


Abstract
We present evidence that Tim establishes a physical and functional interaction with DDX11, a super-family 2 iron-sulfur cluster DNA helicase genetically linked to the chromosomal instability disorder Warsaw breakage syndrome. Tim stimulates DDX11 unwinding activity on forked DNA substrates up to 10-fold and on bimolecular anti-parallel G-quadruplex DNA structures and three-stranded D-loop approximately 4–5-fold. Electrophoretic mobility shift assays revealed that Tim enhances DDX11 binding to DNA, suggesting that the observed stimulation derives from an improved ability of DDX11 to interact with the nucleic acid substrate. Surface plasmon resonance measurements indicate that DDX11 directly interacts with Tim. DNA fiber track assays with HeLa cells exposed to hydroxyurea demonstrated that Tim or DDX11 depletion significantly reduced replication fork progression compared to control cells; whereas no additive effect was observed by co-depletion of both proteins. Moreover, Tim and DDX11 are epistatic in promoting efficient resumption of stalled DNA replication forks in hydroxyurea-treated cells. This is consistent with the finding that association of the two endogenous proteins in the cell extract chromatin fraction is considerably increased following hydroxyurea exposure. Overall, our studies provide evidence that Tim and DDX11 physically and functionally interact and act in concert to preserve replication fork progression in perturbed conditions.
INTRODUCTION
Accurate transfer of genetic information is critical for survival of living organisms. Checkpoint control pathways prevent the transmission of incompletely replicated or damaged DNA to daughter cells (1,2). Stabilization of stalled replication forks in the presence of DNA damage or at difficult-to-replicate templates is necessary in order to prevent their collapse and consequential inability to restart synthesis. In budding/fission yeast stabilization of paused forks is carried out by a complex (‘fork-protection complex’ or ‘pausing complex’) consisting of Tof1/Swi1 and Csm3/Swi3, which act in concert with a third protein factor named Mrc1 and collectively mediate the replication checkpoint (3,4). The fork-protection complex is believed to prevent uncoupling of replicative DNA polymerases from the DNA unwinding machinery, when DNA synthesis is halted at sites of DNA damage or natural replication fork barriers. Moreover, Mrc1, Tof1/Swi1 and Csm3/Swi3 are associated with the replication fork during S phase and assist its smooth progression even in unperturbed conditions (3–5). Similar mechanisms of fork stabilization and coordination of DNA synthesis and unwinding during normal replication are believed to operate also in metazoans, in which Claspin, Tim (also known as Timeless) and Tipin serve as the orthologs of Mrc1, Tof1/Swi1 and Csm3/Swi3, respectively (6–8). Like their yeast counterparts, these proteins act as S phase checkpoint mediators. In human cells, Tipin is required for efficient cell cycle arrest in response to DNA damage (9). Depletion of Tipin or Tim by RNA interference renders human cells sensitive to ionizing radiation (10) and causes a great reduction in Chk1 activation upon hydroxyurea (HU) or ultraviolet radiation exposure (11). In Xenopus laevis egg extracts, Tipin is required for stalled replication forks to recover after removal of the DNA polymerase inhibitor aphidicolin by promoting DNA polymerase α re-priming on the leading strand (12,13). As in yeast (14), the fork-protection complex was demonstrated to be associated with the advancing replication complexes either in human cells (15) or Xenopus egg extracts (12).
Components of the fork-protection complex are also reported to promote sister-chromatid cohesion in various systems, including yeasts (16–19), Caenorhabditis elegans (20), Xenopus laevis egg extracts (12,21) and human cell lines (22,23). Cohesion is the process that ensures tethering of newly replicated sister chromatids until they separate in metaphase. The role played by Tim and Tipin in facilitating the cohesion process has not yet been clarified (24,25). Important clues may derive from genetic studies in yeast that revealed a functional link between Tof1/Swi1-Csm3/Swi3 and the cohesion establishment factor Chl1 (19,26). Chl1, also known as ChlR1 or DDX11 in vertebrates, is a super-family 2 (SF2) ATP-dependent DEAH-box DNA helicase that unwinds DNA with a 5′- to 3′-directionality. DDX11 shares sequence similarity to the Fe-S cluster-containing DNA helicases FANCJ, XPD and RTEL1 (27) that play important roles in genome stability maintenance and are implicated in rare genetic syndromes and cancer development (28,29). Autosomal recessive mutations of the DDX11-encoding gene are responsible for a rare cohesinopathy-related disease named Warsaw breakage syndrome (WABS). Affected individuals display severe microcephaly, facial dysmorphy, pre- and postnatal growth retardation, intellectual disability and at the cellular level, increased drug-induced chromosomal breakage and sister-chromatid cohesion defects (30,31). Interestingly, WABS-related mutations were found to severely impair DDX11 DNA helicase activity, as demonstrated by biochemical analysis of purified recombinant mutated proteins (27,30–31). A role of budding yeast Chl1 in sister-chromatid cohesion was first suggested by genetic studies (32,33). Thereafter, it was reported that downregulation of DDX11 causes cohesion defects and increased sensitivity to various DNA-damaging agents in human cell lines (34–36). Sister-chromatid cohesion is expected to be tightly coordinated with the DNA replication process (24,25). In fact, genetic deficiency in a number of replication factors in addition to Tim and Tipin was found to cause cohesion defects (18,38). A model was proposed in which enzymes involved in Okazaki fragment processing are believed to act in concert with DDX11 at the lagging strand to facilitate cohesion (25). In agreement with these findings, DDX11 was found to be maximally active in vitro on substrates that mimic key intermediates of DNA replication/repair/recombination reactions (37–39).
Herein, we report that human Tim directly interacts with DDX11 and specifically stimulates DDX11 helicase activity on structurally distinct DNA substrates including forked duplex DNA, a three-stranded D-loop substrate and an anti-parallel bimolecular G-quadruplex (G4) DNA structure. Co-immuno-precipitation experiments on chromatin fraction have revealed that interaction between the two proteins is increased upon replication stress induction by treating cells with HU. DNA fiber track analyses have indicated that Tim downregulation by RNA interference in HeLa cells substantially reduces DNA replication rate in unperturbed conditions, whereas DDX11 depletion does not affect replication fork progression. Treatment of Tim- and/or DDX11-depleted cells with HU causes reduced fork rate and impaired fork restart capability. These findings show for the first time a direct functional interaction between a replication fork stabilizing factor and a DNA helicase implicated in the sister-chromatid cohesion process.
MATERIALS AND METHODS
Proteins
Recombinant human FLAG-Tim, Tipin-His6, the His6Tim-HA/His6-Tipin-3xFLAG complex, DDX11–3xFLAG and FANCJ were produced as previously described (8,39–40).
DNA substrates
PAGE-purified oligonucleotides used for the preparation of DNA substrates were purchased from Primm (Milan, Italy). The DNA oligonucleotides were 5′-end-labeled with T4 polynucleotide kinase (Roche) and [γ-32P]ATP (specific activity: 6000 Ci/mmoles) and purified from the unincorporated radionuclide by passage over a Micro Bio-Spin 30 column (Bio-Rad). For the preparation of the forked DNA duplex, the labeled antiM13–39-T40–5′ oligonucleotide (5′-GTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′) was annealed to a 3-fold molar excess of the cold complementary M13–39-T40–3′ oligonucleotide (5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGATTAAGTTGGGTAACGCCAGGGTTTTCCCAGTCACGAC-3′). The three-stranded D-loop substrate was prepared by annealing the 32P-labeled DL4–20NT oligonucleotide (5′-TAAGAGCAAGATGTTCTATAAAAGATGTCCTAGCAAGGCAC-3′) with a 3-fold molar excess of partially complementary oligonucleotides DL1 and DL2 by incubation at 95°C for 5 min and slow cooling below 30°C. The sequence of the above oligonucleotides are as follows: DL1, 5′- GACGCTGCCGAATTCTACCAGTGCCTTGCTAGGACATCTTTGCCCACCTGCAGGTTCACCC-3′; DL2, 5′-GGGTGAACCTGCAGGTGGGCGGCTGCTCATCGTAGGTTAGTTGGTAGAATTCGGCAGCGTC-3′. The D-loop-containing DNA molecules were resolved on a non-denaturing 8% polyacrylamide-bis gel (19:1) in TBE 1× and eluted from gel slices in TE buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA). G4-containing DNA substrate (named OX-1-G2′) was prepared from the OX-1 oligonucleotide (5′-ACTGTCGTACTTGATATTTTGGGGTTTTGGGG-3′, guanine residues that compose the G4 structure are underlined), as previously described (39,41). The oligonucleotide was 5′-end-labeled with T4 polynucleotide kinase (Roche) and [γ-32P]ATP (specific activity: 6000 Ci/mmoles) and purified from the unincorporated radionuclide by passage over a Micro Bio-Spin 30 column (Bio-Rad). Then, NaCl was added at 1 M and the sample was denatured at 95°C for 5 min and then incubated at 60°C for at least 16 h in a PCR thermal cycler. The sample was resolved on a 8% polyacrylamide-bis (19:1) gel in TBE 1× containing 10 mM KCl. The band corresponding to the anti-parallel bimolecular G4 structure was cut out from the gel and the DNA eluted by incubating the crushed gel slice for 16 h at room temperature in TE buffer (50 mM Tris-HCl, pH 7.5, 1 mM EDTA) containing 10 mM KCl. The presence of the G-quartet structure in the eluted sample was verified by DNA band shift assays, which were carried out using a G4 structure-specific antibody, as described in Supplemental data (see Supplementary Figure S1).
