


Abstract
We have developed a new, sequence-specific DNA labeling strategy that will dramatically improve DNA mapping in complex and structurally variant genomic regions, as well as facilitate high-throughput automated whole-genome mapping. The method uses the Cas9 D10A protein, which contains a nuclease disabling mutation in one of the two nuclease domains of Cas9, to create a guide RNA-directed DNA nick in the context of an in vitro-assembled CRISPR-CAS9-DNA complex. Fluorescent nucleotides are then incorporated adjacent to the nicking site with a DNA polymerase to label the guide RNA-determined target sequences. This labeling strategy is very powerful in targeting repetitive sequences as well as in barcoding genomic regions and structural variants not amenable to current labeling methods that rely on uneven distributions of restriction site motifs in the DNA. Importantly, it renders the labeled double-stranded DNA available in long intact stretches for high-throughput analysis in nanochannel arrays as well as for lower throughput targeted analysis of labeled DNA regions using alternative methods for stretching and imaging the labeled long DNA molecules. Thus, this method will dramatically improve both automated high-throughput genome-wide mapping as well as targeted analyses of complex regions containing repetitive and structurally variant DNA.
INTRODUCTION
Two of the major challenges in genome analysis are de novo genome sequence assembly based on ‘short read’ shotgun sequencing and structural variation analysis. Several approaches and combinations of different approaches have been attempted to meet these challenges. The most widely adopted strategy relies on deep sequencing of shotgun libraries and sequencing of mate-pair libraries, which increases the sequence contiguity of short-read sequencing (1). The paired sequencing approach includes conventional mate-pair libraries, labor-intensive fosmid or BAC clone libraries (2), Hi-C read-pairs for chromosome-scale scaffolding (3) and transposase-mediated libraries (4). Another approach relies on the stochastic separation of corresponding genomic or polymerase chain reaction (PCR) fragments into physically distinct pools followed by subsequent fragmentation to generate shorter sequencing template (5–8). With appropriate high-throughput reaction handling and barcoding, this strategy reduces the complexity and thus can improve the quality of assemblies. Longer-read sequencing technologies such as PacBio's SMRT and Oxford Nanopore sequencing promise to eventually further improve assembly contiguity. For example, SMRT sequencing has been successfully applied to closing some gaps and detecting some structural variations in the human reference genome (9). However, their high error rate, low throughput and high cost have thus far prevented widespread adoption.
None of the aforementioned approaches, however, adequately address the problems of long-range de novo assembly contiguity and validation, sequence mis-assembly in complex segmentally duplicated and repetitive regions, and structural variant detection and delineation. Whole genome mapping technologies have been developed for these purposes as complementary tools to provide scaffolds for genome assembly and structural variation analysis. Optical mapping, pioneered by David Schwartz and colleagues has been used to construct restriction maps for various genomes and has proven to be very useful in providing scaffolds for shotgun sequence assembly and detection of structural variations (10,11). More recently, we developed a highly-automated whole genome mapping in a nanochannel array (12,13).
Both of the above-described genome mapping strategies are based on mapping the distribution of short (from 6 bp to 8 bp) sequence motifs across the genome. However, the distribution of the sequence motifs is uneven at different genomic regions. Often, there are no appropriate sequence motifs in repetitive genomic regions, which results in large segments of the genome that cannot be mapped (14) (Figure 1A). Another challenge resides in detecting and typing structural variations or clinical diagnostics of specific structural variants. Target sequence-specific labeling of the structural variations is required to obtain accurate breaking points, but this cannot be achieved by sequence-motif mapping (Figure 1B).
Figure 1.
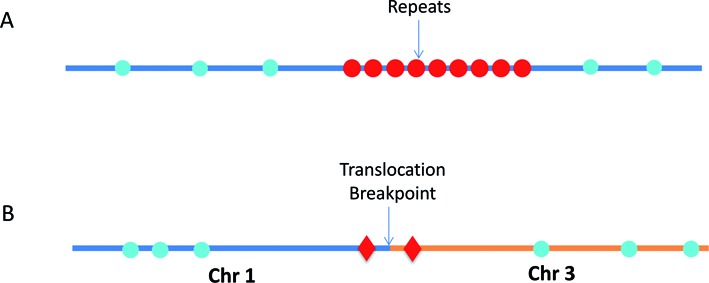
Schematics of sequence motif fluorescent DNA labeling and target sequence specific fluorescent DNA labeling. (A) The repetitive elements contain no recognition motifs for nicking enzymes (blue circles), and therefore this region is undetectable by conventional sequence-motif dependent nick-labeling. However, this region can be mapped by the new Cas9n fluorescent nick-labeling method (red circles). (B) The Cas9n fluorescent nick-labeling system can identify translocations by designing gRNAs to target Cas9n D10A to nick specific sequences near the breakpoints (Red diamonds). These translocation breakpoint regions can then be precisely mapped by visualizing the targeted Cas9n fluorescent nick-labeling system-dependent signals relative to the conventional sequence motif-label-dependent bar codes generated on the breakpoint-adjacent large DNA segments (blue circles).
