


Abstract
Type II restriction-modification (R-M) systems encode a restriction endonuclease that cleaves DNA at specific sites, and a methyltransferase that modifies same sites protecting them from restriction endonuclease cleavage. Type II R-M systems benefit bacteria by protecting them from bacteriophages. Many type II R-M systems are plasmid-based and thus capable of horizontal transfer. Upon the entry of such plasmids into a naïve host with unmodified genomic recognition sites, methyltransferase should be synthesized first and given sufficient time to methylate recognition sites in the bacterial genome before the toxic restriction endonuclease activity appears. Here, we directly demonstrate a delay in restriction endonuclease synthesis after transformation of Escherichia coli cells with a plasmid carrying the Esp1396I type II R-M system, using single-cell microscopy. We further demonstrate that before the appearance of the Esp1396I restriction endonuclease the intracellular concentration of Esp1396I methyltransferase undergoes a sharp peak, which should allow rapid methylation of host genome recognition sites. A mathematical model that satisfactorily describes the observed dynamics of both Esp1396I enzymes is presented. The results reported here were obtained using a functional Esp1396I type II R-M system encoding both enzymes fused to fluorescent proteins. Similar approaches should be applicable to the studies of other R-M systems at single-cell level.
INTRODUCTION
Type II restriction-modification (R-M) systems are highly abundant in bacteria (1). These systems code for two enzymes: a restriction endonuclease that cleaves DNA at specific recognition sites and a methyltransferase that modifies—by adding methyl groups—same sites and thus protects the host genome from degradation by the restriction endonuclease. R-M systems benefit bacteria that host them by protecting from dsDNA bacteriophages. Upon entry of unmodified phage DNA into cell containing an R-M system, restriction endonuclease prevents the infection by locating its recognition sites and cleaving them. Typically, the presence of an R-M system decreases the efficiency of phage plaque formation (EOP) by several orders of magnitude (2–6). Only rare phage genomes that become modified by methyltransferase before restriction endonuclease attack can mount a productive infection. Progeny phages from such an infection contain modified genomes and infect restrictive (i.e. containing the R-M system) cells as efficiently as permissive (without the R-M system) cells. Thus, the protection afforded by R-M systems, if overcome once, becomes ineffective.
The amount of R-M enzymes present in cells should be tightly controlled. Suboptimal levels of methyltransferase may lead to accumulation of unmodified sites in bacterial DNA followed by their cleavage and subsequent cell death (7). Too high a level of restriction endonuclease may have the same consequence. On the other hand, a higher than optimal amount of methyltransferase should increase the probability of modification of infecting phage genomes prior to their degradation by restriction endonuclease, compromising protection and posing a threat to the whole bacterial population (8). Decreased amount of restriction endonuclease can lead to the same outcome.
At least some present-day bacteriophages are under the evolutionary pressure from R-M systems common to their hosts (9,10) and have evolved specific mechanisms to counter restriction. Known mechanisms include recognition site avoidance in phage genomes (11–13), the presence of phage-encoded anti-restriction proteins (14), modification of phage DNA by phage-encoded enzymes and the use of alternative bases such as uridines in phage DNA (15).
While undeniably useful to their host cells at least in some cases, R-M systems also demonstrate features typical for selfish genetic elements or ‘addictive’ modules, with restriction endonuclease playing the role of a toxin and methyltransferase serving as an anti-toxin. Post-segregational killing was observed upon loss of plasmids containing some R-M systems genes (16–20).
Genes coding for R-M systems are occasionally carried on broad host range plasmids capable of horizontal spread through bacterial populations (18,21). Upon the entry of such a plasmid into a naïve host with unmodified recognition sites in the genome, it is crucial, both from the host and plasmid perspective, that the protective methyltransferase is synthesized first, and is given sufficient time to methylate restriction sites in bacterial genome before the toxic restriction endonuclease activity appears. Hence, it is not surprising that R-M systems evolved elaborate regulatory mechanisms that ensure proper synthesis dynamics of their products. The following commonalities have emerged from analyses of transcription regulation in several differently organized R-M systems. First, a promoter from which the methyltransferase gene is transcribed is typically much stronger than the restriction endonuclease gene promoter (2,22,23). Second, unlike the methyltransferase, most type II restriction endonucleases act as homodimers or homotetramers (24,25), meaning that a certain threshold concentration of the gene product needs to be reached before the enzymatic activity appears. In some cases, the methyltransferase polypeptide (26,27) or its enzymatic activity (28–30) provides a negative feedback loop that decreases transcription from methyltransferase promoter as methyltransferase accumulates. The R-M systems are compact, with R-M genes often sharing an intergenic regulatory region. Decreased transcription of methyltransferase gene thus often leads, directly or indirectly, to increased transcription from the restriction endonuclease promoter, which partially overlaps with a divergent methyltransferase promoter and is initially kept silent by active competing transcription (21,30).
A large class of Type II R-M systems relies on a product of additional controller (C) gene for coordinated regulation of its functional genes (2,22,31–33). C-proteins are small DNA binding proteins related to phage λ repressor—a prototypical regulator orchestrating the lysis-to-lysogeny genetic switch of this virus (34,35). C-proteins bind to their binding sites (C-boxes), which are analogous to phage λ operators. A typical C-box contains several binding sites of varying affinity (2,22,36,37). The binding to the high-affinity site usually serves to decrease transcription of the methyltransferase gene by steric hindrance of the bound protein with RNA polymerase (RNAP) binding. Conversely, C-protein bound to this site activates transcription from a divergent restriction endonuclease gene promoter, either indirectly (by preventing interference from a partially overlapping methyltransferase promoter) or directly, by favorable protein–protein interactions with RNAP. The C-protein genes are commonly found in an operon with the restriction endonuclease genes, with the C-protein gene preceding the restriction endonuclease genes. Thus, the initial binding of C-protein to its binding site creates a positive feedback loop that ensures additional synthesis of the C-protein (and, therefore, restriction endonuclease). However, binding to the second, weaker site within the C-box decreases further synthesis of C-protein (and restriction endonuclease) allowing stable maintenance of desired R-M system gene expression levels.