DNA helicase assays
DNA helicase assays were carried out in reaction mixtures (20 μl) containing the indicated proteins in buffer 10 mM sodium phosphate, pH 7.5, 6 mM NaCl, 2.5 mM potassium acetate, 0.1 mM magnesium acetate (when the D-loop and G4 DNA substrates were used, the magnesium acetate was at 0.5 mM), 0.3 mM dithiotreitol, 2% glycerol, 1 mM ATP, 1 nM DNA substrate. Reactions were initiated by addition of ATP and then incubated for 30 min at 37°C. When the three-stranded D-loop was used as the substrate, the assays were carried out at 25°C to reduce DNA spontaneous melting. Reactions were quenched with the addition of 5 μl of 5× stop solution (0.5% SDS, 40 mM EDTA, 0.5 mg/ml proteinase K, 20% glycerol, 0.1% blue bromophenol). Samples were run on a 12% polyacrylamide-bis (19:1) gel in TBE containing 0.1% SDS at a constant voltage of 150 V. An 8% polyacrylamide-bis (19:1) gel was used for the DNA helicase reactions carried out with the bimolecular G-quadruplex DNA as the substrate. After the electrophoresis, the gels were soaked in 20% trichloroacetic acid and analyzed by phosphorimaging. The reaction products were quantified and any free oligonucleotide in the absence of enzyme was subtracted.
Electrophoretic mobility shift assays
DNA band shift assays were carried out using mixtures (volume: 20 μl) containing the DNA substrates (20 fmoles) and DDX11 (0.22 pmoles) either alone or in presence of the indicated amounts of Tim. The DNA binding activity of Tim alone was also assayed. Binding buffer used had the following composition: 10 mM sodium phosphate, pH 7.5, 6 mM NaCl, 2.5 mM potassium acetate, 0.1 mM magnesium acetate, 0.3 mM dithiotreitol, 2% glycerol. Samples were incubated on ice for 30 min and then subjected to electrophoresis on a 5% polyacrylamide/bis (37.5:1) gel in 1× TBE. In EMSAs, where G-quadruplex DNA was used as a ligand, electrophoresis was carried out using the same gel containing 0.5× TBE and 10 mM KCl and the gel was run in 0.5× TBE buffer. Gels were analyzed by phosphorimaging.
ATPase assay
ATPase assay reaction mixtures (10 μl) contained 10 mM sodium phosphate, pH 7.5, 6 mM NaCl, 2.5 mM potassium acetate, 0.2 mM magnesium acetate, 0.3 mM dithiotreitol, 2% glycerol, 40 nM single-stranded M13 mp18(+) DNA, 100 μM [γ-32P] ATP (0.5–1 μCi) and the indicated amounts of DDX11 and/or Tim. Samples were incubated for 1 h at 37°C. Reactions were quenched with 0.5 M EDTA. A 1-μl aliquot of each mixture was spotted onto a polyethylenimine-cellulose thin layer plate developed in 0.5 M LiCl, 1 M formic acid. The amount of ATP hydrolyzed to orthophosphate was quantified by phosphorimaging.
Surface plasmon resonance measurements
Dynamic interaction of Tim with DDX11 was analyzed by using the surface plasmon resonance biosensor system Biacore2000 (Biacore). Tim (7700 resonance units, RU) was immobilized on a CM5 sensor chip in 10 mM sodium acetate buffer, pH 4.0, according to the manufacturer's instructions. To collect the sensorgrams, increasing concentrations of human DDX11 (from 0 to 80 nM) in PBS buffer (50 mM sodium phosphate pH 7.5, 150 mM NaCl) were fluxed over the sensor chip surface at a flow rate of 20 μl/min. Recorded sensorgrams were normalized to a baseline of 0 RU and the relative dissociation constant (KD) value was calculated using the BIA Evaluation software.
Immunoprecipitation experiments
Mixtures (83 μl) containing purified Tim (0.4 μg) and/or recombinant DDX11 (1.5 μg) in binding buffer (25 mM Tris-HCl, pH 7.5, 1 mM EDTA, 150 mM NaCl, 1 mM dithiotreitol, 0.01% Nonidet-P40) were incubated for 1 h at 4°C with gentle shaking. Ten microliters of Protein A-agarose beads (Roche) were mixed with 0.6 μg of polyclonal anti-Tim antibodies (Abcam, ab72458) in 300 μl of PBS buffer for 1 h at 4°C with gentle shaking. After an extensive wash with PBS buffer to remove the unbound antibodies, the beads were re-suspended in 300 μl of binding buffer. One hundred microliters of beads were mixed with the above mixtures containing recombinant Flag-Tim and/or DDX11-Flag and incubated for an additional 2 h at 4°C. The beads were then washed three times with 600 μl of washing buffer (25 mM Tris-HCl, pH 7.5, 1 mM EDTA, 300 mM NaCl, 1 mM dithiotreitol, 0.1% Nonidet-P40) and re-suspended in 30 μl of SDS-PAGE loading buffer (50 mM Tris-HCl, pH 6.8, 10% glycerol, 200 mM β-mercaptoethanol, 0.5% SDS, 0.01% blue bromophenol). Samples were subjected to electrophoresis through 8% polyacrylamide-bis (29:1) gel. The gel was electro-blotted onto a PVDF membrane and proteins on the blot were detected with a monoclonal horseradish peroxidase-conjugated anti-Flag antibody (Abcam, ab49763) detected using the ECL+ system (GE Healthcare).
Immunoprecipitations were carried out on nuclear extracts prepared from HEK 293T cells synchronized in S-phase with a single block in thymidine. Where indicated, cells were treated with HU at 2 mM for 9 h. Cells were re-suspended in 1 ml of osmotic buffer (10 mM Hepes-NaOH, pH 7.9, 0.2 M potassium acetate, 0.34 M sucrose, 10% glycerol, 1 mM dithiotreitol, 0.1% Triton X-100 and protease inhibitors) and the sample was incubated for 5 min on ice. After centrifugation (800 × g for 5 min), the nucleus/chromatin fraction in the pellet was re-suspended in 1 ml of hypotonic buffer (10 mM Hepes-NaOH, pH 7.9, 0.25 M potassium acetate, 1 mM dithiothreitol, 0.1% Triton X-100 and protease inhibitors) containing 10 mM CaCl2 and microccocal nuclease (0.2 units/μl; SIGMA, cat. N3755) for 20 min at 37°C. Reaction was stopped by adding EGTA (2 mM final concentration) to the sample. Insoluble material was removed by centrifugation at 16 000 × g for 30 min. This fraction was used in immunoprecipitation experiments with 4 μg of anti-DDX11 mouse polyclonal antibodies (Abcam ab66971) and control mouse IgG bound to Protein A agarose. Samples were incubated for 2 h at 4°C with gentle shaking. Beads were washed three times with the following buffer: 10 mM Hepes-NaOH pH 7.9, 0.25 M potassium acetate, 1 mM dithiotreitol, 0.1% Triton X-100, 0.25% Nonidet-P40 and protease inhibitors. Samples were analyzed by denaturing gel electrophoresis and western blot as previously described.
