB (2012) Oral transmission of Chagas disease. Clinical Infectious Diseases, 54, 845–852. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M (1999) Multiple comparisons of log‐likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution, 16, 1114–1116. [Google Scholar]
- Stevens J, Noyes H, Dover G et al (1999) The ancient and divergent origins of the human pathogenic trypanosomes, Trypanosoma brucei and T. cruzi . Parasitology, 118, 107–116. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N et al (2011) mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M, Ayala FJ (2012) Reproductive clonality of pathogens: a perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proceedings of the National Academy of Sciences, USA, 109, e3305–e3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M, Ayala FJ (2013) How clonal are Trypanosoma and Leishmania? Trends in Parasitology, 29, 264–269. [DOI] [PubMed] [Google Scholar]
- Waleckx E, Depickere S, Salas R et al (2012) New discoveries of sylvatic Triatoma infestans (Hemiptera: Reduviidae) throughout the Bolivian Chaco. The American Journal of Tropical Medicine and Hygiene, 86, 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherly DB, Boehlke C, Tarleton RL (2009) Chromosome level assembly of the hybrid Trypanosoma cruzi genome. BMC Genomics, 10, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2014) Global Burden of Disease Estimates for 2000–2012. [cited 2014 11/11/2014]; Available from: http://www.who.int/healthinfo/global_burden_disease/estimates/en/index2.html.
- Yang Z (2007) paml 4: a program package for phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yeo M, Acosta N, Llewellyn M et al (2005) Origins of Chagas disease: Didelphis species are natural hosts of Trypanosoma cruzi I and armadillos hosts of Trypanosoma cruzi II, including hybrids. International Journal for Parasitology, 35, 225–233. [DOI] [PubMed] [Google Scholar]
- Yeo M, Lewis MD, Carrasco HJ et al (2007) Resolution of multiclonal infections of Trypanosoma cruzi from naturally infected triatomine bugs and from experimentally infected mice by direct plating on sensitive solid medium. International Journal for Parasitology, 37, 111–120. [DOI] [PubMed] [Google Scholar]
- Yeo M, Mauricio IL, Messenger LA et al (2011) Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi . PLoS Neglected Tropical Diseases, 5, e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingales B, Andrade SG, Briones MR et al (2009) A new consensus for Trypanosoma cruzi intraspecific nomenclature: a second revision meeting recommends TcI to TcVI. Memórias do Instituto Oswaldo Cruz, 104, 1051–1054. [DOI] [PubMed] [Google Scholar]
- Zumaya‐Estrada FA, Messenger LA, Lopez‐Ordonez T et al (2012) North American import? Charting the origins of an enigmatic Trypanosoma cruzi domestic genotype. Parasites and Vectors, 5, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Dataset
Table S1 Panel of Bolivian T. cruzi TcI biological clones assembled for analysis.
Table S2 Panel of microsatellite loci and primers employed in this study.
Data Availability Statement
Strain panel (Table S1, Supporting information), microsatellite primers (Table S2, Supporting information), microsatellite genotypes (Appendix S1, Supporting information), concatenated maxicircle sequence alignment, mitochondrial ML phylogeny and microsatellite NJ phylogeny are all available from Dryad: doi: 10.5061/dryad.b8465.