In addition to the basic type of C-protein-dependent R-M systems described above, many variations on the general theme are known. The object of this study, the Esp1396I R-M system originally found on a Enterobacter sp. RFL1396 plasmid, provides one such example (38). Genes coding for Esp1396I methyltransferase and restriction endonuclease are transcribed convergently (Figure 1A), a deviation from the common divergent organization described above. The Esp1396I C-protein gene is co-transcribed with the restriction endonuclease gene from a common promoter, a standard arrangement. C-protein ensures balanced expression of Esp1396I system genes by interacting with a single binding site overlapping with the strong Esp1396I methyltransferase promoter and a pair of binding sites located upstream of and partially overlapping with the promoter of C-protein/restriction endonuclease genes operon. Binding to the methyltransferase site inhibits activity of the methyltransferase promoter. Binding to the duplicated restriction endonuclease site activates restriction endonuclease promoter at low C-protein concentrations, ensuring increased supply of both restriction endonuclease and C-protein. Higher C-protein levels lead to full occupancy of the duplicated binding site and inhibit transcription.
Figure 1.

Construction of functional fluorescently-labeled Esp1396I restriction-modification system. (A) Genetic organization of the wild-type Esp1396I and Esp1396I_Fluo encoding fusions of the restriction endonuclease and methyltransferase genes to, respectively, mCherry and Venus fluorescent proteins genes is schematically shown. Individual genes are represented by arrows whose directions indicate the direction of transcription. Blue arrows indicate C-protein genes. The three sites of binding of C-protein dimers are shown as light blue rectangles labeled “C". (B) The titers of λvir phage lysate determined on lawns of E. coli cells with or without indicated plasmids are shown. Mean results from three independent measurements with standard deviations are presented. (C) Aliquots of whole-cell lysates corresponding to indicated numbers of E. coli cells transformed with pEsp1396I_Fluo were separated by SDS-PAGE and blotted with anti-Venus or anti-mCherry antibodies. As controls, known amounts of purified Venus and mCherry proteins were loaded on the same gel.
Esp1396I, just like other R-M systems should be able to implement delayed synthesis of restriction endonuclease and initial high level followed by subsequent lower level of methyltransferase synthesis upon its establishment in a naïve host. However, this has not been demonstrated directly in live cells. This is due to experimental complications of such an analysis, which requires synchronous populations of cells transformed with R-M system genes. In fact, the existence of such a delay was demonstrated for only one Type II R-M system, a prototypical C-protein dependent system PvuII (39). The authors placed the entire system on an M13 phage, which allowed them to synchronously introduce the PvuII system genes into naïve cells by phage infection, followed by time-resolved monitoring of restriction endonuclease and methyltransferase genes transcripts and corresponding enzymatic activities accumulation in infected cultures. These experiments directly demonstrated delayed transcription of restriction endonuclease gene relative to methyltransferase gene transcription, delayed appearance of restriction endonuclease activity relative to methyltransferase, and the importance of the C-protein in orchestrating this delay. In addition, it was shown that during the establishment of Type I R-M system hsdK after conjugal transfer into naïve cells, both the restriction endonuclease and methyltransferase promoters express simultaneously (40), but restriction activity appears only after 15 generations, while modification activity appears immediately after conjugation (41).
Unlike bulk assays, single-cell approaches allow the experimenter to monitor accumulation of R-M proteins in real time and follow the fate of individual cells and their progeny. In this work, we report the application of such an approach to the Esp1396I system. We created a functional Esp1396I system encoding restriction endonuclease and methyltransferase proteins fused to fluorescent proteins. Transformation of cells with a plasmid carrying such a modified Esp1396I system allowed us to monitor the appearance and dynamics of synthesis of both fluorescent fusion proteins in individual transformed cells. Our results generally agree with theoretical expectations by showing a significant delay in restriction endonuclease appearance and a sharp early peak in methyltransferase activity that should ensure complete modification of recognition sites in bacterial genome early after the introduction of R-M systems genes in a naïve host. The approach used here opens way for further analysis of genetic and stochastic factors that may influence the establishment of various R-M systems and their protective function.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli strain Top10 was used as a host for cloning; microscopy was conducted using E. coli XL-1Blue strain (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F'proAB lacIqZΔM15 Tn10 (Tetr)]). Chemically competent XL-1Blue cells (efficiency of transformation of 1010 per microgram of plasmid DNA) were purchased from Evrogen (Russia). E. coli Rosetta cells were used to express fluorescent proteins from pET plasmids. Cells were cultivated in LB medium supplemented when appropriate with ampicillin (100 μg/ml). Expression from pET-based plasmids was induced by the addition of 1 mM IPTG to cultures. Phage λvir was propagated as described by Sambrook et al. (42).
A plasmid carrying fusions of Esp1396I genes to fluorescent protein genes was derived from pEsp1396IRM5.6 (38), carrying the entire Esp1396I system. A unique PmlI site was engineered at the end of esp1396I.R coding sequence and was used for blunt-end cloning of a polymerase-chain-reaction fragment with the mCherry coding sequence. The StuI and BamHI sites were added at the end of esp1396I.M gene in the resulting plasmid and were used for subsequent Venus coding sequence insertion. pET21a-based expression plasmids with Venus or mCherry genes cloned under the control of T7 RNAP promoter were created using standard molecular cloning methods.
Restriction assay/phage titering
E. coli XL1-Blue cells without plasmids or harboring pUC19, pEsp1396IRM5.6 or pEsp1396I_Fluo plasmids were grown overnight and then diluted and grown to OD600≈0.6. Onto freshly seeded cell-lawns prepared from exponentially growing cultures, 10 μl aliquots of serial dilutions of λ-vir phage lysate were spotted. Plates were incubated at 37°C overnight and phage titer was determined by calculating individual phage plaques seen in spots corresponding to the most dilute aliquote of phage lysate.
Western blotting
E. coli XL1-Blue cells harboring pEsp1396I_Fluo plasmid were grown to OD600 = 0.6. 5 ml of cell culture was centrifuged and resuspended in 500 μl SDS-PAGE (Laemmli) sample buffer, boiled for 10 min and after centrifugation 5 μl of the sample was loaded on a 10% SDS polyacrylamide gel. After electrophoresis, proteins were transferred to a nitrocellulose blotting membrane (Amersham). Blots were analyzed using primary polyclonal rabbit antibodies specific to mCherry (ab167453, Abcam) or monoclonal mouse antibodies specific to Venus (Living Colors® JL-8, Clontech) and peroxidase-conjugated goat anti-rabbit IgG antibodies in the case of mCherry primary antibodies or peroxidase-conjugated rabbit anti-mouse IgG antibodies antibodies in the case of Venus primary antibodies (Sigma). The detection of peroxidase activity was performed with SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Scientific). To estimate the relative amounts of proteins, blots were scanned using ChemidocTM XRS+ system (Biorad) and individual band densities were measured and compared using Quantity One 1-D analysis software (Biorad). Serial dilutions of purified mCherry and Venus proteins of known concentration were used as calibrants.