Down-regulation of Tim and DDX11 in HeLa cells by RNA interference and DNA fiber track analysis
The siRNAs (Dharmacon) used for Tim and DDX11 depletion experiments in HeLa cells had the following sequence: 5′-CCUGUGUCUGUCUUCUUCCUGCGAA-3′, Tim #1; 5′-CCUCACCAACUACUAUGAG-3′, Tim #2; and 5′-GAGCUAAGAAGCCUAGGGG-3′, DDX11 #2; 5′-GAUAUUCCAGGAACCUAAG-3′, DDX11 #5. The protein levels of Tim and DDX11 were analyzed by Western blot 48 h after siRNA transfection. Briefly, 1 × 106 cells of each sample were re-suspended in SDS lysis buffer (65.8 mM Tris-HCl, pH 6.8, 2.1% SDS, 26.3% (w/v) glycerol, 0.01% bromophenol blue containing 715 μM β-mercapto-ethanol). Cell extracts were cleared after 16 000 × g centrifugation in table top centrifuge for 10 min. Half of the samples were resolved in 4–12% NuPage Bis-Tris gel (ThermoFisher Scientific). Blot was developed with 1:250 dilution of either rabbit anti-DDX11 antibody (Santa Cruz, sc-68855) or mouse anti-Tim antibody (Santa Cruz, sc-393122).
Forty eight hours after siRNA transfection, DNA fiber spreads in undamaged cells were prepared with a modified protocol as described before (11). Briefly, cells were first pulse-chased with CldU at 50 μM for 20 min and then with IdU at 100 μM for the indicated times. Cells were treated with trypsin and re-suspended in ice-cold PBS at a concentration of 0.5 × 106 cells/ml. A sample of cells (volume: 2 μl) was mixed with lysis buffer (10 μl; 200 mM Tris-HCl, pH 7.4, 50 mM EDTA, 0.5% SDS) on a glass slide. Slides were tilted ∼15° to allow cell extract to move down the slide. The DNA spreads were air-dried and fixed in a mixture of methanol/acetic acid (3:1, v:v). The slides were incubated in a solution of HCl (2.5 M) for 60 min, neutralized in a buffer (400 mM Tris-HCl, pH 7.4) for 10 min, washed in PBS and immuno-stained, as described before (42). Staining of the slides with antibodies specific to CldU and IdU was done sequentially. The antibodies and dilutions used for staining were: rat anti-BrdU (CldU), 1:200; Daylight 647 goat anti-rat, 1:100; mouse anti-BrdU (IdU), 1:40 and Daylight 488 goat anti-mouse, 1:100. Imaging of the slides was carried out using a Zeiss Axiovert 200 M microscope with the Axio Vision software packages (Zeiss).
For replication fork analysis in the presence of HU, HeLa cells depleted of DDX11 and/or Tim were CldU-labeled as described above, subsequently incubated with HU during the IdU-labeling period, and DNA fiber spreads were processed.
To measure the frequency of replication fork restart events, cells were transfected with the indicated siRNAs and, 48 h after transfection, were pulse-chased with CldU (red label) at 50 μM for 30 min; then, they were treated (or not) with HU (at 2 mM) for 14 h. Thereafter, cells were washed to remove HU from the medium and pulsed with IdU (green label) at 100 μM for 60 min. Restarting fork events were calculated by dividing the number of red-green tracts (corresponding to restarted forks) by the total number of red-only plus red-green tracts (corresponding to stalled and restarted forks, respectively).
For each condition, 150–250 individual fibers were analyzed in all DNA fiber track assays and a statistical analysis of data was carried out using Student's t-test.
RESULTS
Tim specifically stimulates DDX11 DNA unwinding activity on a forked duplex DNA substrate
The fork-protection complex and DDX11 have been proposed to be involved in the same cellular pathways that preserve chromosomal integrity and genome homeostasis. As a matter of fact, depletion of Tim in mammalian cells causes cohesion defects that can be partially rescued by overexpressing DDX11 (19,22,23). To investigate if a functional interaction exists between purified recombinant human DDX11 and Tim (or Tipin) we assessed the effect of these checkpoint mediators on DDX11 helicase activity. First, we measured the ability of DDX11 to unwind a forked 39-bp duplex DNA in the absence/presence of purified recombinant Tim. DDX11 is expected to efficiently unwind this kind of DNA substrate, as previously reported (37–39). We found that Tim could stimulate the DDX11 DNA unwinding activity on the forked duplex DNA substrate in a protein concentration dependent manner, as shown in Figure 1A. A quantitative analysis revealed that DDX11 displacement of the radio-labeled DNA strand was increased by approximately 10-fold in the presence of Tim (Figure 1B). No detectable stimulation of DDX11 helicase activity by purified recombinant Tipin protein was observed (F. Calì et al., data not shown). To address the specificity of the stimulatory effect of Tim on DDX11, we analyzed whether Tim could enhance the DNA unwinding activity of FANCJ, another Fe-S cluster SF2 DNA helicase having the same 5′-3′ polarity of DDX11 (43). Tim did not exert any effect on FANCJ helicase activity on the same forked duplex DNA substrate that was used for the DDX11 assays (see Figure 1C).
Figure 1.
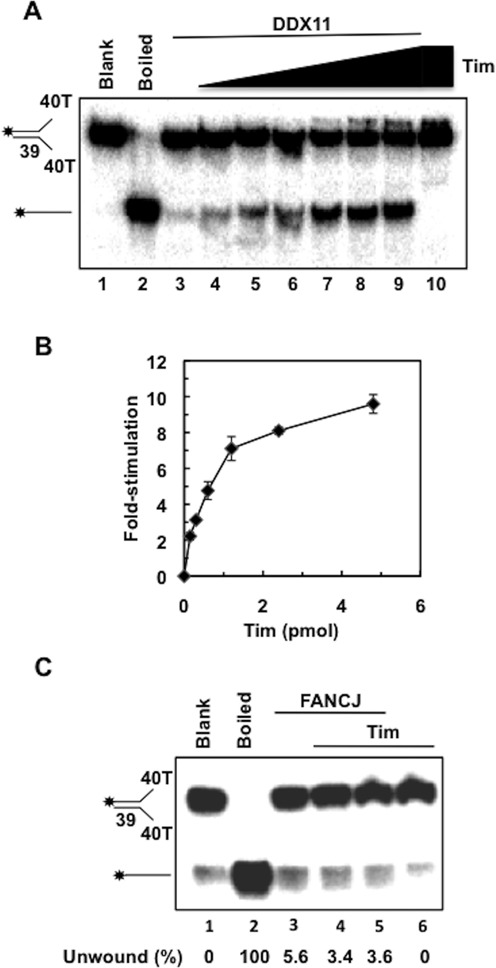
Effect of human Tim on DDX11 DNA helicase activity on a forked duplex DNA. (A) DNA helicase reactions (20 μl) were performed by incubating a fixed amount of DDX11 (27 fmol) with 20 fmol of radio-labeled DNA substrate for 30 min at 37°C (lane 3), as described under Materials and Methods. DDX11 helicase activity was measured in the presence of increasing amounts of recombinant Tim (0.15, 0.3, 0.6, 1.2, 2.4 and 4.8 pmol; lanes 4–9). A control assay was carried out that contained only Tim (4.8 pmol) in the absence of DDX11 (lane 10). A sample without protein (Blank) and heat-denatured DNA substrate control (Boiled) were loaded in lanes 1 and 2, respectively. Reaction products were subjected to electrophoresis on a polyacrylamide gel in TBE containing 0.1% SDS. Asterisk denotes 5′-32P end label. (B) Quantitative analyses of the DNA helicase assays were carried out by phosphorimaging. The amount of radio-labeled oligonucleotide displaced by DDX11 was determined for each lane and normalized to the value calculated for the reaction carried out by DDX11 alone to obtain the DNA helicase activity stimulation folds. Averaged values with error bars from three independent experiments were plotted versus the amounts of Tim used in each assay. (C) FANCJ DNA helicase activity is not stimulated by Tim. DNA helicase reactions (20 μl) were carried out by incubating a fixed amount of human FANCJ (26 fmol) with radio-labeled forked duplex substrate (20 fmol) for 15 min at 30°C, alone (lane 3) or in the presence of human Tim (2 and 4 pmol; lanes 4–5). A control assay was carried out that contained only Tim (4 pmol) in the absence of FANCJ (lane 6). All the reactions, including the sample without protein (Blank, lane 1) and the one containing heat-denatured substrate (Boiled, lane 2) were loaded on an 8% polyacrylamide/bis (19:1) gel in 0.5× TBE. Quantitative analysis of the DNA helicase assays was carried out by phosphorimaging. The amount of radiolabeled oligonucleotide displaced by FANCJ was determined for each lane and the unwound substrate present in the blank sample was subtracted to each value.
