


Abstract
The DNA damage checkpoint pathway is activated in response to DNA lesions and replication stress to preserve genome integrity. However, hyper-activation of this surveillance system is detrimental to the cell, because it might prevent cell cycle re-start after repair, which may also lead to senescence. Here we show that the scaffold proteins Slx4 and Rtt107 limit checkpoint signalling at a persistent double-strand DNA break (DSB) and at uncapped telomeres. We found that Slx4 is recruited within a few kilobases of an irreparable DSB, through the interaction with Rtt107 and the multi-BRCT domain scaffold Dpb11. In the absence of Slx4 or Rtt107, Rad9 binding near the irreparable DSB is increased, leading to robust checkpoint signalling and slower nucleolytic degradation of the 5′ strand. Importantly, in slx4Δ sae2Δ double mutant cells these phenotypes are exacerbated, causing a severe Rad9-dependent defect in DSB repair. Our study sheds new light on the molecular mechanism that coordinates the processing and repair of DSBs with DNA damage checkpoint signalling, preserving genome integrity.
INTRODUCTION
All eukaryotic cells respond to DNA lesions by activating a surveillance network called the DNA damage checkpoint (DDC), which coordinates DNA repair with cell cycle progression (1). Notably, mutations in checkpoint genes lead to genome instability and in higher eukaryotes often give rise to carcinogenesis (2). At double strand DNA breaks (DSBs), the checkpoint is triggered by the formation of long stretches of single-stranded DNA (ssDNA) generated by 5′-3′ nucleolytic degradation (DSB resection) of DNA ends. This action is carried out by multiple conserved factors. In S. cerevisiae, CDK1-phosphorylated Sae2 primes the Mre11-Rad50-Xrs2 (MRX) complex to trim DSB ends (short-range resection), which are afterwards extensively processed by the Exo1 and Dna2 nucleases, together with the Bloom helicase Sgs1 (long-range resection). As resection proceeds, the 3′ ssDNA tail is covered by RPA, which then recruits the checkpoint clamp 9–1–1 complex (Rad17, Mec3 and Ddc1 in budding yeast) and the upstream checkpoint kinase Mec1. Proper cooperation of all these factors is critical to establish appropriate DSB resection, repair and checkpoint signalling (3).
A key player in the DDC is Rad9, an ortholog of human 53BP1. Rad9 acts as an adaptor protein, which mediates checkpoint signalling from the sensor kinase Mec1 to the central transducer kinases Rad53 and Chk1 (2,4). Moreover, Rad9 is recruited to DSBs and to uncapped telomeres, limiting the resection of the 5′ strand (5). More recently, we have also shown that increased Rad9 binding close to DSB ends affects the initiation of resection and the balance between non-homologous end joining and homologous recombination events in sae2Δ cells (6).
Rad9 recruitment to DSBs is mediated by its interactions with a Mec1-dependent phosphorylation site (S129) in histone H2A (γ-H2AX), and with the multi-BRCT domain protein Dpb11 (TopBP1 in human cells), which is itself recruited to DSBs via interaction with yet another Mec1-dependent phosphorylation site (T602) in the 9–1–1 subunit Ddc1. In addition, the Dot1-dependent methylation of Lysine 79 of histone H3 provides a constitutive docking site for the Rad9 Tudor domain (5,7–11). Up to now, the regulation of Rad9 dissociation from DNA lesions after repair is poorly understood.
The role of Rad9 in DDC signalling has been recently shown to be counteracted by the action of Slx4, a protein scaffold with established roles in the coordination of structure-specific nucleases (12–15). Upon replication stress caused by the DNA alkylating agent methyl methanesulfonate (MMS), a complex formed by Slx4 with the multi-BRCT domain protein Rtt107 was shown to compete with Rad9 for interaction with Dpb11 and γ-H2AX. As such, cells lacking Slx4 are hypersensitive to MMS treatment due to Rad53 hyper-activation (16). Interestingly, expression of a chimeric protein, in which the BRCT domains 5 and 6 of Rtt107 were fused to BRCT domains 3 and 4 of Dpb11 (MBD, minimal multi-BRCT-domain module), was shown to completely rescue the sensitivity of slx4Δ cells to MMS (17). The checkpoint dampening function of Slx4-Rtt107 appears to be distinct from Slx4's role in coordinating DNA repair via regulation of the Rad1 and Slx1 nucleases (16). Disruption of the Slx4-Dpb11 interaction prevents the binding to Mus81 nuclease, leading to the accumulation of unresolved DNA joint molecules (JMs) and RPA foci (indicative of ssDNA gaps), after MMS treatment (18). Therefore, an open question is whether the DDC hyper-activation in slx4Δ is primarily due to the defect in dampening checkpoint signalling or the defect in regulation of the JM resolution.
In this study, we investigated the role of the Rtt107-Slx4 complex in the regulation of the DDC in cells responding to an irreparable DSB and to uncapped telomeres. Our results indicate that cooperation between Slx4 and Rtt107 limits Rad9 binding near a DSB, leading to a reduction in DDC signalling and an increase in DNA resection, through a mechanism that does not require the Rad1, Slx1 and Mus81 nucleases. This Slx4-Rtt107 role is critical for the cell to successfully repair DSBs and to survive exposure to MMS and camptothecin (CPT, a topoisomerase-aborting agent), particularly when DSB resection and DDC are already compromised by sae2Δ mutation.
MATERIALS AND METHODS
Yeast strains, Media and Growth conditions
All the strains listed in Table S1 are derivative of JKM179 or W303. To generate strains, standard genetic procedures of transformation and tetrad analysis were followed. Deletions and tag fusions were generated by the one-step PCR system (19). All the strains, except the Y603 derivatives, obtained by direct transformation were outcrossed with the parental to clean the background. For the indicated experiments, cells were grown in YP medium enriched with 2% glucose (YEP+glu), 3% raffinose (YEP+raf) or 3% raffinose and 2% galactose (YEP+raf+gal). Unless specified all the experiments were performed at 28°C.
Measurement of DSB resection at MAT locus
DSB end resection in JKM179 derivative strains was analysed by alkaline agarose gels using a single-stranded RNA probe as described previously (5,20), and by quantitative PCR (qPCR) analysis (6,21). The oligonucleotides used in qPCR analysis are listed in Table S2. The DNA was digested with the RsaI restriction enzyme (NEB), which cuts inside the amplicons at 5 kb and 10 kb from the HO-cut site, but not in the PRE1 control region on chromosome V.
SDS-PAGE and Western blot
TCA protein extracts were prepared as described previously (22), and separated by SDS-PAGE. Western blotting was performed with monoclonal (EL7) or polyclonal (generous gift from C. Santocanale) anti-Rad53 antibodies.
Checkpoint adaptation by micro colony assay
For JKM179 derivative strains, cells were grown O/N in YEP + raf at 28°C. The unbudded cells were micro manipulated on YEP + raf + gal and plates were incubated at 28°C for 24 h. Micro colonies formed by more than 3 cells were scored as ‘adapted’. Standard deviation was calculated on three independent experiments. For cdc13–1 derivative strains, cells were grown O/N in YEP + glu at 23°C and micro manipulated on YEP + glu plates and were incubated at 37°C for 24 h.
ChIP-seq analysis
Cells were grown to log phase in YEP + raffinose and arrested in G2/M with 20 μg/ml nocadozole before addition of galactose to a final concentration of 2%. Cells were sampled immediately (0 h) and at 2, 4 and 6 h after galactose addition. Chromatin immunoprecipitation and sequencing data analysis were performed as previously described (23). Data are presented for chromosome III as a log2 ratio of normalized read counts for each IP:input pair. All sequencing data are deposited in the Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra; Study accession SRP062913).