Fluorescence microscopy
Fluorescence microscopy was performed using an AxioImager.Z1 upright microscope (Zeiss) equipped with a custom incubation system. Zeiss Filter set 10 and Semrock mCherry-40LP filter set were used to detect Venus and mCherry fluorescence, respectively. Image acquisition was performed using Photometrics CascadeII 1024 back-illuminated EM-CCD. Microscope was controlled using MicroManager (43) with custom scripts created to perform multichannel and time-lapse imaging.
Single-molecule intensity calibration
Fluorescence intensities corresponding to single Venus and mCherry molecules were calculated by comparing average fluorescence intensities of E. coli cells obtained from fluorescence microscopy images to the average Venus and mCherry molecule numbers per cell measured using spectrofluorimeter. Overnight cultures of E. coli Rosetta cells carrying pET21a-based expression plasmids with Venus or mCherry genes cloned under the control of T7 RNAP promoter were used. High concentrations of soluble Venus or mCherry proteins produced from these plasmids allowed reliable measurements of concentrations of each fluorescent proteins using spectrofluorimeter. Known concentrations of purified mCherry and Venus were used as fluorescence standards for spectrofluorimetric measurements. Bacterial titers of cultures were determined by direct counting under the microscope.
Transformation procedure
E. coli XL-1 Blue highly-competent cells (Evrogen, Russia) were transformed according to manufacturer instructions. Briefly, 20 μl of thawed cells suspension was gently mixed with 3 μl of pEsp1396I_Fluo plasmid in microcentrifuge tube and incubated on ice for 30 min. Transformation mixture was heat-shocked for 50 s at 42°C and then placed on ice for 2 min. Cells were diluted 1:1 (as opposed to 1:10) in preheated LB medium (Amresco) and were grown at 37°C with 500 rpm shaking for 30 min prior to microscopy.
Microscope slide preparation
Two types of coverslips were used to construct sample chambers. The first coverslip (24 х 24 mm, Menzel-Gläser) was attached to a double-sided adhesive frame (1.5 х 1.6 cm Gene Frame, Thermo Scientific). To form smooth agarose surface inside the frame, 70 μl of 1.5% agarose (Helicon) diluted in 0.25X LB medium (Amresco) with the addition of 100 μg/ml ampicillin was placed in the center of the coverslip attached to the frame and pressed with the second similar coverslip. After solidification of agarose the second coverslip was removed. To provide oxygen supply for the cells in a sealed chamber, 1.5 х 0.5 cm strips of agarose gel from each of two sides of the gel slab were removed. Transformation mixture (1 μl) was placed on the resulting agarose surface (1.5 х 0.6 mm). When transformation mixture was completely absorbed, the chamber was sealed using a longer coverslip (24 х 60 mm, Menzel-Gläser). The sealed chamber was fixed in custom-build holder and mounted in the microscope.
Automated fluorescence microscopy
Custom MicroManager script was used to control the set-up during time-lapse imaging of cells after transformation. A number of fields of view (FOV) were monitored in parallel (typically from 10 20 FOV) with autofocusing performed at each FOV and time point by locating the local minimum of the standard deviation of pixels intensities in the transmitted light image. Each FOV was imaged once per 15 min (after 2 h this delay was doubled) for at least 8 h. To minimize photobleaching halogen lamp intensity was set as low as possible to still ensure reliable focusing and transmitted light shutter was closed whenever possible.
Image analysis
All image analyses were performed using ImageJ (44) (Fiji package (45)). Venus and mCherry concentrations in individual cells after transformation were quantified manually using custom circular selection tool with the radius of 4 pixels with the use of single molecule intensity calibration. Selection volume was calculated using cell diameter assumed to be equal to 1 μm (46). Stationary concentrations of R.Esp1396I::mCherry and M.Esp1396I::Venus were determined using automatic segmentation based on Venus channel images. Background fluorescence in mCherry channel was negligible, while in the Venus channel it reached about 20% of the total signal. Therefore the background intensity distribution in Venus channel, obtained from images of cells, harboring non-fluorescent Esp1396I plasmid, was deconvoluted from the Venus intensity distribution of Esp1396I_Fluo cells to obtain background-corrected M.Esp1396I::Venus distribution. Nonlinear contrast was used in Figure 3 to promote simultaneous visualization of bright and dim features (ImageJ, Gamma = 0.7).
Figure 3.

Dynamics of Esp1396I enzymes accumulation in individual transformed E. coli cells. (A and B) A representative kinetic series of images showing Venus (green) and mCherry (magenta) fluorescence in a microcolony growing from a single transformed cell. The initial part of the sequence is shown in A, in B, the full duration of experiment is shown. A nonlinear contrast was used in B to allow simultaneous visualization of bright and dim signals (see Materials and Methods). White arrows in A show transformed cell at early stages before Venus accumulation is clearly visible. Another kinetic series is shown in Supplementary Figure S1. (C) Atypical cells in growing microcolonies transformed with pEsp1396I_Fluo. See text for details. Atypical cells are indicated by white arrows. In each panel, two different examples of identified atypical cell types are shown. (D) Quantification of a representative kinetic series showing changes in Venus and mCherry fluorescence intensities per individual cell in microcolony growing from a single transformed cell over time. Dashed lines at infinity show mean stationary R.Esp1396I::mCherry and M.Esp1396I::Venus levels in cells from exponentially growing cultures.