In order to demonstrate that Tim truly stimulated a DDX11 ATPase-dependent motor function, we carried out various control assays using the forked duplex DNA substrate. First we observed no stimulation of the DDX11 DNA helicase activity by Tim when ATP was omitted from the reaction mixture (see Figure 2A) or substituted with the poorly hydrolyzable analog ATP-γ-S (see Figure 2B) in the reaction mixtures. Moreover, no stimulatory effect of Tim was observed when we used an ATPase/helicase-dead mutant of DDX11, in which the Walker A invariant lysine residue was replaced with arginine (DDX11 K50R; 38; see Figure 2C). To rule out the possibility that our purified samples of Tim were contaminated by a non-specific helix-destabilizing activity, control assays were performed in which the maximal amount of Tim was tested in the absence of DDX11 (i.e. see lane 10 of the gel in Figure 1A and lanes 7–8 and 5 of the gels in Figure 2A–C).
Figure 2.
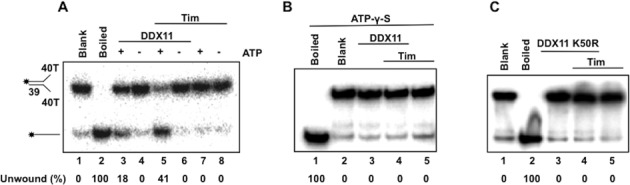
Tim stimulates an ATPase-dependent motor function of DDX11. (A) and (B) DNA helicase reactions (20 μl) were carried out by incubating a fixed amount of DDX11 with 20 fmol of radio-labeled forked duplex substrate for 30 min at 37°C. (A) The activity of DDX11 (0.15 pmol) was assayed with ATP (lanes 3 and 5) and without ATP (lanes 4 and 6) in the absence (lanes 3–4) or presence (lanes 5–6) of Tim (1.1 pmol). Control assays were carried out containing only Tim (1.1 pmol) with and without ATP (lanes 7–8). (B) The activity of DDX11 (0.15 pmol) was assayed using ATP-γ-S instead of ATP in the absence of Tim (lane 3) or in the presence of Tim (1.1 pmol, lane 4). Control assays were also carried out containing only Tim without DDX11 (lane 5). (C) The DDX11 ATPase-dead mutant (K50R DDX11; 0.15 pmol) was assayed alone (lane 3) or in the presence of Tim (1.1 pmol; lane 4). A control assay was performed containing only Tim (1.1 pmol; lane 5). A sample without protein (Blank) and a heat-denatured DNA substrate control (Boiled) were loaded in lanes 1 and 2 of each gel. Quantitative analysis of the DNA helicase assays was carried out by phosphorimaging. The amount of radio-labeled oligonucleotide displaced by DDX11 was determined for each lane and the unwound substrate present in the blank sample was subtracted from each value.
Tim enhances the DDX11 ability to unwind G-quadruplex and D-loop DNA substrates
Next we examined whether Tim had any effect on the ability of DDX11 to resolve G4 DNA structures. DDX11 was reported to unwind G4-containing DNA molecules with a strong preference for a two-stranded anti-parallel DNA substrate (G2’), whereas it was only marginally active on a four-stranded parallel G4 and not active at all on an uni-molecular G4 DNA substrate (39,41). The anti-parallel bimolecular G-quartet structure can occur when hairpin dimers of two anti-parallel strands form Hoogsteen hydrogen bonds between guanine residues (44–46). We tested the activity of DDX11 on a bimolecular anti-parallel G4 DNA in the presence of increasing amounts of Tim. As shown in Figure 3A and B, Tim was able to stimulate the ability of DDX11 to resolve this G-quadruplex structure approximately 4-fold.
Figure 3.
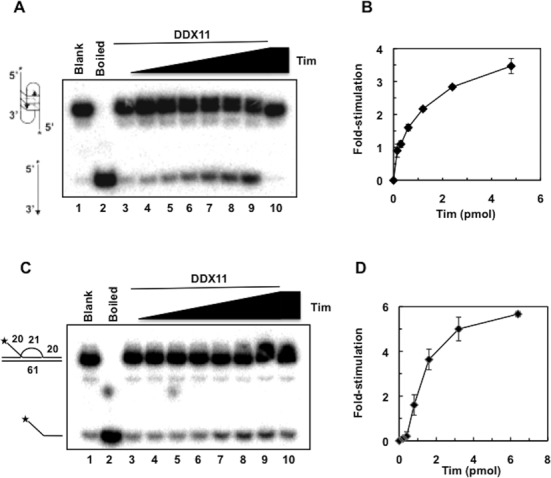
Tim enhances the ability of DDX11 to resolve anti-parallel bimolecular G4 structures and three-stranded D-loop DNA substrates. (A) DNA helicase reactions (20 μl) were carried out by incubating a fixed amount of DDX11 (45 fmol) with radio-labeled G-quadruplex-containing DNA substrate for 30 min at 37°C (lane 3), as described under Materials and Methods. DDX11 helicase activity was measured in the presence of increasing amounts of recombinant Tim (0.15, 0.3, 0.6, 1.2, 2.4, 4.8 pmol; lanes 4–9). A control assay was carried out that contained only Tim (4.8 pmol) in the absence of DDX11 (lane 10). C) DNA helicase reactions (20 μl) were carried out by incubating a fixed amount of DDX11 (28 fmol) with the D-loop DNA substrate that contained an invading strand with a 20-nt 5′ single-strand DNA tail for 30 min at 25°C (lane 3), as described under Materials and Methods. DDX11 helicase activity was measured in the presence of increasing amounts of recombinant Tim (0.2, 0.4, 0.8, 1.6, 3.2, 6.4 pmol; lanes 4–9). A control assay was carried out that contained only Tim (6.4 pmol) in the absence of DDX11 (lane 10). Reaction products were subjected to electrophoresis on polyacrylamide gels in TBE containing 0.1% SDS (+ 10 mM KCl in the gel shown in (A). Samples without protein (Blank) and heat-denatured DNA substrate controls (Boiled) were loaded in lanes 1 and 2, respectively, of the gels in (A) and (C). Asterisk denotes 5′-32P end label. (B) and (D) Quantitative analyses of the DNA helicase assays were carried out by phosphorimaging. The amount of radiolabeled oligonucleotide displaced by DDX11 was determined for each lane and normalized to the value calculated for the reaction carried out by DDX11 alone to obtain the DNA helicase activity stimulation folds. Averaged values with error bars from three independent experiments were plotted versus the amounts of Tim used in each assay.
We next assessed if Tim had any effect on the ability of DDX11 to unwind a three-stranded D-loop DNA substrate that contained an invading strand with a 20-nt 5′ single-strand DNA tail, as depicted in Figure 3C. This structure can be considered a model for an early intermediate of homologous recombination that is formed when a DNA strand from one chromatid invades the sister chromatid DNA duplex to anneal with its complementary strand. As shown in Figure 3C and D, the ability of human DDX11 to displace the third invading strand was enhanced more than 5-fold in the presence of Tim. No stimulatory effect by Tipin was detected on either anti-parallel bimolecular G4 or three-stranded D-loop-containing DNA substrates (F. Calì et al., data not shown).
Taken together, these data suggest that Tim, but not Tipin, enhances the ATPase-dependent helicase activity of DDX11 in a dose-dependent and specific manner. The stimulatory effect of Tim on DDX11 helicase activity is observed on structurally diverse DNA substrates that represent key intermediates of various processes implicated in genome maintenance.
Tim enhances DDX11 DNA binding and ATPase activity
The effect of Tim on the DNA binding ability of DDX11 was analyzed. We performed band-shift assays of purified recombinant DDX11 in the absence and presence of Tim using radiolabeled forked duplex or anti-parallel bimolecular G4 or three-stranded D-loop DNA structures (see Figure 4A–C). In these EMSAs a fixed amount of DDX11 was used alone or in the presence of two amounts of Tim. We found that DDX11 alone formed a single DNA–protein complex; whereas, Tim alone was almost completely unable to shift the radiolabeled probes due to its low DNA binding affinity (see lane 2 and 5 of each gel in Figure 4A–C), as previously observed (8). When the DNA binding activity of DDX11 was assayed in the presence of Tim, we found that the amount of shifted DDX11:DNA binary complex was substantially increased for all the DNA molecules used in the mobility shift assays (see lane 3 and 4 of each gel in Figure 4A–C). This result suggests that Tim facilitates DDX11 binding to these DNA molecules, although a Tim:DDX11:DNA ternary complex does not appear to be present in our gel-shift assays. This phenomenon, which was observed for other protein:DNA interactions (47) is likely due to the fact that association of Tim to the DDX11:DNA complex is not stable enough to be preserved during the electrophoretic run. Besides, visual inspection and quantitative analysis of the EMSAs data showed that a greater percentage of the forked duplex DNA substrate was bound by DDX11 compared to the G-quadruplex or D-loop DNA substrates. Moreover, a greater percentage of DDX11:DNA complex in the presence of Tim was observed for the forked duplex compared to the G4 or D-loop substrates. This finding is consistent with the higher level of stimulation by Tim of the DDX11 helicase activity on forked duplex DNA molecules in comparison with G-quadruplex or D-loop substrates.