Recently, a new genome editing tool based on a bacterial CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-associated protein-9 nuclease (Cas9) from Streptococcus pyogenes has been developed for generating double strand DNA breaks in vivo (15). To achieve site-specific DNA recognition and cleavage, the protein Cas9 must form a complex with a duplex consisting of a crRNA and a trans-activating crRNA (tracrRNA), which is partially complementary to the crRNA. The HNH and RuvC-like nuclease domains of Cas9 cut both DNA strands, generating double-stranded breaks (DSBs) at sites defined by a 20-nucleotide seed sequence within an associated crRNA transcript (16,17). Mutations of both sites generate nuclease-deficient Cas9 (dCas9) that is still capable of binding to the crRNA:tracrRNA duplex and moving to the target sequence (18), and has been used to visualize repetitive DNA sequences (19,20). A mutant form known as Cas9 D10A, which lacks just the RuvC-like nuclease domain activity, only nicks the DNA strand complementary to its crRNA, and is characterized as a Cas9 nickase (Cas9n). This mutant of Cas9 has been used with paired singled guide RNA (sgRNA) targeting opposite strands of the same locus to generate DSBs with great precision (16,21). Here, we adapt the Cas9n-dependent nicking protocol to fluorescently label specific sequences for whole genome mapping through in vitro nick-labeling. Such Cas9n fluorescent nick-labeling based sequence-specific labeling methods can be used to target repetitive regions which often lack appropriate restriction site motifs. This method can also help to precisely map the breaking points of structural variations such as translocations, by designing guide RNAs (gRNAs) to recognize and direct labeling of sequences near these breakpoints prior to high throughput single molecule analysis (Figure 1B). Novel combinations of conventional sequence-motif mapping and target-specific mapping will unleash the full potential of our nanochannel array-based genome mapping method (12,13).
MATERIALS AND METHODS
DNA samples
Target sequence-specific labeling with Cas9n fluorescent nick-labeling was carried out on the BAC clone CH17–353B19, fosmids carrying cloned telomere-terminal DNA fragments ending in several hundred bases of (TTAGGG)n (Stong et al., 2014), an HIV-1 entire genome-containing plasmid pEcoHIV-NL4–3-eLuc (gift from Dr Won-Bin Young at University of Pittsburgh) and genomic DNA isolated from human B-Lymphocyte cells NA12878 (Corriel Research Institute, NJ, USA).
Guide RNA preparation
The seed sequence of 20 nucleotides complementary to the 3′–5′ strand of the target template DNA were designed via a gRNA design tool (Feng Lab CRISPR Design Web Tool at http://crispr.mit.edu.). Each seed sequence was incorporated into the crRNA. Two crRNAs for the genomic sequences of DUF1220 domain (in BAC clone), 1 for the telomere repeat sequence (TTAGGG)n and 7 for subtelomeric sequences, along with the universal tracrRNA, were synthesized by GE Dharmacon. The fosmid and CH17–353B19 gRNAs were created by pre-incubating the tracrRNA (0.5 nmol) and corresponding crRNA (0.5 nmol) with 1X NEB Buffer 3 and 1X BSA at 4°C for 30 min.
Three single guide RNAs (sgRNAs) containing seed sequence targeting HIV-1 structural gene regions (Gag, Pol and Env) were designed for efficiency and specificity using bioinformatics analysis tools. All the oligonucleotides for each target sequence (Table 1) were synthesized in Alpha DNA (Montreal, Canada) and cloned into pKLV-WG-sgRNA vector modified from pKLV-U6gRNA(BbsI)-PGKpuro2ABFP vector, a gift from Kosuke Yusa (Addgene plasmid # 50946) (21). The vector was digested with BbsI and treated with Antarctic Phosphatase, and the linearized vector was purified with the QIAquick nucleotide removal kit (QIAGEN). A pair of oligonucleotides for each targeting site was annealed, phosphorylated and ligated to the linearized vector. The sgRNA expression cassette was validated by sequencing with U6 sequencing primer in GENEWIZ. The validated vector was used as template for PCR with forward T7-U6 and reverse sgRNA primer to generate T7 promoter-driven gRNA expression cassette. Then the sgRNA for each target was in vitro transcribed using MEGAshortscript™ T7 transcription kit (Life Technology). The quality of HIV-1 sgRNAs was verified by electrophoresis in 5% denaturing polyacrylamide gel.