RESULTS
Fluorescently labeled Esp1396I restriction-modification system retains its biological activity
To enable the study of Esp1396I restriction-modification system at the single-cell level using fluorescence microscopy, sequences coding for Venus and mCherry fluorescent proteins were added in frame to the 3′ termini of, respectively, the esp1396I.M and esp1396I.R genes cloned on the pUC19 plasmid (28). The resulting plasmid was named Esp1396I_Fluo (Figure 1A). To assess functional activity of the fluorescently-labeled Esp1396I R-M system, E. coli cells harboring pEsp1396I_Fluo were analyzed for their ability to restrict the growth of bacteriophage λ. As can be seen from Figure 1B, the Esp1396I_Fluo plasmid protects cells against bacteriophage infection as efficiently, as the pEsp1396I plasmid containing wild-type Esp1396I system (a decrease of phage plaque formation efficiency of four orders of magnitude or more compared to cells harboring the pUC19 vector).
Western-blot analysis of lysates of cells transformed with Esp1396I_Fluo using antibodies specific to Venus and mCherry proteins (Figure 1C) showed that both the mCherry and Venus antigens migrated as bands corresponding to ≈60 kDa fusions with Esp1396I enzymes. The degradation of either fusion protein was undetectable. Western blots shown on Figure 1C also included lanes with known amounts of pure Venus and mCherry proteins, which allowed to come up with an estimate of ≈6000 M.Esp1396I::Venus and ≈30 000 R.Esp1396I::mCherry monomer molecules per cell in exponentially growing cultures.
Measuring the amounts of Esp1396I restriction endonuclease and methyltransferase in individual cells using fluorescence microscopy
The results from the previous section suggest that fusion proteins M.Esp1396I::Venus and R.Esp1396I::mCherry retain their biological activities. Therefore, fluorescence intensities of Venus and mCherry can serve as faithful reporters of intracellular concentrations of Esp1396I methyltransferase and restriction endonuclease, respectively. Fluorescence microscopy of cells transformed with the pEsp1396I_Fluo plasmid revealed, as expected, fluorescence in both the Venus and mCherry channels (Figure 2A–C). Using a single molecule fluorescence intensity calibrations for Venus and mCherry (see Materials and Methods) absolute numbers of mCherry and Venus molecules in individual cells were determined based on fluorescence images. Mean numbers of M.Esp1396I::Venus and R.Esp1396I::mCherry were determined to be 12 700 ± 400 (Mean ± SEM) and 25 600 ± 700 molecules per cell (Figure 2E). There was a strong (r = 0.67, N = 2069) correlation between the amounts of restriction endonuclease and methyltransferase in individual cells (Figure 2F).
Figure 2.

Determining amount of Esp1396I enzymes in individual E. coli cells. (A–C) Fluorescence images of bacteria harboring pEsp1396I_Fluo plasmid in Venus (A) and mCherry (B) channels and their overlay (C). (D) A representative intensity profile in Venus (black line) and mCherry (red line) channels through the dashed line of enlarged images of cells from A and B (shown to the left) demonstrating different localization of M.Esp1396I::Venus and R.Esp1396I::mCherry. (E) Histograms of R.Esp1396I::mCherry and M.Esp1396I::Venus molecule numbers per cell (N = 2069). (F) Correlation plot for R.Esp1396I::mCherry and M.Esp1396I::Venus concentrations in individual E. coli cells analyzed in E. The correlation coefficient between R.Esp1396I::mCherry and M.Esp1396I::Venus concentrations is r = 0.67.
The availability of multiple images of cells transformed with pEsp1396I_Fluo allowed us to investigate the localization of Esp1396I enzyme fusions in individual cells. The results revealed that R.Esp1396I was uniformly distributed through the cell (Figure 2D). In contrast, M.Esp1396I distribution was non-uniform (Figure 2D) and resembled that of the nucleoid. This effect was most pronounced in rapidly dividing cells, suggesting that it may be caused by the appearance of hemimethylated Esp1396I recognition sites during replication that recruit M.Esp1396I. Another possible source of non-uniform distribution of M.Esp1396I could be the tendency of high-copy number plasmids to preferentially localize in nucleoid-free space at cell poles (47), however, this explanation would require a significant difference in diffusion coefficients of R.Esp1396I and M.Esp1396I to account for their different localization patterns.
Dynamics of R.Esp1396I and M.Esp1396I accumulation upon entry of pEsp1396I_Fluo into a naïve cell
E. coli cells are transformed by plasmids pEsp1396I, pEsp1396I_Fluo, or control pUC19 vector with the same efficiency (data not shown). The result indicates that upon the introduction of Esp1396I genes into a naïve host no significant penalty for inappropriate expression of R.Esp1396I is sustained. The establishment of Esp1396I_Fluo restriction-modification system in naïve cells after transformation was followed at the single-cell level using automated fluorescence microscopy (Figure 3A,B, see also Supplementary Figure S1). Highly competent E. coli cells were transformed by Esp1396I_Fluo plasmid, transformation mixtures were deposited onto agar blocks soaked in 0.25X LB medium with ampicillin and growth of microcolonies from individual fluorescent (and, therefore, transformed) cells was observed. Despite the fact that only small fraction of cells acquired the plasmid (less than 0.01%), highly parallel acquisition allowed up to a dozen of transformation events to be captured in a single experiment and to monitor the growth of transformed cell progeny over the course of several hours. Experiments with transformed cell cultures treated with rifampicin and/or chloramphenicol showed that photobleaching of Venus and mCherry was negligible during the time course of the experiment (Supplementary Figure S5). During microcolony formation, Venus fluorescence reporting the synthesis of M.Esp1396I::Venus fusion protein was the first to appear and could be detected as early as at the stage of 1–2 transformed cells. At the earliest time when Venus fluorescence became detectable, the calculated amount of M.Esp1396I::Venus molecules per cell was ≈400. Up to a certain point (usually when 16–32 cells were present in the colony), only Venus fluorescence was detectable. The mCherry fluorescence indicative of accumulation of R.Esp1396I::mCherry fusion protein appeared with a delay of 30–60 min (Figure 3A,B and Supplementary Figure S1). At the earliest time when the mCherry fluorescence became detectable, the calculated amount of R.Esp1396I::mCherry was also ≈400.
Changes in fluorescence of individual cells within the microcolony were determined as a function of time. A striking kinetics of Venus fluorescence intensity was revealed: after a rapid increase reaching a peak value, a sharp decline to a steady-state value was observed (Figure 3D and Supplementary Figure S1). The accumulation of restriction endonuclease fusion was much slower and monotonous. The peak of Venus fluorescence was reached just before the time restriction endonuclease fusion became detectable.