Figure 4.
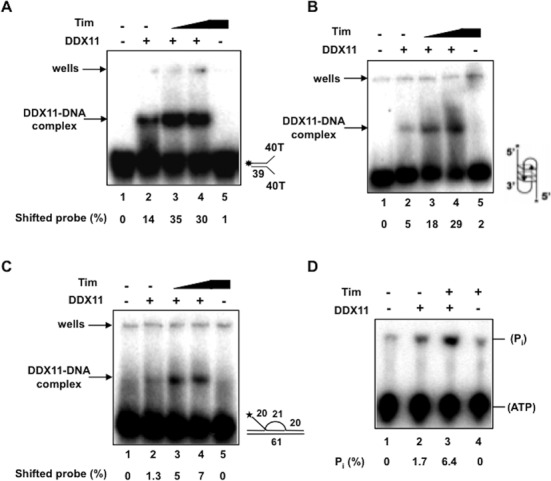
Tim stimulates DDX11 DNA binding and ATPase activity. EMSAs were carried out using a forked duplex (A) or anti-parallel bimolecular G4 (B) or three-stranded D-loop (C) DNA structures and DDX11 (0.22 pmol) either alone (lane 2) or in presence of Tim (2 and 4 pmol of protein in lanes 3 and 4, respectively), as described in Materials and Methods. The DNA binding activity of Tim alone was also assayed (4 pmol of protein in lane 5). A blank mixture with no protein was loaded in lane 1. Samples were run on a 5% polyacrylamide/bisacrylamide (37.5:1) gel in 1× TBE. Radioactive signals were detected by phosphorimaging. The amount of shifted probe was determined for each lane and the shifted DNA present in the blank sample was subtracted to each value. (D) ATP hydrolysis catalyzed by DDX11 was measured using [γ-32P] ATP in the presence of single-stranded M13 mp18(+) DNA and analyzed by thin layer chromatography, as described under Materials and Methods. A fixed amount of DDX11 (0.9 pmol) was assayed alone (lane 2) or in presence of human Tim (3.4 pmol; lane 3). A control assay was carried out which contained only Tim (3.4 pmol) in the absence of DDX11 (lane 4). The amount of spontaneously hydrolyzed ATP was determined using blank reactions without protein (lane 1) and subtracted.
We tested the effect of Tim on DDX11 ATPase activity. The ATP hydrolysis catalyzed by DDX11 was reported to be strictly dependent on DNA and a maximal stimulation was observed in the presence of single-stranded M13 DNA (37). As shown in Figure 4D, DDX11 ATPase activity was enhanced approximately 4-fold in the presence of Tim, consistent with the observed stimulation of its DNA binding activity.
Tim directly interacts with DDX11
The finding that Tim stimulates DDX11 DNA helicase activity prompted us to investigate whether a direct physical interaction could be detected between these proteins by using surface plasmon resonance measurements. For these analyses, purified recombinant DDX11 was fluxed over a sensor-chip where recombinant Tim was immobilized. Figure 5 shows the overlaid sensorgrams obtained by testing five increasing concentrations of DDX11 (from 0 to 80 nM) over a Tim-immobilized sensor-chip (Panel A). These results indicated that DDX11 and Tim physically interact with a calculated binding affinity, KD, of 4.32 × 10−9 M. A similar analysis performed with a Tipin-immobilized sensor-chip revealed that DDX11 directly interacts also with Tipin, but the calculated binding affinity is markedly lower than the one measured for Tim (KD = 6.55 × 10−7 M; see Supplementary Figure S2).
Figure 5.
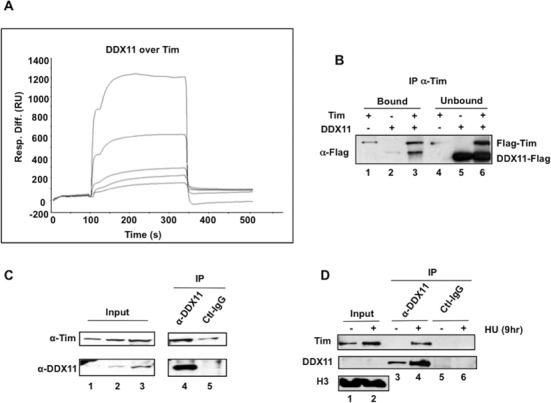
Interaction of DDX11 with Tim. (A) Surface plasmon resonance measurements were carried out to analyze the interaction of DDX11 with Tim using a Biacore2000 instrument. Purified recombinant DDX11 was fluxed at increasing concentrations (from 0 to 80 nM, lower to upper curves) over a Tim-immobilized sensor-chip. Overlaid plots of sensorgrams were obtained corresponding to the recorded resonance units (RU) versus time (s). (B) Immuno-precipitation experiments were carried out using Protein A-agarose beads conjugated with anti-Tim antibodies and mixtures of purified recombinant Flag-Tim and DDX11-Flag, as described in Materials and Methods. The samples were subjected to SDS-PAGE and western blot analysis with anti-Flag antibodies to detect recombinant Flag-Tim and DDX11-Flag, as indicated. The bound (100% of the total sample, lanes 1–3) and unbound proteins (15% of the total sample, lanes 4–6) were analyzed. (C) and (D) Immuno-precipitations (IP) were carried out on nuclear extracts (protein: 1.5 mg in Panel C and 0.5 mg in Panel D) prepared from HEK 293T cells as described in Materials and Methods. Where indicated in Panel D, cells were treated with HU at 2 mM for 9 h. Immuno-precipitated samples with anti-DDX11 antibody and control IgG (IP, lanes 4 and 5 in Panel C and lanes 3–6 in Panel D) and input nuclear extract (samples of 20, 40 and 80 μg of total protein loaded in lanes 1, 2 and 3, respectively, in Panel C; samples of 10 μg of total protein loaded in lanes 1 and 2 in Panel D) were analyzed by western blot with the indicated primary antibody. Peroxidase-conjugated secondary antibodies were used and chemiluminescent signals detected using the ECL+ kit (GE Healthcare).
The direct interaction between Tim and DDX11 was tested also by co-immuno-precipitation of the recombinant proteins (Figure 5B). In these experiments, we used Protein A-agarose beads conjugated with anti-Tim antibodies that were incubated with mixtures containing recombinant DDX11 and Tim proteins. The results of this analysis confirmed that the two proteins interact each other. On the other hand, we were unable to co-immuno-precipitate DDX11 and Tipin from mixtures of the two purified recombinant proteins using either anti-DDX11 or anti-Tipin antibodies in similar pull-down experiments (Calì et al., data not shown). This might depend on the weaker interaction between DDX11 and Tipin or on the intrinsic properties of the antibodies used for the pull-down assays.
We also analyzed whether Tim and DDX11 are associated in cells, as suggested by our in vitro analysis using purified recombinant proteins. Co-immuno-precipitation experiments, carried out on nuclear extracts of HEK 293T cells using anti-DDX11 polyclonal antibodies bound to Protein A agarose beads, revealed that endogenous Tim interacts with DDX11 (see Figure 5C). When cells were treated with HU to induce replication stress, a substantially greater amount of Tim was co-pulled down with DDX11 from the chromatin fraction using anti-DDX11-specific antibodies (see Figure 5D). This finding reveals that in replication stressful conditions recruitment of Tim and DDX11 onto chromatin is markedly enhanced.