Table 1. Sequences for gRNAs.
| Loci | Loci Number | Sequence 5′- 3′ | Type |
|---|---|---|---|
| DUF1220 | 1–12 | AAGUUCCUUUUAUGCAUUGG | gRNA |
| HIV plasmid | 1 | CACCGCAGGATATGTAACTGACAG | sgRNA |
| 2 | CACCGGCCAGATGAGAGAACCAAG | sgRNA | |
| 3 | CACCGAGAGTAAGTCTCTCAAGCGG | sgRNA | |
| Chr1q telomere | 1 | UUAGGGUUAGGGUUAGGGUU | gRNA |
| Chr1q subtelomere | 1 | CCCCUGUUGCCAGAGCCAGU | gRNA |
| 2 | GUAUUUAGUCAGAGGGCUAG | gRNA | |
| 3 | AUACAGUAGGAUAACCGCAA | gRNA | |
| Chr15q subtelomere | 1 | ACCUUGCUACCACGAGAGCA | gRNA |
| 2 | UCCAUUGGUUUAAUUAGGAA | gRNA | |
| Chr11q subtelomere | 1 | GGUCCACCCUACAGAUGUGC | gRNA |
| 2 | AGAUCAGCAGCCACGUGUGC | gRNA | |
| Chr12p subtelomere | 1 | ACCUUGCUACCACGAGAGCA | gRNA |
| 2 | UCCAUUGGUUUAAUUAGGAA | gRNA | |
| Alu | 1 | UGUAAUCCCAGCACUUUGGG | gRNA |
Cas9n fluorescent nick-labeling of fosmids, HIV-1 plasmid and BAC clone CH17–353B19
The gRNAs or sgRNAs(5 μM) were incubated with 600 ng of Cas9n D10A (PNA Bio Inc), 1X NEB Buffer 3 and 1X BSA (NEB) at 37°C for 15 min. The DNA (500ng) was added to the mixture and incubated at 37°C for 60 min. The nicked DNA was then labeled with 4.12 units of DNA Taq Polymerase (NEB), 0.1 μM of ATTO- 532 dUTP dAGC and 1X Thermopol Buffer (NEB) at 72°C 60 min. The labeled fosmids and BACs were cut and linearized with 5 units of NotI enzyme (NEB) at 37°C for 60 min. The labeled pecoHIV-NL4–3-eLuc plasmid (17,099 bp) was digested with 20 units of a unique restriction enzyme EcoRI (at 5744 bp) (NEB). NotI and EcoRI were inactivated at 65°C for 20 min.
The two color genome mapping with Cas9n fluorescent nick-labeling and sequence-motif labeling
After nicking with Cas9n D10A as previously described in the Cas9n fluorescent nick-labeling section, the sample was digested with RNAseA (190 ng/μL, QIAGEN) at 37°C for 20 min. After digestion, the sample was labeled with ATTO 532-dATP, dTGC (100 nM) and 2.5 units of DNA Taq Polymerase (NEB) in the presence of 1X Thermopol Buffer (NEB) at 72°C for 1 h. The sample was treated with 1 unit of SAP (USB Products) and RNAseA (100 ng/μL) at 37°C for 20 min and then 65°C for 15 min. The nicks were repaired with 500 μM NAD+, 100 nM dNTPs and 20 kU of Taq DNA Ligase at 45°C for 20 min. The sample was then treated with 6 mAU of QIAGEN Protease at 56°C for 10 min and 70°C for 15 min. The sample was dialyzed in TE on a 0.1 μm membrane (Millipore) for 2 h. After dialysis, the sample was nicked with 10 units of Nt. BspQI (NEB) at 72°C for 2 h. The nicked DNA was then labeled with 2.5 units of Taq DNA Polymerase (NEB), 0.1 μM ATTO-647 dUTP dAGC and 1X Thermopol Buffer (NEB) for 60 min at 72°C. The DNA backbone was stained with YOYO-1, and is shown in blue in all figures. The stained samples were loaded and imaged inside the nanochannels following the established protocol (13).
Data analysis
We calculated the distances between spots using ImageJ. The histogram of the label distributions were plotted in Excel. If the pattern matched the predicted pattern we considered the labels as true positives. Missing labels were used for the calculation of labeling efficiency and the extra labels were used for calculating the false positive percentage.