ChIP analysis
ChIP analysis was performed as described previously (6). The oligonucleotides used are listed in Table S2. Data are presented as fold enrichment at the HO cut site (5 kb from DSB) over that at the PRE1 locus on chromosome V (for Slx4) or ARO1 locus on chromosome IV (for Rad9), and normalized to the corresponding input sample.
Ectopic recombination assay
We used derivatives of the tGI354 strain (Table S1). The percentage of cell viability of the indicated mutants after HO induction was calculated as a ratio between the number of colonies grown on YEP + raf + gal medium and the number of colonies grown on YEP + raf medium after 2–3 days of incubation at 28°C.
Physical analysis of DSB repair kinetics during ectopic gene conversion was performed with DNA samples isolated at different time points from HO induction. Genomic DNA was digested with EcoRI enzyme and separated on a 0.8% agarose gel. Southern blotting was done using a 1000 bp MATa probe. To calculate DSB repair values we normalized DNA amount using a DNA probe specific for IPL1 gene (unprocessed locus).
Drug sensitivity assay
Logarithmically growing cell cultures were serially diluted and spotted on media containing different dosages of MMS or CPT, as indicated. Plates were incubated at 28°C for 3 days.
RESULTS
The Slx4-Rtt107 complex contributes to checkpoint adaptation to one irreparable DSB and to uncapped telomeres
We asked whether the competition between Slx4 and Rad9 for Dpb11 binding might affect the cellular response to DSBs. In particular, we hypothesized that in the absence of Rtt107 or Slx4, the Rad9-dependent checkpoint signalling should be hyper-activated in the presence of one DSB. To address this question, we induced a persistent DSB at the MAT locus by over-expression of HO endonuclease in a JKM139 yeast background (20,24). This genetic system is ideal to correlate the DNA damage checkpoint signalling with the formation of ssDNA. Indeed, in these cells, the DSB induced by HO is extensively 5′-to-3′ resected, and the lack of homology elsewhere in the genome prevents the formation of any recombination intermediate. Thus, G1 unbudded cells were micro-manipulated in galactose containing medium to induce the HO-break. In this condition, the activation of the DNA damage checkpoint blocked cell cycle progression at the G2/M transition for several hours (24). However, wild type cells undergo checkpoint adaptation proceeding through 3–4 divisions after 24 h (25), when we scored the percentage of micro-colonies of 4–8 cells formed (Figure 1A,B). Strikingly, the percentage of cells that underwent adaptation and re-started the cell cycle was severely reduced in slx4Δ and rtt107Δ cells. A similar result was observed in the slx4-S486A mutant (Figure 1B), which specifically affects the Slx4-Dpb11 interaction (16,18), supporting the model that the interaction with Dpb11 is a key event in this regulatory mechanism. Moreover, as an additional indication of the central role of the interaction with Dpb11, the expression of the chimera MBD rescued the checkpoint adaptation defect of slx4Δ (Supplementary Figure S1). Importantly, the percentage of cells that underwent adaptation and re-started the cell cycle was not affected in the slx1Δ rad1Δ mus81Δ triple mutant (Figure 1B). These results, obtained after an irreparable DSB, indicate that the Slx4-Rtt107 complex, likely interacting with Dpb11, may have a role in checkpoint adaptation that is distinct from a role in DSB repair and JM resolution, which requires the Slx1, Rad1 or Mus81 nucleases (12–15).
Figure 1.
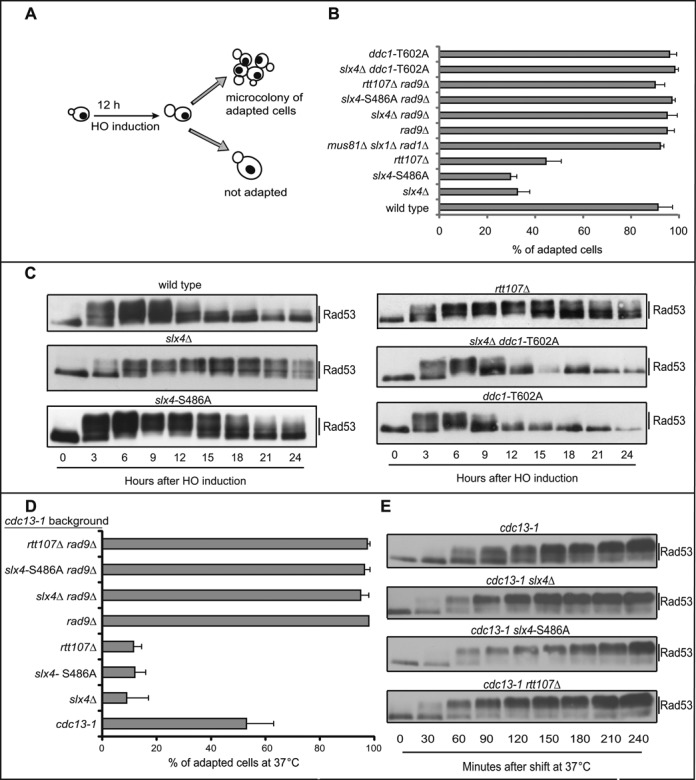
The Slx4-Rtt107 complex is required for checkpoint adaptation to one irreparable DSB and uncapped telomeres. (A) Schematic illustration of the HO-cut checkpoint-adaptation assay. (B) Graph shows the percentage of adapted cells for each mutant 24 h after plating on galactose containing medium to induce one irreparable HO cut. Values are the mean of three independent experiments ± standard deviation. (C) Rad53 phosphorylation analysis by Western Blot in JKM179 derivative strains after HO induction. (D) Adaptation assay in cdc13–1 derivative strains. Graph shows the percentage of adapted cells after 24 h at 37°C. Values are the mean of three independent experiments ± standard deviation. (E) Rad53 phosphorylation analysis by Western blot in cdc13–1 derivative strains after telomere uncapping at 37°C.
Consistent with the hypothesis that the Rad9 pathway is hyper-activated in the absence of Slx4 and Rtt107, the deletion of the RAD9 gene completely by-passed the prolonged cell cycle block of slx4Δ, slx4-S486A and rtt107Δ mutants (Figure 1B). The same by-pass was observed in ddc1-T602A cells, in which Dpb11 and Rad9 cannot be recruited by the 9–1–1 complex onto chromatin (11,26,27) (Figure 1B).
To monitor DSB-induced checkpoint signalling in the absence of a functional Slx4-Rtt107 pathway, we analysed Rad53 phosphorylation following formation of one irreparable HO-cut. In wild type cells, Rad53 was dephosphorylated 12–15 h after the DSB formation, as expected (28). On the contrary, Rad53 phosphorylation was prolonged and more robust in slx4Δ and rtt107Δ mutants (Figure 1C), consistent with the checkpoint adaptation defect shown in Figure 1B. The slx4-S486A mutant cells also show a robust and prolonged Rad53 phosphorylation, although slightly less than slx4Δ cells. Interestingly, Rad53 was only transiently phosphorylated in ddc1-T602A and ddc1-T602A slx4Δ mutants (Figure 1C), consistent with the defect in Rad9 stable association with DNA in ddc1-T602A mutant cells (11,18,26,27).