Effect of Tim and/or DDX11 depletion in HeLa cells on DNA replication dynamics
We next analyzed the effect of depleting Tim and/or DDX11 in HeLa cells on the replication fork progression by DNA fiber track assays. Cells were transfected with distinct sets of siRNAs, which were already reported to efficiently downregulate expression of Tim (9) or DDX11 (36), in order to rule out possible off-target effects. When the labeling protocol depicted in Figure 6A was used, transfected cells were pulse-labeled with the thymidine analog CldU (red label) for 20 min, followed by labeling with a second thymidine analog, IdU, (green label) for additional 20 min. To evaluate the efficacy of gene silencing Tim and DDX11 protein level was analyzed by western blot of whole cell extracts. As reported in Figure 6B, HeLa cells depleted of DDX11 showed a reduction in Tim protein level; similarly, HeLa cells depleted of Tim showed a reduction in DDX11 protein level. Similar results were also obtained when Tim and/or DDX11 depletion was carried out by an additional couple of siRNAs (see Supplementary Figure S5). IdU-tract length (or CldU-tract length) was then measured in DNA fibers containing either red or green label (as shown in Figure 6C and D and in Supplementary Figure S3). These analyses revealed that a deficiency in Tim reduced fork tract length 2-fold, whereas loss of DDX11 had only a modest effect on fork progression. Cells that were depleted of both Tim and DDX11 displayed a shortened tract length that was comparable to Tim depletion alone. Based on these results, we conclude that the marked reduction in replication as a consequence of Tim depletion is not further exasperated by co-depletion of DDX11, suggesting that the residual Tim protein in DDX11-depleted cells is enough to maintain near normal replication. We then tested the effect of HU treatment on advancement rate of replication forks that were active at the time of drug administration (length of green tract in fibers labeled with both thymidine analogs, CldU and IdU) in Tim- and/or DDX11-depleted cells. The results revealed that replication forks progression was impaired with respect to control cells, when either Tim or DDX11 were downregulated; whereas no additive effect was observed by co-depletion of both Tim and DDX11 (see Figure 6E and F). This finding suggests that Tim and DDX11 operate in concert in the same cellular pathway required for efficient progression of DNA replication forks in perturbed S-phase conditions.
Figure 6.
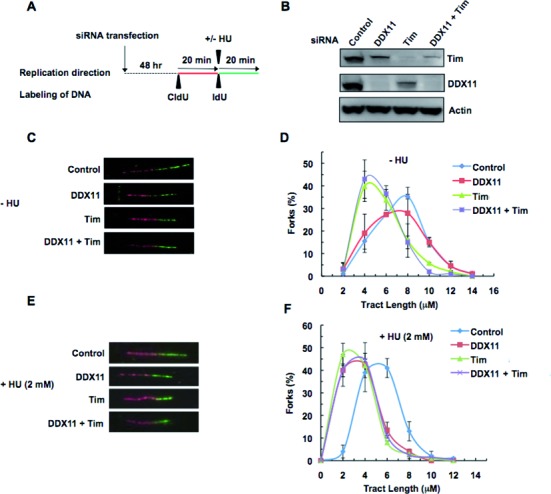
DNA fiber track assays on Tim- and/or DDX11-depleted HeLa cells. (A) Schematic representation of the protocol used to track DNA fiber replication. Labeling of cells were done 48 h after siRNA transfection (Tim #1 and/or DDX11 #2). First, cells were pulse-chased with CldU (red label); then, they were labeled with IdU (green label) for the indicated times. (B) Western blot analysis of HeLa cell extracts to show the knock-down of DDX11 and/or Tim as compared to control siRNAs. (C) and (E) Representative of fluorescently labeled DNA fibers from cells treated with the indicated siRNAs without and with HU treatment, respectively. (D) and (F) Distribution of replication fork tract lengths of cells transfected with the indicated siRNAs, as measured by IdU tract length in a DNA fiber labeled with CldU and IdU, without and with HU treatment with HU, respectively. The IdU tract length represents mean of at least three experiments with SD and analysis is based on 150–250 DNA fibers from each experiment. IdU mean track length for the cells treated with the indicated siRNA in the absence of HU are 6.4 ± 0.13 μM (control), 5.5 ± 0.79 μM (DDX11), 4.3 ± 0.85 μM (Tim), 4.4 ± 0.65 μM (Tim + DDX11); whereas, in the presence of HU, the mean track lengths are 4.4 ± 0.38 μM (control), 2.4 ± 0.11 μM (DDX11), 2.4 ± 0.25 μM (Tim), 2.6 ± 0.11 μM (Tim + DDX11).
Our analyses also revealed that Tim downregulation increased newly fired origins and reduced termination events either in normal or in stressful conditions; whereas DDX11 depletion caused similar, although smaller, effects only upon HU exposure of cells (see Supplementary Figure S4). These results appear to be consistent with the finding that Tim and/or DDX11 depletion causes a slight reduction in Chk1 phosphorylation (less than 30% with respect to control cells, as shown in Supplementary Figure S5) in response to our mild genotoxic treatment protocol (addition of HU at 2 mM into the cell culture medium for 20 min).
Moreover, we analyzed the effect of longer IdU-labeling time (40 min) and we found that in unperturbed conditions Tim downregulation still caused shorter IdU-tract relative to control siRNAs treated cells; whereas, following HU exposure, either Tim- or DDX11-depleted cells still showed IdU-tract length reduction (see Supplementary Figure S6).
In a subsequent set of experiments, we monitored the ability of replication forks to restart synthesis after HU treatment. Cells, depleted of Tim and/or DDX11, were pulse-labeled with CldU for 30 min and then treated with HU for 14 h before a second labeling pulse with IdU for 60 min. In this setting, forks arrested by HU administration incorporated only the first nucleotide analog, whereas those able to restart synthesis were labeled with both analogs (see Figure 7A and B). Loss of either Tim or DDX11 impaired the ability of cells to rescue HU-challenged replication forks. Co-depletion of Tim and DDX11 did not result in an additive effect on reduction of restarted DNA replication forks upon HU treatment suggesting an epistatic relationship between the two proteins in the forks rescue process in stressful conditions (see Figure 7C). Analysis of IdU tract length in DNA fibers labeled with CldU and IdU revealed that Tim and DDX11 are both required for fork progression following rescue from HU treatment (see Supplementary Figure S7).
Figure 7.
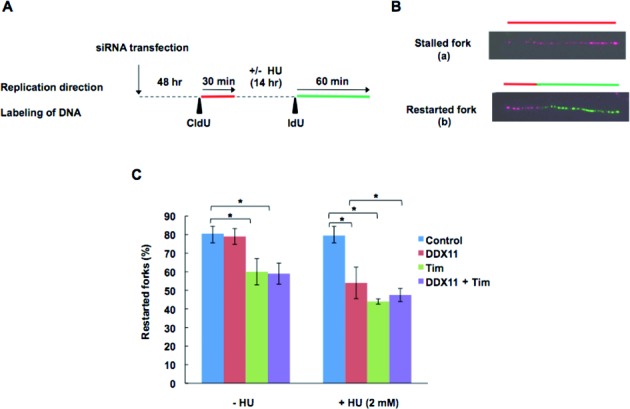
Replication fork restart in Tim- and/or DDX11-depleted cells following HU treatment. (A) Schematic representation of the protocol used to analyze replication restart events. Forty eight hours after transfection of siRNAs (Tim #2 and/or DXX11 #5), HeLa cells were labeled for 30 min with CldU (red label); then they were treated with (or without) HU at 2 mM for 14 h and then labeled for 60 min with IdU (green label). (B) Representative of fluorescently labeled DNA fibers: fibers containing only a red tract correspond to stalled forks (a); those containing a red-green contiguous tract represent restarting forks (b), as indicated. (C) Percentage of restarted forks were calculated by dividing the number of restarting forks (i.e., b forks) by the total number of forks (i.e., a + b forks). Analyses were performed on 150–250 individual DNA fibers. Data are presented as mean of at least three experiments with SD. *P <0.05 (Student's t-test).
DISCUSSION
In this study, we report evidence of a functional interaction between DDX11, a human SF2 Fe-S cluster DNA helicase, and Tim, a component of the replication fork protection complex. We determined that Tim enhances the DNA unwinding activity of DDX11 in vitro on substrates that mimic key intermediates of DNA replication/repair/recombination reactions, including forked duplex, anti-parallel bimolecular G-quadruplex and three-stranded D-loop DNA molecules. Moreover, we found that Tim directly interacts with DDX11 as demonstrated by Biacore measurements and co-immuno-precipitation experiments performed on the purified recombinant proteins. Interaction between endogenous Tim and DDX11 was also observed in the chromatin fraction of cell extracts by co-immuno-precipitation analysis. This interaction was noticeably increased following prolonged HU-treatment of cell cultures suggesting that two proteins are co-recruited on chromatin upon replication stress induction. Furthermore, stability of Tim and DDX11 proteins in human cells is mutually interdependent since depletion of either one will lead to a significant reduction of its interacting partner as demonstrated by western blot analyses of whole cell extracts, a result consistent with a direct interaction between the two proteins in a cellular context. These findings are in accordance with previous analyses performed in HEK 293T cells by the Noguchi's group that reported disappearance of DDX11 from cell extracts as a consequence of RNAi-dependent downregulation of Tim (22). Overall, our findings are consistent with the phenotypic similarities between cells lacking either Tim or DDX11 suggesting that both proteins are involved in the same cellular pathways to preserve chromosomal integrity and genome homeostasis.