RESULTS AND DISCUSSIONS
We incorporated the CRISPR-Cas9 technology into our nick-labeling procedure for targeted, sequence-specific nicking and fluorescent labeling in one color, followed by global nickase enzyme motif-dependent labeling in a second color. We used two different forms of guide RNAs. For the HIV- plasmid experiments, the target sequence was cloned, amplified and then in vitro transcribed to result in an expressed single guide RNA (sgRNA). For all other experiments, the CRISPR RNAs (crRNAs) and trans-activating CRISPR RNA (tracrRNA) were purchased from GE Dharmacon and pre-incubated to form each guide RNA (gRNA). In both cases, the gRNA-directed Cas9n D10A makes a precise single cut in one strand of the target double-strand DNA three nucleotides upstream of a protospacer adjacent motif (PAM) and fluorescent nucleotides are directly incorporated to these specific nick sites using Taq DNA Polymerase (Figure 2). This approach promises to dramatically improve DNA mapping in complex and structurally variant genomic regions, as well as facilitate high-throughput automated whole-genome mapping.
Figure 2.
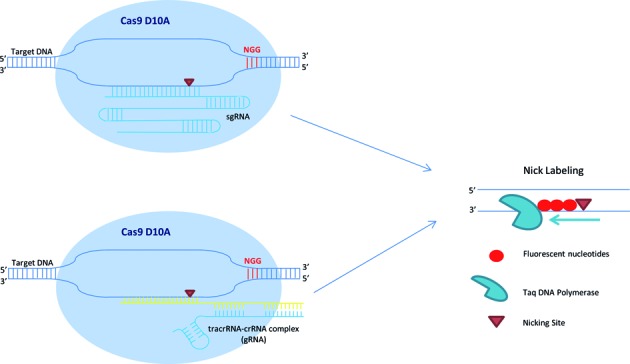
Schematics of Cas9n/gRNA target sequence specific fluorescent labeling for whole genome DNA mapping. The Cas9n fluorescent nick-labeling system uses a guide RNA (gRNA) to direct the Cas9 nuclease to a targeted site. The gRNA is composed of a trans-activating crRNA (tracrRNA) and a crRNA that contains a 20 nucleotide sequence that is complementary to the site of interest. A mutation in the RuvC-like domain nuclease alters the Cas9 enzyme to make only a single cut three nucleotides upstream of a protospacer adjacent motif (PAM) of the 3′–5′ strand of the target DNA. In the nick labeling method, after Cas9n D10A generates a nick, fluorophores are directly incorporated to the nick sites using Taq DNA Polymerase. These fluorophores can be detected using fluorescence microscopy.
We first established the Cas9n fluorescent nick-labeling conditions and investigated the labeling efficiency with BAC clones, fosmids and plasmids as model systems. Figure 3A shows the Cas9n fluorescent nick-labeling results of HLS DUF1220 triplets on a BAC clone. The histogram of the label distribution is shown in the bottom graph of Figure 3A. Genome sequences encoding DUF1220 protein domains have undergone an exceptional human lineage-specific (HLS) increase in copy number, which is implicated in human brain size, pathology and evolution (22). A single copy of HLS DUF1220 triplet spans about 4.7 kb, and there are 12 copies on the BAC clone CH17–353B19. A gRNA probe was designed to target one unit of the triplets. Clearly, there are 12 copies detected on this 240 kb BAC clone and the distance between the each triplet measures 4.7 kb, which is in a good agreement with the clone sequence. The labeling efficiency of each locus was determined by evaluating 192 labeled BAC molecules, ranging from 87% to 98%. Interestingly, the middle copies (#6–8 labels from the left most label in Figure 3A) are labeled at lower labeling efficiency. The Cas9n fluorescent nick-labeling is very specific.
Figure 3.