Checkpoint adaptation was previously observed at uncapped telomeres in thermo sensitive cdc13–1 mutant cells (29). Therefore we analysed cell cycle block and re-start in cdc13–1 derivative strains incubated at the restrictive temperature. Unbudded cells, grown at the permissive temperature, were micro-manipulated on a plate and immediately shifted to 37°C. As expected (29), cdc13–1 cells remained blocked at the G2/M transition for several hours, but after 24 h incubation a significant percentage of cells have re-started the cell cycle, producing micro-colonies of 4–8 cells (Figure 1D). Interestingly, cdc13–1 slx4Δ, cdc13–1 slx4-S486A and cdc13–1 rtt107Δ cells did not divide after the shift to 37°C (Figure 1D), suggesting that checkpoint adaptation following telomere uncapping was compromised in the absence of a functional Slx4-Rtt107 pathway, similarly to what we found in the presence of one irreparable DSB (Figure 1B). Importantly, the prolonged cell cycle block was rescued by deleting RAD9 (Figure 1D). Furthermore, by analysing protein extracts from cells shifted to the restrictive temperature, we found that Rad53 phosphorylation in cdc13–1 slx4Δ, cdc13–1 slx4-S486A and cdc13–1 rtt107Δ mutants occurred earlier than in cdc13–1 cells (Figure 1E), indicating that DNA damage checkpoint signalling was hyper-activated in the absence of Slx4 and Rtt107, consistent with the defect in cell cycle re-start by adaptation.
Taking the cellular and molecular results in Figure 1 together, we concluded that the Slx4-Rtt107 pathway plays a regulatory role in dampening the Rad9-dependent checkpoint signalling after one irreparable DSB and after telomere uncapping, leading to checkpoint adaptation and re-start of cell cycle progression after a prolonged G2/M arrest.
The Slx4-Rtt107 complex modulates Rad9 binding to one irreparable DSB
As we and others have recently shown that Rad9 plays a role in preventing DNA resection at a DSB (1,6,30,31), the results in Figure 1 prompted us to test the amount of Rad9 bound near a DSB in the absence of a functional Slx4-Rtt107 pathway. Recruitment of Rad9 oligomers near a DSB is a critical event to elicit a fully active DDC, also affecting DSB processing and repair. Interestingly, Rad9 oligomers are recruited through the interaction with modified histones and Dpb11 soon after DSB formation (6,10).
For high-resolution analysis of Rad9 binding along chromosome III after HO cutting at the MAT locus, in wild type and slx4Δ JKM139 cells, we used chromatin immunoprecipitation coupled to deep sequencing (ChIP-seq). Interestingly, before the induction of the HO-cut, we found several regions of Rad9 binding along the entire chromosome III, both in wild type and slx4Δ cells (Supplementary Figure S2). This result is in agreement with previous findings, indicating that Rad9 is recruited to several genome loci through the interaction with the transcription factor Aft1, even in the absence of exogenously induced DNA damage (32). After the induction of the HO cut at MAT locus (at ∼0.2 Mb on chromosome III), Rad9 binding increased around the DSB, both in wild type and slx4Δ cells (Figure 2A and Supplementary Figure S2), and the binding signal increased and spread along the flanking regions over time. Deletion of SLX4 resulted in an increase Rad9 binding proximal to the DSB (Figure 2A).
Figure 2.
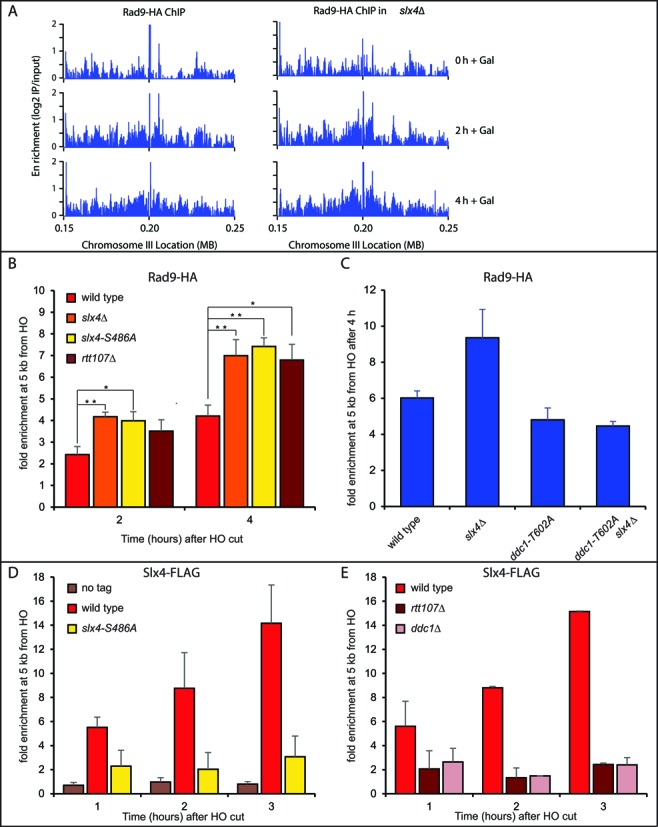
The Slx4-Rtt107 complex modulates Rad9 binding to one irreparable DSB. (A) ChIP-seq analysis of Rad9 following induction of a DSB on chromosome III. Rad9 was subjected to chromosome immunoprecipitation at the indicated times after induction of HO endonuclease, in wild type and slx4Δ strains. The enrichment scores (the log2 ratio of immunoprecipitate : input) across 100 kb flanking the HO cut site on chromosome III are plotted. (B–E) ChIP-qPCR analysis showing DSB-induced binding of Rad9, Slx4 or the Slx4-S486A variant at 5 kb from the HO site at the indicated times. All the experiments were performed in nocodazole-blocked cells of the indicated JKM179 derivative strains. In (B), (D) and (E), plotted values are the mean ± SEM of three independent experiments while in (C) two experiments were analyzed. Where indicated, significance was determined by single-tailed Student's t test (*for P < 0.05 and ** for P < 0.01).
To quantify the difference in Rad9 binding at the irreparable DSB in the presence and absence of Slx4, we performed ChIP followed by quantitative PCR (qPCR) with primers specific for a region 5 kb from the break. We found that deletion either of SLX4 or RTT107, as well as the slx4-S486A mutation, led to a significant increase of Rad9 binding 5 kb from the HO cut (Figure 2B). Strikingly, the ddc1-T602A mutation, which affects binding to Dpb11 (11,26,27), totally eliminated the increased binding of Rad9 in slx4Δ cells (Figure 2C). These results indicate that the Slx4-Rtt107 pathway is critical to limit the accumulation of Rad9, bound to Dpb11 at a persistent DSB, and may provide a molecular explanation for the prolonged checkpoint signalling observed in slx4Δ, rtt107Δ and slx4-S486A cells (Figure 1). Consistent with the proposed model, we also found by ChIP that the Slx4 protein was recruited 5 kb from an HO-induced DSB (Figure 2D). Interestingly, the binding of the Slx4-S486A protein variant was greatly lowered (Figure 2D), according to the effects on Rad9 binding, Rad53 phosphorylation and checkpoint adaptation that we found in slx4-S486A cells (Figures 1B,C and 2C). Moreover, deletion of the DDC1 gene abrogated Slx4 binding near a DSB (Figure 2E), further suggesting that Slx4 binding depends upon the interaction with Dpb11, which in turn is recruited through the 9–1–1 complex (33). We also found that Slx4 binding was severely reduced in rtt107Δ (Figure 2E), in agreement with recent findings indicating Rtt107 recruits Slx4 to stressed replication forks (23), and that Rtt107 stabilizes the interaction between Slx4 and Dpb11 (16,34).