As was the case with individual cells from transformed cultures (Figure 2F), after the cell number in microcolonies increased to several dozens, there was a correlation between the levels of fluorescence in both channels for most individual cells. However, three types of atypical cells were observed. The first kind of cells contained abnormally high amounts of restriction endonuclease fusion (Figure 3C images labeled “i"). The cells formed filaments and may thus have undergone DNA damage caused by excess restriction endonuclease. The second type of cells contained unusually high amounts of methyltransferase fusion (Figure 3C images labeled “ii"). These cells remained short and yet failed to divide. Earlier, it was reported that excess M.Esp1396I is toxic to cells for an unknown reason (31), abnormal cells like those shown in shown in Figure 3C images labeled “ii" may have undergone arrest due to M.Esp1396I toxicity. Finally, there were cells that contained unusually high concentrations of both proteins (Figure 3C images labeled “iii"). They too formed filaments and eventually disappeared from the colony apparently due to lysis. The atypical cells were infrequent (less than 1% of the population). We speculate that they may have arisen due to stochastic variation of plasmid copy number (47), or misregulation of expression of Esp1396I genes caused by ‘intrinsic’ and ‘extrinsic’ genetic noise (48).
Quantitative modeling of R-M protein dynamics
The Esp1396I methyltranferase (M), and the restriction endonuclease (R) show two clearly distinct temporal patterns of expression in the first time interval of the experiment (≈first 200 min after transformation): the concentration of R is low and gradually increases; while the concentration of M increases rapidly early on followed by a somewhat slower (but still fast) decrease at later times, so that a pronounced peak—and significant enzyme amounts—are accumulated early after the entry of R-M system carrying plasmid. We asked if such a pattern can be reproduced by a minimal quantitative model, which takes into account the available knowledge on the Esp1396I system transcription regulation by the C-protein (C), and the dynamics of the transcript and protein expression through the experiment. Specifically, we incorporated (i) a thermodynamic model, which takes into account the activation and the repression of M gene and CR operon promoters by the C-protein, by calculating probabilities of relevant promoter configurations, and (ii) a kinetic model, which takes into account R, C and M transcript and protein expression. The model is minimal, in a sense that it takes into account only the experimentally established regulatory mechanisms (i.e. the transcription control by C) that we summarize below, and that all model parameters are considered as time-independent.
The experimental information on Esp1396I transcription control, used in the model, is based on (2), and can be briefly summarized as follows: promoters of both the CR operon and the M gene each contain single RNAP binding sites. The CR promoter has two C-protein binding sites (the distal and the proximal one), each of them binding one C-protein dimer, where binding to the distal site leads to activation, while binding to both the distal and the proximal site leads to repression of transcription. Note that in the absence of RNAP, a C-protein dimer cannot be bound only to one of the two binding sites, since binding of C-protein dimer to the stronger distal site immediately recruits another dimer to the adjacent site. The M promoter has a single C-protein binding site, where one dimer can bind to repress transcription.
An important factor in the protein dynamics is the cell division rate, which controls the extent of transcripts and proteins dilution, since even an inherently highly stable protein will be effectively ‘degraded’ through dilution caused by cell division if its synthesis stops. As can be seen from Figure 4A, there are clearly two intervals in the system dynamics observed during the growth of microcolonies of transformed cells: the cell division time is constant before ≈160 min, and also after ≈200 min, but there is an abrupt change between these two time intervals, so that the division time is ≈40 min in the first interval, and ≈320 min in the second interval. The reason of this change is unknown and could be related to early use of strongly preferred substrates from the LB medium followed by a shift to other nutrients (49,50).
Figure 4.

Dynamic modeling of Esp1396I enzymes accumulation in individual transformed E. coli cells. (A) Logarithm of the experimentally measured number of cells, as a function of time. The first and the second time interval, characterized respectively by the fast and slow cell division rate, are clearly visible. As the time dependences in these two intervals are nearly linear, the respective linear fits are indicated, with the slopes corresponding to the cell division rate. (B and C) Model vs. experiment for the expression dynamics of Esp1396I restriction endonuclease (B) and methyltransferase (C) as a function of time. Circles correspond to experimentally measured concentrations of protein fusions (expressed in relative units), while the full lines correspond to the model predictions. Note that the time is set to zero at the point of the first available measurement.
Below, we provide the main steps and results of the model, with detailed description provided in the Supplementary section. Our thermodynamic model of CR and M promoter regulation is based on the classical Shea-Ackers model (51), which assumes the transcription activity to be proportional to the equilibrium binding probability of RNAP to promoter. Consequently, the transcription activity of a promoter is proportional to the ratio of statistical weights (probabilities) of active configurations—those in which RNAP is bound to the promoter—and all the statistical weights corresponding to both active and repressed configurations. Each of the statistical weights includes protein (C and RNAP) concentrations, and protein–DNA and protein–protein interaction energies, as detailed in the Supplementary section. Accordingly, the transcription activity of CR operon promoter can be written in the following form:
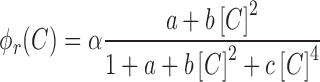 |
(0.1) |
where 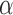 is the proportionality constant; a corresponds to the probability of RNAP alone being bound to the promoter;
is the proportionality constant; a corresponds to the probability of RNAP alone being bound to the promoter; 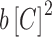 —to the probability of the activating complex formation, i.e. a C-protein dimer bound to the distal site and RNAP bound to promoter; while
—to the probability of the activating complex formation, i.e. a C-protein dimer bound to the distal site and RNAP bound to promoter; while 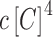 corresponds to the probability of the repressing complex formation, i.e. when two C-protein dimers are bound to both the distal and the proximal binding sites, excluding RNAP binding. Note that constants a, b and c absorb the relevant interaction energies and RNAP concentration (see Supplementary section) and are used below as fitting parameters in comparing the model with the data. Similarly, the transcription activity of M promoter can be written as:
corresponds to the probability of the repressing complex formation, i.e. when two C-protein dimers are bound to both the distal and the proximal binding sites, excluding RNAP binding. Note that constants a, b and c absorb the relevant interaction energies and RNAP concentration (see Supplementary section) and are used below as fitting parameters in comparing the model with the data. Similarly, the transcription activity of M promoter can be written as:
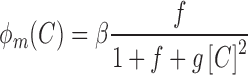 |
(0.2) |
where 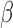 is the proportionality constant, f corresponds to the probability of RNAP alone binding to the promoter and
is the proportionality constant, f corresponds to the probability of RNAP alone binding to the promoter and 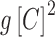 —to the probability of C-protein dimer binding to its site and excluding RNAP (repressed configuration, (2,38)).