Our analysis has revealed that human Tim (but not Tipin) is able to enhance the DNA helicase activity of DDX11 on structurally diverse DNA substrates. The observed stimulation appears to derive from a direct interaction of Tim with DDX11, as suggested by the fact that the two proteins establish a tight physical association either in vitro or in cells. We believe that it is unlikely Tim stimulates DDX11 in a nonspecific manner, simply by stabilizing melted single-stranded DNA. In fact, we previously reported that the DNA-binding affinity of Tim is very low (8). Moreover, Tim stimulated DNA unwinding activity catalyzed by DDX11, but not the sequence-related FANCJ DNA helicase (Figure 1C; 28,29). The high Tim:DDX11 molar ratio required to detect the stimulatory effect on the DNA helicase activity might depend on the fact that Tim forms various oligomeric assemblies in solution when it is produced in isolated form, as we observed by a glycerol gradient ultracentrifugation analysis of the recombinant human protein (Calì et al., data not shown). This result is consistent with a biochemical study of the mouse Tim ortholog (48). Nonetheless, in our experiments Tim concentration values were calculated assuming that it has a monomeric structure. Besides, we cannot rule out that the high Tim:DDX11 molar ratio required to detect the DNA helicase stimulation might depend on any cell cycle dependent post-translational modification, which would be retained only by a fraction of the purified recombinant proteins used in our in vitro enzymatic assays. We found that Tim enhances the capability of DDX11 to bind various DNA substrates (forked duplex, anti-parallel bimolecular G-quadruplex and three-stranded D-loop DNA molecules) and also its DNA-dependent ATP-hydrolysis activity (see Figure 4). Based on these findings, we postulate that the observed stimulation of DDX11 unwinding activity by Tim derives from an improved ability of the DNA helicase to bind and utilize the nucleic acid substrate.
Our data are fully consistent with the proposal that human Tim and DDX11 participate in the same functional network that operates at the crossroad between genome maintenance and sister-chromatid cohesion (22–23). We speculate that Tim acts in concert with DDX11 at the replication fork and enhances its ability to unwind difficult to replicate DNA structures, such as hairpins present in the 5′-flaps of the Okazaki fragments and/or other DNA secondary structures (including bimolecular anti-parallel G-quartets) that potentially form between the two duplicated DNA strands and could interfere with smooth interaction of the replisome with the cohesin ring. Further studies are required to determine the precise molecular mechanism by which the interaction of DDX11 and Tim with the cohesin ring and/or other components of the replication machinery promotes sister-chromatid cohesion and genomic stability. In this context it is worth mentioning that Caenorhabditis elegans Chl1, the ortholog of mammalian DDX11, was implicated in chromosomal stability, G-quadruplex DNA metabolism and repair of unresolved structures during DNA replication (49).
The stimulation of DDX11 helicase activity on the D-loop DNA substrate with an invading 3′-strand may be relevant to recombinational DNA repair transactions that occur when sister chromatids are closely paired to one another (27). In this context, DDX11 was reported to be required for efficient sister-chromatid exchange in mammalian cells (36). The ability of Tim to stimulate DDX11 to resolve D-loop-containing DNA molecules might be important for replication or repair of the chromosome telomeric regions, if we consider that the above substrate is structurally similar to a T-loop. A direct role of DDX11 in telomere metabolism can be inferred from the finding that DDX11 downregulation caused a significant reduction of chromosomal length in human melanoma cells compared to control experiments (50). Similarly, in human cells Tim depletion was found to cause a dramatic delay of telomere replication, as well as increased level of DNA damage leading to telomere aberrations (51). Therefore, it is plausible that Tim and DDX11 may cooperate to metabolize DNA structures that can be recognized and bound by the telomeric capping proteins or favor DNA synthesis through telomeric ends by the replication machinery.
Tim and Tipin are components of the fork-protection complex that is known to have a role in allowing the smooth passage of the replication fork at difficult-to-replicate sequences. Presumably, an important aspect of Tim and Tipin in this capacity is to coordinate the activities of various proteins of the replication machinery that bind and/or remodel DNA structures to maintain genomic stability. DNA fiber analysis confirmed that Tim depletion results in shorter DNA tracks of nascent DNA synthesis; however, DDX11 depletion alone or in co-depletion with Tim did not lead to further reduction in tract length, suggesting that Tim is the primary determinant of how a cell deals with endogenous impediments to replication and this function of Tim is independent of the binary complex it forms with DDX11. On the other hand, efficient rescue of replicons in stressful conditions requires the concerted action of both proteins, because loss of either one substantially reduced fork rate displacement after HU treatment. This finding is consistent with previous reports suggesting a role for DDX11 in replication recovery from DNA damage (35). Interestingly, it has been recently demonstrated that in fission yeast during meiosis the Tim–Tipin complex recruits the Dbf4-dependent kinase (DDK) to the replication fork and the fork-associated DDK phosphorylates the meiosis-specific factors Mer2, which in turns recruits other proteins responsible for DNA double-strand break formation (52,53). This Tim-Tipin-mediated mechanism that allows a temperospatial coordination of meiotic DNA replication with recombination could be paradigmatic for how other chromosomal processes can be locally and timely regulated and connected with the replication fork passage. Future studies will address the importance of Tim-DDX11 interaction for supporting DNA damage repair and/or S-phase stress checkpoints and the role of the fork-protection complex in coordinating all these chromosomal transactions with smooth replisome progression.
Supplementary Material
Acknowledgments
Shankar Balasubramanian (University of Cambridge, Cambridge, United Kingdom) is acknowledged for sending the anti-G4 DNA antibody. Daniela Corda and Alberto Luini (IBP-CNR, Naples, Italy) are acknowledged for continuous help and encouragement.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
PON projects from Italian ‘Ministero dell'Istruzione, Università e Ricerca’ (MIUR) [PON01_00862; PON01_00117 and PON01_01585 to F. M. P.]; Project ‘FaReBio di Qualità’ from Italian ‘Ministero della Economia’ [PhD grant to F. C.]; ‘Progetto CREME’ from Regione Campania [to R. D. P.]; Intramural Research Program of the National Institutes of Health, National Institute on Aging [to R. B. in part]. Funding for open access charge: The publication fee will be charged on grant from Italian MIUR.
Conflict of interest statement. None declared.