Remarkably, Slx4 and Rtt107 were not detectable by ChIP very close to the DSB (35,36), although Dpb11 was recruited soon after the break formation, through the interaction with the 9–1–1 complex (33,37). As Rtt107 interacts with γ-H2AX (36,38), a possible explanation of this discrepancy might be related to the low amount of modified histones close to the break (10,39–42).
The Slx4-Rtt107 complex modulates long-range DSB resection
Rad9 oligomers bound around a DSB represents a physical barrier towards 5′-3′ resection (4). Thus, we hypothesized that DSB resection should be affected in the absence of a functional Slx4-Rtt107 pathway, which leads to increased Rad9 binding (Figure 2). To address this issue, we tested the formation of 3′ ssDNA at one irreparable DSB by Southern blotting of denatured DNA after restriction enzyme digestion (6). The HO-cut was induced in G2/M blocked cells to avoid any possible interference with cell cycle progression. Interestingly, we found that the formation of long 3′ ssDNA tail (specifically the r7 fragment in Figure 3A) was delayed in slx4Δ, slx4-S486A and rtt107Δ mutants, compared to wild type (Figure 3B,C). This may indicate that resection at distal regions from DSB is affected in the absence of a functional Slx4-Rtt107 pathway.
Figure 3.
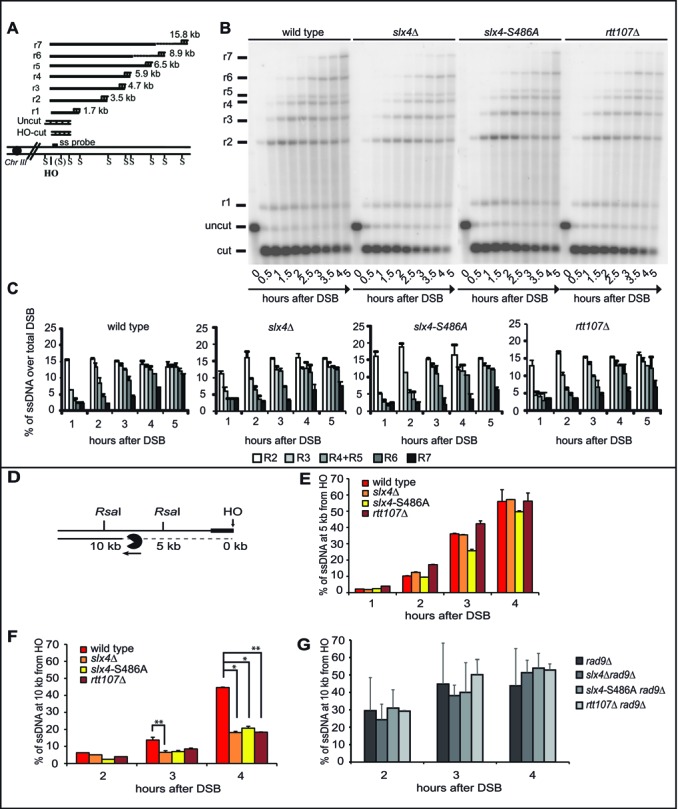
The Slx4-Rtt107 complex modulates long-range DSB resection. (A) Schematic illustration of MAT locus in JKM179 strains showing positions of HO-cut site, SspI restriction sites and the RNA probe used. (B) ssDNA visualization using RNA probe as described in (A) in indicated mutants after inducing HO in nocodazole arrested cells. (C) Resection products analysis by densitometry. Plotted values are the mean values ± SEM from two independent experiments performed as in (B). (D) Schematic illustration of DSB resection at the indicated RsaI sites, 5 kb and 10 kb distal from the irreparable HO cut at the MAT locus on chromosome III. (E–G) DSB resection analysis by qPCR in nocodazole-arrested JKM179 derivative strains. Plotted values are the mean of at least two independent experiments ± SEM. Where indicated, significance was determined by single-tailed Student's t test (*for P < 0.05, ** for P < 0.01 and *** for P < 0.001).
We also analysed DSB resection at different distances from an HO-cut using a more accurate quantitative PCR-based method (Figure 3D) (6,21). Using the same experimental conditions described for the Southern blot (Figure 3B), we found that the percentage of ssDNA accumulated at 5 kb far from the break in slx4Δ, slx4-S486A and rtt107Δ cells was comparable to what found in the wild-type cells (Figure 3E), although Rad9 binding was increased at this site (Figure 2B). Strikingly, a higher amount of un-resected DNA was detected 10 kb from the break in slx4Δ, slx4-S486A and rtt107Δ cells (Figure 3F). A possible explanation might be that, as resection was proceeding, the discrepancy between the amount of resected DNA in wild type and slx4-rtt107 mutant cells increased and became evident only at long distances from the DSB, consistent with what we found by Southern blot (Figure 3B,C).
Importantly, the DSB resection delay observed in slx4Δ, slx4-S486A and rtt107Δ cells at 10 kb from the break was completely rescued by deleting RAD9 (Figure 3G), in agreement with the proposed model that the Rad9-dependent DSB resection barrier is higher in slx4 and rtt107 mutants.
Taking all the results in Figures 2 and 3 together, we propose that a functional Slx4-Rtt107 pathway contributes to maintaining efficient DSB resection, likely limiting the Rad9 barrier and Rad53 signalling.
DSB resection and DDC inactivation are severely compromised in the absence of both Sae2 and Slx4-Rtt107
Based on the results in Figure 3, we reasoned that deletion of SLX4 might exacerbate a resection delay in those mutants already defective in DSB processing, particularly short-range resection (3,43). Indeed it is known that double mutants affecting both the short- and long-range resection steps, such as sae2Δ exo1Δ, show a severe DSB resection defect (6,44–46). To this end, we generated a sae2Δ slx4Δ double mutant strain and we analysed DSB resection by qPCR, after induction of HO in G2/M blocked cells. In agreement with the hypothesis, the sae2Δ slx4Δ double mutant cells showed a severe delay in DSB resection (Figure 4A), further supporting our previous conclusion that Slx4 plays a significant role in the long-range DSB resection. Interestingly, we also found that sae2Δ slx4Δ double mutant cells hyper-activated Rad53 after an irreparable HO-cut, blocking the cell cycle re-start by checkpoint adaptation, even more than the respective single mutants (Figure 4B,C).
Figure 4.
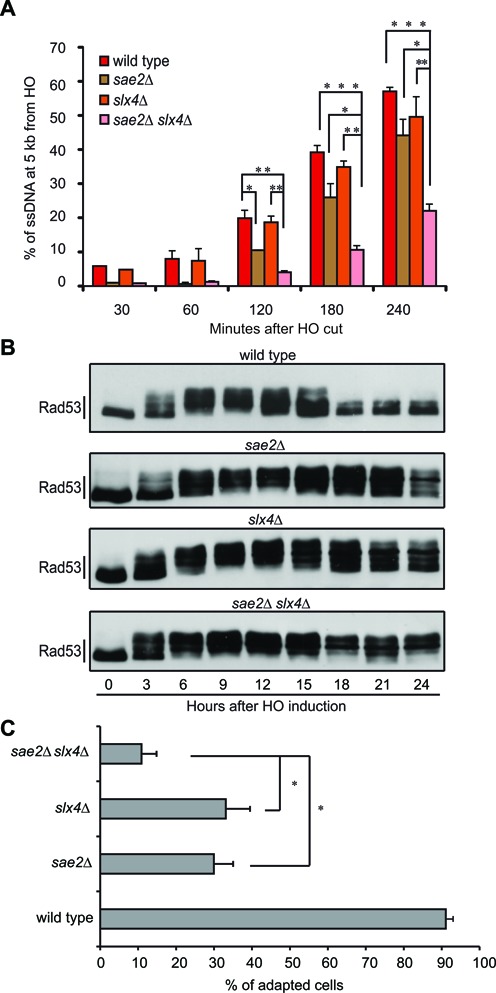
Deletion of SLX4 exacerbates sae2Δ cells phenotypes. (A) DSB resection analysis by qPCR in nocodazole-arrested JKM179 derivative strains. Plotted values are the mean of at least two independent experiments ±SEM. Where indicated, significance was determined by single-tailed Student's t test (*for P < 0.05, ** for P < 0.01 and *** for P < 0.001). (B) Rad53 phosphorylation analysis by Western Blot in JKM179 derivative strains after HO induction. (C) Graph shows the percentage of adapted cells for each mutant 24 h after plating on galactose containing medium. Values are the mean of three independent experiments ± standard deviation. Where indicated, significance was determined by double-tailed Student's t test (* for P < 0.05).