—to the probability of C-protein dimer binding to its site and excluding RNAP (repressed configuration, (2,38)).
The equations above specify how the transcription activity of CR and M promoters depends on C-protein concentration, which further allows modeling the dynamics of transcript and protein expression. Specifically, expression of the CR transcript (r), and changes in C and R protein amounts are given by the following equations:
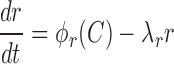 |
(0.3) |
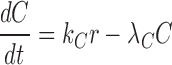 |
(0.4) |
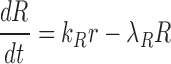 |
(0.5) |
where 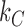 and
and 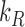 are the translation rates for C and R proteins, while
are the translation rates for C and R proteins, while 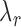 ,
, 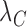 and
and 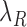 denote the decay rates for the CR transcript, and the C and the R protein, respectively. The first terms on the right hand side of the equations denote transcript or protein synthesis, while the second terms denote their degradation. Note that
denote the decay rates for the CR transcript, and the C and the R protein, respectively. The first terms on the right hand side of the equations denote transcript or protein synthesis, while the second terms denote their degradation. Note that 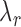 ,
, 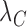 and
and 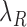 also account for the effective decay due to cell division (see Supplementary data).
also account for the effective decay due to cell division (see Supplementary data).
Similarly, the methyltransferase transcript and protein expression is described by the following equations:
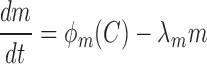 |
(0.6) |
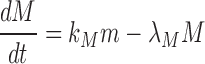 |
(0.7) |
where 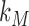 is the protein translation rate, and
is the protein translation rate, and 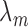 and
and 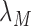 are, respectively, the transcript and the protein decay rates.
are, respectively, the transcript and the protein decay rates.
Equations (0.1)–(0.7) determine the transcript and the protein expression dynamics. For a given set of the model parameters the equations lead to the solution for the dependence of the protein amounts on time; the parameter values are then determined through the best fit (minimal 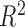 , sum of error squares) of the model solution to experimentally measured data. This is a computationally intensive problem, as it corresponds to parameter search in 12 dimensions; to simplify the problem, we applied a strategy described below. Equations (0.3)–(0.5) can be solved independently from Equations (0.6) and (0.7), as M does not regulate the CR operon promoter. Therefore, we first solve only Equations (0.3)–(0.5) and determine the respective 7 parameters through the best fit to R experimental data (see Supplementary section). As can be seen in Figure 4B there is a very good, both quantitative and qualitative, agreement of the model with experimentally observed R protein dynamics. This agreement requires that both the transcript and the protein are highly stable, i.e. that their effective decay is determined only by dilution due to cell division. Such stability underlies the observed quadratic increase of R with time and is supported by experiments with chloramphenicol referred to above. We next use the determined R protein dynamics, i.e. the quadratic dependence of R with time, to solve Equations (0.6) and (0.7) that determine M protein dynamics. We again determine the remaining 5 parameters of the model (see Supplemental section) through the best fit to the experimentally measured M dynamics. As can be seen from Figure 4C, we obtained a good agreement with the data in the first time period (first ≈160 min characterized by fast cell division). The model correctly describes the abrupt increase and the gradual decrease in the amount of M protein in the cell. However, we observe a different trend with respect to the measured data during the second time interval, when the cell division rate becomes lower. That is, in the model, the amount of methyltransferase increases somewhat in the interval between 160 and 300 min, which is a consequence of the abrupt (and significant) increase of the effective transcript and protein stability due to increase in the cell division time and consequent decrease of dilution factor. The amount of M in the model starts to decrease after ≈300 min, as a consequence of the large increase in C amount at later times, which represses M.
, sum of error squares) of the model solution to experimentally measured data. This is a computationally intensive problem, as it corresponds to parameter search in 12 dimensions; to simplify the problem, we applied a strategy described below. Equations (0.3)–(0.5) can be solved independently from Equations (0.6) and (0.7), as M does not regulate the CR operon promoter. Therefore, we first solve only Equations (0.3)–(0.5) and determine the respective 7 parameters through the best fit to R experimental data (see Supplementary section). As can be seen in Figure 4B there is a very good, both quantitative and qualitative, agreement of the model with experimentally observed R protein dynamics. This agreement requires that both the transcript and the protein are highly stable, i.e. that their effective decay is determined only by dilution due to cell division. Such stability underlies the observed quadratic increase of R with time and is supported by experiments with chloramphenicol referred to above. We next use the determined R protein dynamics, i.e. the quadratic dependence of R with time, to solve Equations (0.6) and (0.7) that determine M protein dynamics. We again determine the remaining 5 parameters of the model (see Supplemental section) through the best fit to the experimentally measured M dynamics. As can be seen from Figure 4C, we obtained a good agreement with the data in the first time period (first ≈160 min characterized by fast cell division). The model correctly describes the abrupt increase and the gradual decrease in the amount of M protein in the cell. However, we observe a different trend with respect to the measured data during the second time interval, when the cell division rate becomes lower. That is, in the model, the amount of methyltransferase increases somewhat in the interval between 160 and 300 min, which is a consequence of the abrupt (and significant) increase of the effective transcript and protein stability due to increase in the cell division time and consequent decrease of dilution factor. The amount of M in the model starts to decrease after ≈300 min, as a consequence of the large increase in C amount at later times, which represses M.
We used our model to quantitatively address the biological relevance of Esp1396I expression control by C-protein. This question is impossible to address experimentally, due to a toxicity of the M.Esp1936I protein that is overexpressed in the absence of C-protein (38), but can be directly addressed by simulation, i.e. by predicting the R and M dynamics with the same parameters that provide the best fit to the data, but in the absence of C-protein control. The results are shown in Supplementary Figure S4: as can be seen, the mutant system allows for very high M expression, a consequence of the absence of repression of the strong M promoter, and a smaller R expression level, as the weak CR operon promoter is no longer activated by C-protein. While methyltransferase toxicity may be a specific feature of the Esp1936I R-M system, even in the absence of such toxicity inactivation of C-protein control is predicted to abolish the protective function of the R-M system due to a very large M to R ratio that would cause rapid methylation of foreign DNA before it can be attacked by restriction endonuclease.