REFERENCES
- 1.Branzei D., Foiani M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell. Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 2.Branzei D., Foiani M. The checkpoint response to replication stress. DNA repair. 2009;8:1036–1046. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Katou Y., Kanoh Y., Bando M., Noguchi H., Tanaka H., Ashikari T., Sugimoto K., Shirahige K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–1083. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 4.Calzada A., Hodgson B., Kanemaki M., Bueno A., Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–1919. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodgson B., Calzada A., Labib K. Mrc1 and Tof1 regulate DNA replication forks in different ways during normal S phase. Mol. Biol. Cell. 2007;18:3894–3902. doi: 10.1091/mbc.E07-05-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aze A., Zhou J.C., Costa A., Costanzo V. DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma. 2013;5:401–413. doi: 10.1007/s00412-013-0411-3. [DOI] [PubMed] [Google Scholar]
- 7.Cho W.H., Kang Y.H., An Y.Y., Tappin I., Hurwitz J., Lee J.K. Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc. Natl. Acad. Sci. USA. 2013;110:2523–2527. doi: 10.1073/pnas.1222494110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aria V., De Felice M., Di Perna R., Uno S., Masai H., Syväoja J.E., van Loon B., Hübscher U., Pisani F.M. The human Tim-Tipin complex interacts directly with DNA polymerase epsilon and stimulates its synthetic activity. J. Biol. Chem. 2013;288:12742–12752. doi: 10.1074/jbc.M112.398073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshizawa-Sugata N., Masai H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 2007;282:2729–2740. doi: 10.1074/jbc.M605596200. [DOI] [PubMed] [Google Scholar]
- 10.Chou D.M., Elledge S.J. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. USA. 2006;103:18143–18147. doi: 10.1073/pnas.0609251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unsal-Kacmaz K., Chastain P.D., Qu P.P., Minoo P., Cordeiro-Stone M., Sancar A., Kaufmann W.K. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell Biol. 2007;27:3131–3142. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Errico A., Costanzo V., Hunt T. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc. Natl. Acad. Sci. USA. 2007;104:14929–14934. doi: 10.1073/pnas.0706347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Errico A., Cosentino C., Rivera T., Losada A., Schwob E., Hunt T., Costanzo V. Tipin/Tim1/And1 protein complex promotes Pol alpha chromatin binding and sister chromatid cohesion. EMBO J. 2009;28:3681–3692. doi: 10.1038/emboj.2009.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gambus A., Jones R.C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R.D., Labib K. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 2006;8:358–366. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- 15.Gotter A.L., Suppa C., Emanuel B.S. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J. Mol. Biol. 2007;366:36–52. doi: 10.1016/j.jmb.2006.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rabitsch K.P., Tóth A., Gálová M., Schleiffer A., Schaffner G., Aigner E., Rupp C., Penkner A.M., Moreno-Borchart A.C., Primig M., et al. A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr. Biol. 2001;11:1001–1009. doi: 10.1016/s0960-9822(01)00274-3. [DOI] [PubMed] [Google Scholar]
- 17.Warren C.D., Eckley D.M., Lee M.S., Hanna J.S., Hughes A., Peyser B., Jie C., Irizarry R., Spencer F.A. S-phase checkpoint genes safeguard high-fidelity sister chromatid cohesion. Mol. Biol. Cell. 2004;15:1724–1735. doi: 10.1091/mbc.E03-09-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer M.L., Pot I., Chang M., Xu H., Aneliunas V., Kwok T., Newitt R., Aebersold R., Boone C., Brown G.W., Hieter P. Identification of protein complexes required for efficient sister chromatid cohesion. Mol. Biol. Cell. 2004;15:1736–1745. doi: 10.1091/mbc.E03-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ansbach A.B., Noguchi C., Klansek I.W., Heidlebaugh M., Nakamura T.M., Noguchi E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol. Biol. Cell. 2008;19:595–607. doi: 10.1091/mbc.E07-06-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan R.C., Chan A., Jeon M., Wu T.F., Pasqualone D., Rougvie A.E., Meyer B.J. Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature. 2003;423:1002–1009. doi: 10.1038/nature01697. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka H., Kubota Y., Tsujimura T., Kumano M., Masai H., Takisawa H. Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells. 2009;14:949–963. doi: 10.1111/j.1365-2443.2009.01322.x. [DOI] [PubMed] [Google Scholar]
- 22.Leman A.R., Noguchi C., Lee C.Y., Noguchi E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 2010;123:660–670. doi: 10.1242/jcs.057984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith-Roe S.L., Patel S.S., Simpson D.A., Zhou Y.C., Rao S., Ibrahim J.G., Kaiser-Rogers K.A., Cordeiro-Stone M., Kaufmann W.K. Timeless functions independently of the Tim-Tipin complex to promote sister chromatid cohesion in normal human fibroblasts. Cell Cycle. 2011;10:1618–1624. doi: 10.4161/cc.10.10.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leman A.R., Noguchi E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle. 2012;11:3945–3955. doi: 10.4161/cc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudra S., Skibbens R.V. Sister chromatid cohesion establishment occurs in concert with lagging strand synthesis. Cell Cycle. 2012;11:2114–2121. doi: 10.4161/cc.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H., Boone C., Brown G.W. Genetic dissection of parallel sister-chromatid cohesion pathways. Genetics. 2007;176:1417–1429. doi: 10.1534/genetics.107.072876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bharti S.K., Khan I., Banerjee T., Sommers J.A., Wu Y., Brosh R.M., Jr Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome. Cell. Mol. Life Sci. 2014;71:2625–2639. doi: 10.1007/s00018-014-1569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brosh R.M., Jr DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer. 2013;13:542–558. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Y., Brosh R.M., Jr DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 2012;40:4247–4260. doi: 10.1093/nar/gks039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Lelij P., Chrzanowska K.H., Godthelp B.C., Rooimans M.A., Oostra A.B., Stumm M., Zdzienicka M.Z., Joenje H., de Winter J.P. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Capo-Chichi J.M., Bharti S.K., Sommers J.A., Yammine T., Chouery E., Patry L., Rouleau G.A., Samuels M.E., Hamdan F.F., Michaud J.L., et al. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum. Mutat. 2013;34:103–107. doi: 10.1002/humu.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holloway L. CHL1 is a nuclear protein with an essential ATP binding site that exhibits a size-dependent effect on chromosome segregation. Nucleic Acids Res. 2000;28:3056–3064. doi: 10.1093/nar/28.16.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skibbens R.V. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parish J.L., Rosa J., Wang X., Lahti J.M., Doxsey S.J., Androphy E.J. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J. Cell. Sci. 2006;119:4857–4865. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 35.Shah N., Inoue A., Lee S.W., Beishline K., Lahti J.M., Noguchi E. Roles of Chlr1 DNA helicase in replication fork recovery from DNA damage. Exp. Cell Res. 2013;319:2244–2253. doi: 10.1016/j.yexcr.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue A., Li T., Roby S.K., Valentine M.B., Inoue M., Boyd K., Kidd V.J., Lahti J.M. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle. 2007;6:1646–1654. doi: 10.4161/cc.6.13.4411. [DOI] [PubMed] [Google Scholar]
- 37.Hirota Y., Lahti J.M. Characterization of the enzymatic activities of ChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000;28:917–924. doi: 10.1093/nar/28.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farina A., Shin J.H., Kim D.H., Bermudez V.P., Kelman Z., Seo Y.S., Hurwitz J. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J. Biol. Chem. 2008;283:20925–20936. doi: 10.1074/jbc.M802696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y., Sommers J.A., Khan I., de Winter J.P., Brosh R.M., Jr Biochemical characterization of Warsaw breakage syndrome helicase. J. Biol. Chem. 2012;287:1007–1021. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y., Sommers J.A., Loiland J.A., Kitao H., Kuper J., Kisker C., Brosh R.M., Jr The Q motif of Fanconi anemia group J protein (FANCJ) DNA helicase regulates its dimerization, DNA binding, and DNA repair function. J. Biol. Chem. 2012;287:19188–19198. doi: 10.1074/jbc.M112.351338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bharti S.K., Sommers J.A., George F., Kuper J., Hamon F., Shin-Ya K., Brosh R.M., Jr Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 2013;288:28217–28229. doi: 10.1074/jbc.M113.496463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merrick C.J., Jackson D., Diffley J.F. Visualization of altered replication dynamics after DNA damage in human cells. J. Biol. Chem. 2004;279:20067–20075. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 43.Gupta R., Sharma S., Sommers J.A., Jin Z., Cantor S.B., Brosh R.M., Jr Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J. Biol. Chem. 2005;280:25450–25460. doi: 10.1074/jbc.M501995200. [DOI] [PubMed] [Google Scholar]
- 44.Smith F.W., Feigon J. Quadruplex structure of Oxytricha telomeric DNA oligonucleotides. Nature. 1992;356:164–168. doi: 10.1038/356164a0. [DOI] [PubMed] [Google Scholar]
- 45.Haider S., Parkinson G.N., Neidle S. Crystal structure of the potassium form of an Oxytricha nova G-quadruplex. J. Mol. Biol. 2002;320:189–200. doi: 10.1016/S0022-2836(02)00428-X. [DOI] [PubMed] [Google Scholar]
- 46.Horvath M.P., Schultz S.C. DNA G-quartet in a 1.86 resolution structure of an Oxytricha nova telomeric protein-DNA complex. J. Mol. Biol. 2001;310:367–377. doi: 10.1006/jmbi.2001.4766. [DOI] [PubMed] [Google Scholar]
- 47.Sharma S., Sommers J.A., Gary R.K., Friedrich-Heineken E., Hübscher U., Brosh R.M., Jr The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–6781. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gotter A.L. Tipin, a novel timeless-interacting protein, is developmentally co-expressed with timeless and disrupts its self-association. J. Mol. Biol. 2003;331:167–176. doi: 10.1016/s0022-2836(03)00633-8. [DOI] [PubMed] [Google Scholar]
- 49.Chung G., O'Neil N.J., Rose A.M. CHL-1 provides an essential function affecting cell proliferation and chromosome stability in Caenorhabditis elegans. DNA Repair. 2011;10:1174–1182. doi: 10.1016/j.dnarep.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 50.Bhattacharya C., Wang X., Becker D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. Mol. Cancer. 2012;11:82. doi: 10.1186/1476-4598-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leman A.R., Dheekollu J., Deng Z., Lee S.W., Das M.M., Lieberman P.M., Noguchi E. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle. 2012;11:2337–2347. doi: 10.4161/cc.20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murakami H., Keeney S. Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell. 2014;158:861–873. doi: 10.1016/j.cell.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borde V., Lichten M. A Timeless but timely connection between replication and recombination. Cell. 2014;158:697–698. doi: 10.1016/j.cell.2014.07.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.