We thought that the Slx4-Rtt107 role in the regulation of DDC and DSB processing might contribute to the DSB repair, especially in sae2Δ cells. To this end, we took advantage of a genetic system in which interchromosomal recombination between two homologous cassettes on chromosome III and V can occur (47,48). Briefly, in these cells, an HO-induced DSB at an additional MAT sequence inserted in chromosome V is repaired by copying the information from the homologous MATa-inc locus on chromosome III (Figure 5A), through a gene conversion process that requires 6–8 h and is coupled with DDC activation (47–50). Importantly, Sae2, Rtt107, Rad1 and Mus81 are almost dispensable for DSB repair and cell viability in this assay (47,51,52). By plating the cells in the presence of galactose to induce the HO-cut, we found that the viability of the slx4Δ, slx4-S486A, rtt107Δ and sae2Δ single mutants was almost similar to the wild type, while the viability of the slx4Δ sae2Δ, slx4-S486A sae2Δ and rtt107Δ sae2Δ double mutants was severely reduced (Figure 5B). Strikingly, by Southern blotting analysis, we found that the total repair product (as a summary of crossovers and noncrossovers) was reduced in the slx4Δ sae2Δ double mutant after the HO-cut induction in G2/M blocked cells, although it is not affected in the single mutants (Figure 5C,D). Moreover, Rad53 phosphorylation by western blotting was very robust and prolonged in the slx4Δ sae2Δ cells during the ectopic recombination assay (Figure 5C), consistent with a persistent DSB. Therefore, our results suggest that the interchromosomal recombination is reduced in slx4Δ sae2Δ cells mainly as a consequence of their defect in dampening the Rad9-dependent checkpoint and resecting the break.
Figure 5.
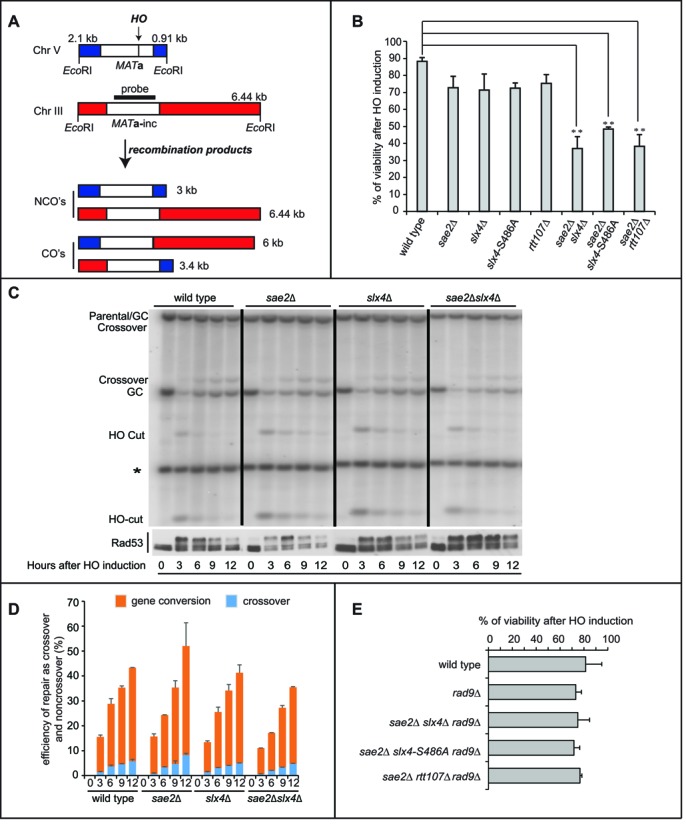
Deletion of SLX4 affects interchromosomal recombination in sae2Δ cells. (A) Schematic illustration of MATa-inc locus in Chromosome III and the additional MATa locus in Chromosome V in tGI354 strain, showing positions of HO-cut site, EcoR1 restriction sites and the probe used to test the interchromosomal recombination. (B) Viability of the tGI354 derivatives after the induction of the HO-cut. (C) Southern blotting analysis of the interchromosomal recombination using the probe as described in (A), in indicated tGI354 derivatives after inducing HO in nocodazole-arrested cells. The intensity of each band was normalized respect to unprocessed IPL1 locus (*). GC is for Gene Conversion. Western blot analysis shows Rad53 phosphorylation of the same experiment. (D) Percentage of crossovers and non-crossovers among all cells in the interchromosomal recombination assay described in (C). (E) Viability of the tGI354 derivatives after the induction of the HO-cut.
Supporting the hypothesis that the Rad9 binding near the break, DSB resection and checkpoint signalling are critical events during the interchromosomal recombination in the slx4Δ sae2Δ, slx4-S486A sae2Δ and rtt107Δ sae2Δ cells, the deletion of RAD9 strikingly rescued the cell lethality in all those double mutants, after DSB induction (Figure 5B,E).
Of importance, slx4Δ sae2Δ, slx4-S486A sae2Δ and rtt107Δ sae2Δ double mutant cells are hypersensitive to both MMS and CPT, even more than the respective single mutant strains (Figure 6), whose sensitivity was already known (16,34,53–55). In particular, rtt107Δ cells are reported to be more sensitive to MMS and CPT than slx4Δ cells (56,57), therefore in Figure 6C we plated the cells in the presence of lower doses of the drugs, to better show the additive sensitivity of the rtt107Δ sae2Δ double mutant. Strikingly, the deletion of RAD9 almost completely rescued the sensitivity of single and double mutants (Figure 6), further suggesting that the hyper-activation of the Rad9-dependent DDC and the slow DNA resection can cause the severe sensitivity to MMS and CPT in these cells.
Figure 6.