DISCUSSION
The principal result of this work is the demonstration, for the first time, of a time delay between restriction endonuclease appearance and methyltransferase appearance in single cells transformed with a plasmid encoding a Type II R-M system. The methodology developed here, of creating functional fusion between R-M system enzymes and fluorescent proteins followed by time-lapse fluorescent microscopy of cells transformed with plasmids expressing functional R-M systems genes, should be applicable to other R-M systems. This approach should allow comparison of time delays between endonuclease and methyltransferase component appearance, the intracellular amounts of each enzyme and their enzymatic and biological activities for different systems. In the case of the Esp1396I R-M system studied here, our main findings can be summarized as follows. (i) Upon transformation with a plasmid containing genes for the Esp1396I system, a very significant delay in the appearance of the restriction endonuclease is observed. The extent of this delay appears to vary in colonies formed from individual transformed cells but is never less than 90 min under our conditions. (ii) The methyltransferase protein becomes detectable very early in single transformed cells. The intracellular concentration of methyltransferase increases sharply up to a stage of 16–32 cells and then starts to decline, together with the appearance of restriction endonuclease, which accumulates gradually. Mathematical modeling suggests that the unusual dynamics of methyltransferase is due to the negative feedback loop provided by the Esp1396I C-protein and the dilution of excess synthesized methyltransferase that results from cell division. The dynamics of Esp1396I enzymes observed here is substantially different from the much more rapid kinetics previously observed for the PvuII enzymes when the PvuII genes are introduced on an infecting phage (39). The difference could be due to (i) different architectures of regulatory circuits of these C-protein dependent systems, (ii) slower rate of growth of cells observed during prolonged microscopic observation compared to growth rate in liquid culture and/or (iii) physiological consequences of the plasmid transformation procedure used to introduce the R-M system genes in our experiments. While both the sharp increase of Esp1396I methyltransferase amounts (and, presumably, its total activity) immediately after the entry of R-M plasmid in naïve cell and subsequent decline can be biologically meaningful, comparative studies or other R-M systems where the M gene transcription is not subject to negative control by C-proteins will be needed to determine whether such behavior is general.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The work was supported by the Ministry of Education and Science of the Russian Federation [grant 14.B25.31.0004 to K.S.], the National Institutes of Health [grant GM59295 to K.S.], and the graduate program in Biotechnology of Skolkovo Institute of Science and Technology. MD and AR are, in part, supported by Ministry of Science of the Republic of Serbia under project No. ON173052, and by the SNSF SCOPES project IZ73Z0_152297. Funding for open access charge: Russian ministry of science and education grant, and the Skolkovo Institute of Science and Technology.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pingoud A., Wilson G.G., Wende W. Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res. 2014;42:7489–7527. doi: 10.1093/nar/gku447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogdanova E. Transcription regulation of restriction-modification system Esp1396I. Nucleic Acids Res. 2009;37:3354–3366. doi: 10.1093/nar/gkp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagornykh M. Regulation of gene expression in restriction-modification system Eco29kI. Nucleic Acids Res. 2011;39:4653–4663. doi: 10.1093/nar/gkr055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semenova E. Transcription regulation of the EcoRV restriction-modification system. Nucleic Acids Res. 2005;33:6942–6951. doi: 10.1093/nar/gki998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gingeras T.R., Brooks J.E. Cloned restriction/modification system from Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 1983;80:402–406. doi: 10.1073/pnas.80.2.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiss A. Nucleotide sequence of the BsuRI restriction-modification system. Nucleic Acids Res. 1985;13:6403–6421. doi: 10.1093/nar/13.18.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichige A., Kobayashi I. Stability of EcoRI restriction-modification enzymes in vivo differentiates the EcoRI restriction-modification system from other postsegregational cell killing systems. J. Bacteriol. 2005;187:6612–6621. doi: 10.1128/JB.187.19.6612-6621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Enikeeva F.N., Severinov K.V., Gelfand M.S. Restriction-modification systems and bacteriophage invasion: who wins? J. Theor. Biol. 2010;266:550–559. doi: 10.1016/j.jtbi.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tock M.R., Dryden D.T.F. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005;8:466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Kruger D.H., Bickle T.A. Bacteriophage survival: multiple mechanisms for avoiding the deoxyribonucleic acid restriction systems of their hosts. Microbiol. Rev. 1983;47:345–360. doi: 10.1128/mr.47.3.345-360.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawamura F. Unusually infrequent cleavage with several endonucleases and physical map construction of Bacillus subtilis bacteriophage phi 1 DNA. J. Virol. 1981;37:1099–1102. doi: 10.1128/jvi.37.3.1099-1102.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korona R., Korona B., Levin B.R. Sensitivity of naturally occurring coliphages to type I and type II restriction and modification. J. Gen. Microbiol. 1993;139 Pt 6:1283–1290. doi: 10.1099/00221287-139-6-1283. [DOI] [PubMed] [Google Scholar]
- 13.Rocha E.P., Danchin A., Viari A. Evolutionary role of restriction/modification systems as revealed by comparative genome analysis. Genome Res. 2001;11:946–958. doi: 10.1101/gr.gr-1531rr. [DOI] [PubMed] [Google Scholar]
- 14.Makino O., Saito H., Ando T. Bacillus subtilis-phage phi 1 overcomes host-controlled restriction by producing BamNx inhibitor protein. Mol. Gen. Genet. 1980;179:463–468. doi: 10.1007/BF00271735. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi I., Marmur J. Replacement of thymidylic acid by deoxyuridylic acid in the deoxyribonucleic acid of a transducing phage for Bacillus subtilis. Nature. 1963;197:794–795. doi: 10.1038/197794a0. [DOI] [PubMed] [Google Scholar]