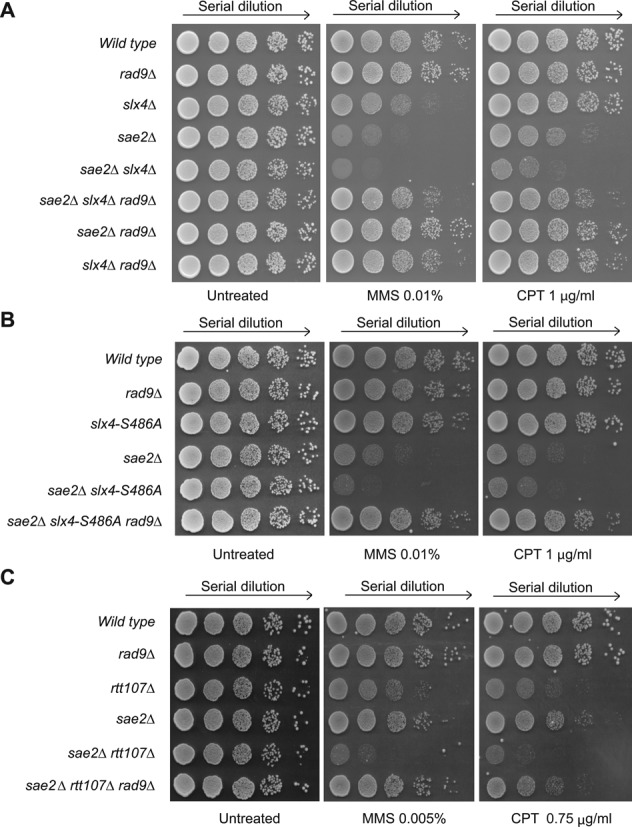
Deletion of RAD9 rescues the sensitivity to MMS and CPT of sae2Δ, slx4Δ, slx4-S486A and rtt107Δ mutant combinations. Exponentially growing cell cultures of the indicated JKM139 derivatives were serially diluted (1:10), and each dilution was spotted out into YPD, YPD + MMS and YPD + CPT plates. Plates were incubated 3 days at 28°C. In panel C, we used lower concentration doses of MMS and CPT (see text for details).
DISCUSSION
The 53BP1-ortholog Rad9 is crucial for DDC signalling and regulation of DNA end resection in S. cerevisiae. Recruitment of Rad9 to DNA lesions is a key aspect of both of these functions, and is mediated by its interaction with modified histones and Dpb11.
Recently, it was shown that the Slx4-Rtt107 complex is in competition with Rad9 for the interaction with Dpb11, contributing to dampen the DDC signalling in the presence of MMS (16). Accordingly, slx4Δ cells hyper-activate the Rad9-dependent checkpoint. More recently, it was shown that slx4Δ cells accumulate DNA lesions (ssDNA) during stressful replication, and that Slx4-Dpb11 interaction is critical to coordinate the Mus81 nuclease, promoting JM resolution (18). Therefore, these data open a debate on how to discriminate the Slx4 role in checkpoint dampening from its role in DNA replication/recombination. To further understand this issue, here we studied the interplay between the Slx4-Rtt107 complex and Rad9 after the formation of one irreparable HO-cut in the MAT locus on chromosome III, in a strain in which the homologous HML and HMR sequences were deleted (24). In fact, the Slx4 role in DNA repair and its cooperation with Mus81 and other resolvases is dispensable in this assay, because no recombination intermediate is formed. Therefore, this experimental setup provided us with a defined system to directly investigate the role Slx4 plays independently of these factors.
Strikingly, slx4Δ and rtt107Δ cells had an increased binding of Rad9 near the DSB, which is dependent on Ddc1 and Dpb11 pathway (Figure 2). As a consequence, the resection of the 5′ strand was slower in slx4Δ and rtt107Δ cells (Figure 3), Rad53 was hyper-activated and checkpoint adaptation was impaired (Figure 1). Of note, although others showed that Slx4 does not bind close to the DSB (35), we found that Slx4 was recruited within a few Kb from a persistent DSB (Figure 2). Remarkably, the slx4-S486A mutation, which prevents Slx4 phosphorylation by CDK1 and its interaction with Dpb11 (16), abolished Slx4 binding to the DSB (Figure 2D), and caused most of the defects found in slx4Δ cells (Figures 1–3), suggesting that Dpb11 and CDK1 are important components of this pathway.
Interestingly, we found that slx4Δ, slx4-S486A and rtt107Δ exacerbated the sensitivity of sae2Δ cells to HO-breaks, MMS and CPT (Figures 4–6). This additive effect is particularly relevant for the slx4Δ and slx4-S486A mutations, which per sé do not cause sensitivity to CPT at the dosage tested. Moreover, by using a specific HO-based assay, we found that slx4Δ sae2Δ cells, but not the single mutants, are defective in interchromosomal recombination (Figure 5C,D). Of note, we recently found that increased binding of Rad9 near a DSB causes all the relevant defects in sae2Δ cells: prolonged binding of Mre11, resection delay, reduced recruitment of Rad52 and defect in DSB end-tethering (6). Our data in Figures 4–6 suggest that in the slx4Δ sae2Δ double mutant the persistent binding of Rad9 limits DSB resection, repair and checkpoint inactivation, even more than the single mutants. In fact, the deletion of RAD9 rescued the DNA damage sensitivity of cells with dysfunctional Slx4-Rtt107 and Sae2 pathways very well (Figures 5E and 6), strongly suggesting that the defects in dampening the DDC and resecting the DSBs cause cell lethality.
Our results reinforce and expand the notion that Rad9 binding near a DSB is critical for the cell to properly respond and repair DSBs. Indeed, in recent literature there are examples in which the increased Rad9 binding has been associated with a slow DSB resection and a prolonged checkpoint signalling, such as mec1-ad and fun30Δ cells, which neither recover from, nor adapt to a DSB (58–61). Possibly, the increased Rad9 binding close to DSB ends may affect the balance between NHEJ and HR events, as we showed in sae2Δ cells (6). Similar function has been shown for 53BP1 in human cells (62–66).
In conclusion, we show that the Slx4-Rtt107 complex acts as an antagonist of Rad9 binding at DSBs, limiting both the Rad9 checkpoint signalling and DSB resection barrier. Altogether, our findings suggest a working model (Figure 7), in which Dpb11 and Rad9 play a role in the early step of the response to a DSB, activating the DDC. Once extensive resection is on going, the Slx4-Rtt107 complex (likely phosphorylated by Mec1 and CDK1 (16,36,54,67)) competes with Rad9 for Dpb11 binding, dampening DDC and allowing further progression of resection, especially in the presence of nucleosomes. Importantly, the novelties described in our work, after the formation of one persistent DSB, indicate that the role of the Slx4-Rtt107 complex to dampen the DDC is active not only during replication in the presence of MMS (16), but also at DSB lesions. Therefore, it will be important to test in the future whether this mechanism is functional at any types of DNA damage in which the Dpb11-Rad9 axes is engaged. It remains also to be investigated if the Slx4-Rtt107 pathway takes over to counteract Rad9 particularly at persistent DNA lesions. Interestingly, persistent or slowly repaired DSBs re-localize to the nuclear periphery, where they are anchored to the nuclear pore complex (68–70). This phenomenon has been proposed to affect the choice of the repair pathways at persistent DSBs (68). An interesting hypothesis to address is whether the checkpoint dampening and adaptation controlled by the Slx4-Rtt107 pathway occur at the nuclear periphery. This may also correlate with the reduction of DSB repair found in slx4Δ sae2Δ cells during an ectopic recombination assay (Figure 5C,D), which notably occurs at the nuclear periphery (68).
Figure 7.
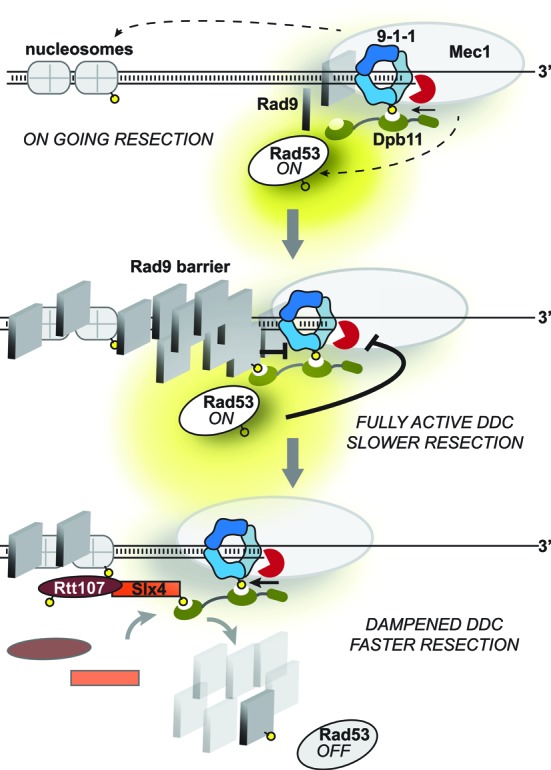
A model for the interplay between Rad9 and the Slx4-Rtt107 complex at a DSB. See text for the details. Yellow circles indicate phosphorylation events.
Considering our data in a wider context, it will be interesting to test if Rad9 may limit ssDNA accumulation during stressful replication in the absence of a functional Dpb11-Slx4-Rtt107 complex (18). In this condition, avoiding the formation of long ssDNA gaps, we can speculate that Rad9 may protect chromosomes from breakages and unscheduled recombination events, preserving genome integrity.
Importantly, mutations in human SLX4 increase sensitivity to DNA damage and are linked with Fanconi Anemia, a genetic disorder associated with high checkpoint marker activation, which could be a cause of bone marrow failure (14,71). Taking that into consideration, in the future it will be relevant to investigate whether SLX4, in addition to its functions in DSB repair, might have a role in controlling DDC and DSB resection in human cells too. Remarkably, we showed that in yeast Slx4 plays an important role in regulating DDC at uncapped telomeres too (Figure 1). An additional open question to address in the future is whether Slx4 might also regulate 53BP1 binding and DDC at eroded telomeres in human cells, where SLX4 localizes to telomeres through TRF2 binding (72,73).
Supplementary Material
Acknowledgments
We thank all the members of our laboratories and M. Neale for helpful discussions. J. Haber generously provided the JKM139 strain, and G. Ira the tGI354 strain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Associazione Italiana per la Ricerca sul Cancro [AIRC_IG Grant 15488 to A.P.]; CARIPLO [2013-0790 to A.P.]; Canadian Institutes of Health Research and the Canadian Cancer Society Research Institute [to G.W.B.]; and a fellowship from Fondazione VERONESI [to M.F]. Funding for open access charge: Associazione Italiana per la Ricerca sul Cancro (AIRC) [IG Grant 15488 to A.P.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Lazzaro F., Giannattasio M., Puddu F., Granata M., Pellicioli A., Plevani P., Muzi-Falconi M. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Rep. (Amst) 2009;8:1055–1067. doi: 10.1016/j.dnarep.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 2.Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 4.Granata M., Panigada D., Galati E., Lazzaro F., Pellicioli A., Plevani P., Muzi-Falconi M. To trim or not to trim: progression and control of DSB end resection. Cell Cycle. 2013;12:1848–1860. doi: 10.4161/cc.25042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazzaro F., Sapountzi V., Granata M., Pellicioli A., Vaze M., Haber J.E., Plevani P., Lydall D., Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–1512. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrari M., Dibitetto D., De Gregorio G., Eapen V.V., Rawal C.C., Lazzaro F., Tsabar M., Marini F., Haber J.E., Pellicioli A. Functional Interplay between the 53BP1-Ortholog Rad9 and the Mre11 Complex Regulates Resection, End-Tethering and Repair of a Double-Strand Break. PLoS Genet. 2015;11:e1004928. doi: 10.1371/journal.pgen.1004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giannattasio M., Lazzaro F., Plevani P., Muzi-Falconi M. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J. Biol. Chem. 2005;280:9879–9886. doi: 10.1074/jbc.M414453200. [DOI] [PubMed] [Google Scholar]
- 8.Hammet A., Magill C., Heierhorst J., Jackson S.P. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007;8:851–857. doi: 10.1038/sj.embor.7401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granata M., Lazzaro F., Novarina D., Panigada D., Puddu F., Abreu C.M., Kumar R., Grenon M., Lowndes N.F., Plevani P., et al. Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle-regulated by CDK1 activity. PLoS Genet. 2010;6:e1001047. doi: 10.1371/journal.pgen.1001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wysocki R., Javaheri A., Allard S., Sha F., Cote J., Kron S.J. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell Biol. 2005;25:8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfander B., Diffley J.F. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 2011;30:4897–4907. doi: 10.1038/emboj.2011.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Princz L.N., Gritenaite D., Pfander B. The Slx4-Dpb11 scaffold complex: -coordinating the response to replication fork stalling in S-phase and the subsequent mitosis. Cell Cycle. 2014;14:488–494. doi: 10.4161/15384101.2014.989126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouse J. Control of genome stability by SLX protein complexes. Biochem. Soc. Trans. 2009;37:495–510. doi: 10.1042/BST0370495. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y. Nuclease delivery: versatile functions of SLX4/FANCP in genome maintenance. Mol. Cell. 2014;37:569–574. doi: 10.14348/molcells.2014.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz E.K., Heyer W.D. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma. 2011;120:109–127. doi: 10.1007/s00412-010-0304-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohouo P.Y., Bastos de Oliveira F.M., Liu Y., Ma C.J., Smolka M.B. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature. 2013;493:120–124. doi: 10.1038/nature11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cussiol J.R., Jablonowski C.M., Yimit A., Brown G.W., Smolka M.B. Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J. 2015;34:1704–1717. doi: 10.15252/embj.201490834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gritenaite D., Princz L.N., Szakal B., Bantele S.C., Wendeler L., Schilbach S., Habermann B.H., Matos J., Lisby M., Branzei D., et al. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014;28:1604–1619. doi: 10.1101/gad.240515.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longtine M.S., McKenzie A. 3rd, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 20.White C.I., Haber J.E. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990;9:663–673. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zierhut C., Diffley J.F. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008;27:1875–1885. doi: 10.1038/emboj.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muzi Falconi M., Piseri A., Ferrari M., Lucchini G., Plevani P., Foiani M. De novo synthesis of budding yeast DNA polymerase alpha and POL1 transcription at the G1/S boundary are not required for entrance into S phase. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10519–10523. doi: 10.1073/pnas.90.22.10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balint A., Kim T., Gallo D., Cussiol J.R., Bastos de Oliveira F.M., Yimit A., Ou J., Nakato R., Gurevich A., Shirahige K., et al. Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J. 2015;34:2182–2197. doi: 10.15252/embj.201591190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S.E., Moore J.K., Holmes A., Umezu K., Kolodner R.D., Haber J.E. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 25.Serrano D., D'Amours D. When genome integrity and cell cycle decisions collide: roles of polo kinases in cellular adaptation to DNA damage. Syst. Synthetic Biol. 2014;8:195–203. doi: 10.1007/s11693-014-9151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navadgi-Patil V.M., Burgers P.M. The unstructured C-terminal tail of the 9–1–1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell. 2009;36:743–753. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puddu F., Granata M., Di Nola L., Balestrini A., Piergiovanni G., Lazzaro F., Giannattasio M., Plevani P., Muzi-Falconi M. Phosphorylation of the budding yeast 9–1–1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol. Cell Biol. 2008;28:4782–4793. doi: 10.1128/MCB.00330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pellicioli A., Lee S.E., Lucca C., Foiani M., Haber J.E. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 29.Toczyski D.P., Galgoczy D.J., Hartwell L.H. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell. 1997;90:1097–1106. doi: 10.1016/s0092-8674(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 30.Ngo G.H., Lydall D. The 9–1–1 checkpoint clamp coordinates resection at DNA double strand breaks. Nucleic Acids Res. 2015;43:5017–5032. doi: 10.1093/nar/gkv409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonetti D., Villa M., Gobbini E., Cassani C., Tedeschi G., Longhese M.P. Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep. 2015;16:351–361. doi: 10.15252/embr.201439764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andreadis C., Nikolaou C., Fragiadakis G.S., Tsiliki G., Alexandraki D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014;42:12650–12667. doi: 10.1093/nar/gku915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogiwara H., Ui A., Onoda F., Tada S., Enomoto T., Seki M. Dpb11, the budding yeast homolog of TopBP1, functions with the checkpoint clamp in recombination repair. Nucleic Acids Res. 2006;34:3389–3398. doi: 10.1093/nar/gkl411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohouo P.Y., Bastos de Oliveira F.M., Almeida B.S., Smolka M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell. 2010;39:300–306. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 35.Toh G.W., Sugawara N., Dong J., Toth R., Lee S.E., Haber J.E., Rouse J. Mec1/Tel1-dependent phosphorylation of Slx4 stimulates Rad1-Rad10-dependent cleavage of non-homologous DNA tails. DNA Rep. (Amst) 2010;9:718–726. doi: 10.1016/j.dnarep.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ullal P., Vilella-Mitjana F., Jarmuz A., Aragon L. Rtt107 phosphorylation promotes localisation to DNA double-stranded breaks (DSBs) and recombinational repair between sister chromatids. PLoS One. 2011;6:e20152. doi: 10.1371/journal.pone.0020152. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Donnianni R.A., Ferrari M., Lazzaro F., Clerici M., Tamilselvan Nachimuthu B., Plevani P., Muzi-Falconi M., Pellicioli A. Elevated levels of the polo kinase Cdc5 override the Mec1/ATR checkpoint in budding yeast by acting at different steps of the signaling pathway. PLoS Genet. 2010;6:e1000763. doi: 10.1371/journal.pgen.1000763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X., Liu K., Li F., Wang J., Huang H., Wu J., Shi Y. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J. Biol. Chem. 2012;287:9137–9146. doi: 10.1074/jbc.M111.311860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shroff R., Arbel-Eden A., Pilch D., Ira G., Bonner W.M., Petrini J.H., Haber J.E., Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C.S., Lee K., Legube G., Haber J.E. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat. Struct. Mol. Biol. 2014;21:103–109. doi: 10.1038/nsmb.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shim E.Y., Hong S.J., Oum J.H., Yanez Y., Zhang Y., Lee S.E. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol. Cell Biol. 2007;27:1602–1613. doi: 10.1128/MCB.01956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsukuda T., Fleming A.B., Nickoloff J.A., Osley M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Symington L.S., Rothstein R., Lisby M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014;198:795–835. doi: 10.1534/genetics.114.166140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mimitou E.P., Symington L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clerici M., Mantiero D., Lucchini G., Longhese M.P. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 2006;7:212–218. doi: 10.1038/sj.embor.7400593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ira G., Malkova A., Liberi G., Foiani M., Haber J.E. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaze M.B., Pellicioli A., Lee S.E., Ira G., Liberi G., Arbel-Eden A., Foiani M., Haber J.E. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell. 2002;10:373–385. doi: 10.1016/s1097-2765(02)00593-2. [DOI] [PubMed] [Google Scholar]
- 49.Trovesi C., Falcettoni M., Lucchini G., Clerici M., Longhese M.P. Distinct Cdk1 requirements during single-strand annealing, noncrossover, and crossover recombination. PLoS Genet. 2011;7:e1002263. doi: 10.1371/journal.pgen.1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saponaro M., Callahan D., Zheng X., Krejci L., Haber J.E., Klein H.L., Liberi G. Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet. 2010;6:e1000858. doi: 10.1371/journal.pgen.1000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prakash R., Satory D., Dray E., Papusha A., Scheller J., Kramer W., Krejci L., Klein H., Haber J.E., Sung P., et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shim E.Y., Chung W.H., Nicolette M.L., Zhang Y., Davis M., Zhu Z., Paull T.T., Ira G., Lee S.E. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010;29:3370–3380. doi: 10.1038/emboj.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flott S., Alabert C., Toh G.W., Toth R., Sugawara N., Campbell D.G., Haber J.E., Pasero P., Rouse J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol. Cell. Biol. 2007;27:6433–6445. doi: 10.1128/MCB.00135-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts T.M., Kobor M.S., Bastin-Shanower S.A., Ii M., Horte S.A., Gin J.W., Emili A., Rine J., Brill S.J., Brown G.W. Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol. Biol. Cell. 2006;17:539–548. doi: 10.1091/mbc.E05-08-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munoz-Galvan S., Tous C., Blanco M.G., Schwartz E.K., Ehmsen K.T., West S.C., Heyer W.D., Aguilera A. Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol. Cell. Biol. 2012;32:1592–1603. doi: 10.1128/MCB.00111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leung G.P., Aristizabal M.J., Krogan N.J., Kobor M.S. Conditional genetic interactions of RTT107, SLX4, and HRQ1 reveal dynamic networks upon DNA damage in S. cerevisiae. G3. 2014;4:1059–1069. doi: 10.1534/g3.114.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leung G.P., Lee L., Schmidt T.I., Shirahige K., Kobor M.S. Rtt107 is required for recruitment of the SMC5/6 complex to DNA double strand breaks. J. Biol. Chem. 2011;286:26250–26257. doi: 10.1074/jbc.M111.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clerici M., Trovesi C., Galbiati A., Lucchini G., Longhese M.P. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J. 2014;33:198–216. doi: 10.1002/embj.201386041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eapen V.V., Sugawara N., Tsabar M., Wu W.H., Haber J.E. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol. Cell. Biol. 2012;32:4727–4740. doi: 10.1128/MCB.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costelloe T., Louge R., Tomimatsu N., Mukherjee B., Martini E., Khadaroo B., Dubois K., Wiegant W.W., Thierry A., Burma S., et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581–584. doi: 10.1038/nature11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X., Cui D., Papusha A., Zhang X., Chu C.D., Tang J., Chen K., Pan X., Ira G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012;489:576–580. doi: 10.1038/nature11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng L., Fong K.W., Wang J., Wang W., Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013;288:11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Escribano-Diaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkac J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D., et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 66.Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A.D., Feldhahn N., Resch W., Oliveira T.Y., Chait B.T., et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rouse J. Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J. 2004;23:1188–1197. doi: 10.1038/sj.emboj.7600129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oza P., Jaspersen S.L., Miele A., Dekker J., Peterson C.L. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009;23:912–927. doi: 10.1101/gad.1782209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalocsay M., Hiller N.J., Jentsch S. Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell. 2009;33:335–343. doi: 10.1016/j.molcel.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 70.Nagai S., Dubrana K., Tsai-Pflugfelder M., Davidson M.B., Roberts T.M., Brown G.W., Varela E., Hediger F., Gasser S.M., Krogan N.J. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science. 2008;322:597–602. doi: 10.1126/science.1162790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kottemann M.C., Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wan B., Yin J., Horvath K., Sarkar J., Chen Y., Wu J., Wan K., Lu J., Gu P., Yu E.Y., et al. SLX4 assembles a telomere maintenance toolkit by bridging multiple endonucleases with telomeres. Cell Rep. 2013;4:861–869. doi: 10.1016/j.celrep.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilson J.S., Tejera A.M., Castor D., Toth R., Blasco M.A., Rouse J. Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 2013;4:853–860. doi: 10.1016/j.celrep.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.