- 16.Kulakauskas S., Lubys A., Ehrlich S.D. DNA restriction-modification systems mediate plasmid maintenance. J. Bacteriol. 1995;177:3451–3454. doi: 10.1128/jb.177.12.3451-3454.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Handa N., Kobayashi I. Post-segregational killing by restriction modification gene complexes: observations of individual cell deaths. Biochimie. 1999;81:931–938. doi: 10.1016/s0300-9084(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 18.Mruk I., Kobayashi I. To be or not to be: regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res. 2014;42:70–86. doi: 10.1093/nar/gkt711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asakura Y., Kobayashi I. From damaged genome to cell surface: transcriptome changes during bacterial cell death triggered by loss of a restriction-modification gene complex. Nucleic Acids Res. 2009;37:3021–3031. doi: 10.1093/nar/gkp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naito T., Kusano K., Kobayashi I. Selfish behavior of restriction-modification systems. Science. 1995;267:897–899. doi: 10.1126/science.7846533. [DOI] [PubMed] [Google Scholar]
- 21.Karyagina A. Specific binding of sso II DNA methyltransferase to its promoter region provides the regulation of sso II restriction-modification gene expression. Nucleic Acids Res. 1997;25:2114–2120. doi: 10.1093/nar/25.11.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bogdanova E. Transcription regulation of the type II restriction-modification system AhdI. Nucleic Acids Res. 2008;36:1429–1442. doi: 10.1093/nar/gkm1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vijesurier R.M. Role and mechanism of action of C. PvuII, a regulatory protein conserved among restriction-modification systems. J. Bacteriol. 2000;182:477–487. doi: 10.1128/jb.182.2.477-487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson G.G. Organization of restriction-modification systems. Nucleic Acids Res. 1991;19:2539–2566. doi: 10.1093/nar/19.10.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pingoud A. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
- 26.Som S., Friedman S. Autogenous regulation of the EcoRII methylase gene at the transcriptional level: effect of 5-azacytidine. EMBO J. 1993;12:4297–4303. doi: 10.1002/j.1460-2075.1993.tb06114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler D., Fitzgerald G.F. Transcriptional analysis and regulation of expression of the ScrFI restriction-modification system of Lactococcus lactis subsp. cremoris UC503. J. Bacteriol. 2001;183:4668–4673. doi: 10.1128/JB.183.15.4668-4673.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christensen L.L., Josephsen J. The methyltransferase from the LlaDII restriction-modification system influences the level of expression of its own gene. J. Bacteriol. 2004;186:287–295. doi: 10.1128/JB.186.2.287-295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beletskaya I.V. DNA methylation at the CfrBI site is involved in expression control in the CfrBI restriction-modification system. Nucleic Acids Res. 2000;28:3817–3822. doi: 10.1093/nar/28.19.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zakharova M. Regulation of RNA polymerase promoter selectivity by covalent modification of DNA. J. Mol. Biol. 2004;335:103–111. doi: 10.1016/j.jmb.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 31.Knowle D. Nature of the promoter activated by C.PvuII, an unusual regulatory protein conserved among restriction-modification systems. J. Bacteriol. 2005;187:488–497. doi: 10.1128/JB.187.2.488-497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin R.N., McGeehan J.E., Kneale G. Structural and mutagenic analysis of the RM controller protein C.Esp1396I. PLoS One. 2014;9:e98365. doi: 10.1371/journal.pone.0098365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ball N.J. The structural basis of differential DNA sequence recognition by restriction-modification controller proteins. Nucleic Acids Res. 2012;40:10532–10542. doi: 10.1093/nar/gks718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawaya M.R. Crystal structure of the restriction-modification system control element C.Bcll and mapping of its binding site. Structure. 2005;13:1837–1847. doi: 10.1016/j.str.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 35.Stayrook S. Crystal structure of the lambda repressor and a model for pairwise cooperative operator binding. Nature. 2008;452:1022–1025. doi: 10.1038/nature06831. [DOI] [PubMed] [Google Scholar]
- 36.Mruk I., Rajesh P., Blumenthal R.M. Regulatory circuit based on autogenous activation-repression: roles of C-boxes and spacer sequences in control of the PvuII restriction-modification system. Nucleic Acids Res. 2007;35:6935–6952. doi: 10.1093/nar/gkm837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorokin V., Severinov K., Gelfand M.S. Large-scale identification and analysis of C-proteins. Methods Mol. Biol. 2010;674:269–282. doi: 10.1007/978-1-60761-854-6_17. [DOI] [PubMed] [Google Scholar]
- 38.Cesnaviciene E. Esp1396I restriction-modification system: structural organization and mode of regulation. Nucleic Acids Res. 2003;31:743–749. doi: 10.1093/nar/gkg135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mruk I., Blumenthal R.M. Real-time kinetics of restriction-modification gene expression after entry into a new host cell. Nucleic Acids Res. 2008;36:2581–2593. doi: 10.1093/nar/gkn097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prakash-Cheng A., Chung S.S., Ryu J. The expression and regulation of hsdK genes after conjugative transfer. Mol. Gen. Genet. 1993;241:491–496. doi: 10.1007/BF00279890. [DOI] [PubMed] [Google Scholar]
- 41.Prakash-Cheng A., Ryu J. Delayed expression of in vivo restriction activity following conjugal transfer of Escherichia coli hsdK (restriction-modification) genes. J. Bacteriol. 1993;175:4905–4906. doi: 10.1128/jb.175.15.4905-4906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edn. NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 43.Edelstein A. Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. 2010 doi: 10.1002/0471142727.mb1420s92. Chapter 14, Unit 14.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindelin J. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trueba F.J., Woldringh C.L. Changes in cell diameter during the division cycle of Escherichia coli. J. Bacteriol. 1980;142:869–878. doi: 10.1128/jb.142.3.869-878.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reyes-Lamothe R. High-copy bacterial plasmids diffuse in the nucleoid-free space, replicate stochastically and are randomly partitioned at cell division. Nucleic Acids Res. 2014;42:1042–1051. doi: 10.1093/nar/gkt918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swain P.S., Elowitz M.B., Siggia E.D. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12795–12800. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baev M.V. Growth of Escherichia coli MG1655 on LB medium: determining metabolic strategy with transcriptional microarrays. Appl. Microbiol. Biotechnol. 2006;71:323–328. doi: 10.1007/s00253-006-0392-8. [DOI] [PubMed] [Google Scholar]
- 50.Sezonov G., Joseleau-Petit D., D'Ari R. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 2007;189:8746–8749. doi: 10.1128/JB.01368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shea M.A., Ackers G.K. The OR control system of bacteriophage lambda. A physical-chemical model for gene regulation. J. Mol. Biol. 1985;181:211–230. doi: 10.1016/0022-2836(85)90086-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.