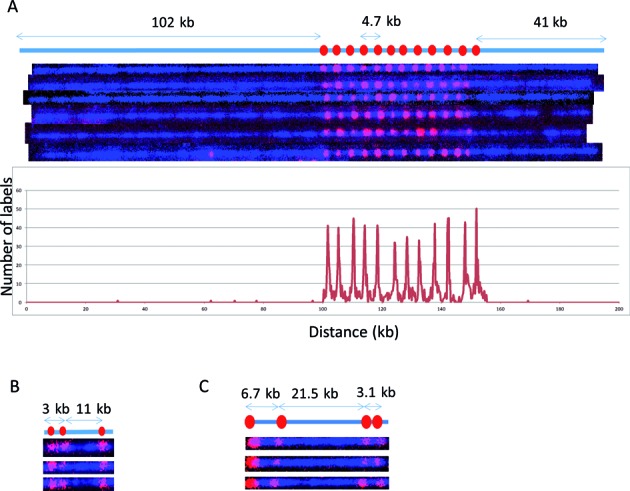
Cas9n-nick labeling. The expected labeling pattern and distances between labels is shown above corresponding fluorescent microscopy images. (A) A single copy of HLS DUF1220 triplet is approximately 4.7 kb, which cannot be identified with the sequence-motif labeling method due to lack of recognition sites within the repeat region. The Cas9n fluorescent nick-labeling method was able to label this region using a gRNA designed to the DUF1220 triplet. These recognition sites are labeled with red labels. The tandem repeats are separated approximately 4.7 kb as predicted. The histogram of the DUF1220 gRNA labels is shown below the molecules. A total of 192 molecules were evaluated for the labeling efficiency. (B) Three sgRNAs were designed to target three different locations (Gag, Pol, Env) within the HIV-1 genome. The sites were correctly labeled with the expected distances between each sgRNA. The image shows the linearized DNA after EcoRI digestion. (C) Three gRNAs designed for the chromosome 1q subtelomere generated the expected pattern. A fourth gRNA, targeting the repetitive telomere sequence (TTAGGG)n, correctly labeled the telomere region on the left end of this telomere-terminal 1q DNA fragment.
The extra labels outside of the DUF domain were used to calculate the false positives in Figure 3A. The spurious labels inside the DUF domain are harder to define because the DNA molecules inside the nanochannel are not static. Their slight movements and the limitation of optical resolution may cause inaccurate measurement of 4.7 kb repetitive sequences during the imaging time (200ms). When all of the spurious spots inside and outside of the DUF domain were used, the false positive percentage is 0.6%.
Next, we applied the Cas9n fluorescent nick-labeling method to a plasmid containing the HIV-1 genome. HIV-1 integrates into the human genome at different locations (23), each of which may be identifiable directly through an appropriate integration site labeling and global genome mapping strategy. Identification of such sites in HIV-1 latent cells is essential for understanding the molecular and epigenetic mechanisms underlying HIV-1 latency (24). Similarly, this sequence-specific mapping approach could be used to identify lentivirus random integration sites in the host cells, which may disrupt both endogenous gene expression and vector gene expression patterns (25). Multiple sgRNAs were designed and tested to target HIV-1 structural region (Gag, Pol, Env) to determine the most effective gRNA that labels the HIV-1 genome. The sites were correctly labeled with the expected distances between each sgRNA (Figure 3B). However, the labeling efficiencies at each site of 36%, 58% and 44% from the left most label respectively, suggesting that labeling efficiency may be sequence- or region-dependent. It could also be the difference in labeling efficiency of sgRNA versus gRNA. The labeling efficiencies of the gRNAs were much higher than those achieved using sgRNAs. In a third model system, a fosmid containing a subtelomeric segment of human 1q ending in 100 bases of (TTAGGG)n was used to test the Cas9n fluorescent nick-labeling. We designed four guide RNA probes to target the (TTAGGG)n tract and three distinct loci on the subtelomere. The labeling pattern matches very closely with the positions of the gRNA seed sequences in the 1q reference sequence (Figure 3C). However, the labeling efficiency is relatively lower for the telomere with 30%, while the labeling efficiency of subtelomeric markers are 95%,79% and 99% respectively from the left most left label on 1q (Figure 3C). This may be due to non-Watson-Crick pairings of the hexameric repeats, the high G+C content or the secondary structure of guide RNA probes targeting the (TTAGGG)n repeat.
We next tested the combination of Cas9n fluorescent nick-labeling with nicking endonuclease based sequence motif labeling. This approach has the potential to find wide applications in whole genome mapping of repetitive sequences as well as genotyping of structural variations and identification/mapping of viral integration sites. In Figure 4A, the DUF1220 triplet repeats were first labeled with Cas9n fluorescent nick-labeling of red fluorescent nucleotides. These labeled DNA molecules were then globally nick-labeled with green nucleotides using Nt.BspQI to target the GCTCTTC motif. Twelve copies of DUF1220 triplets were detected spanning about 52 kb. Only the flanking regions of this 52 kb were shown to have the GCTCTTC motif, which can be mapped to reference genome to indicate the genomic locations of the DUF1220 triplet array. The histogram of the label distribution is shown in the bottom graph of Figure 4A. Clearly, the combination of Cas9n fluorescent nick-labeling and nicking endonuclease sequence motif labeling, not only can detect the copy numbers of the DUF1220 triplets, but also can map the locations of the repeats.
Figure 4.
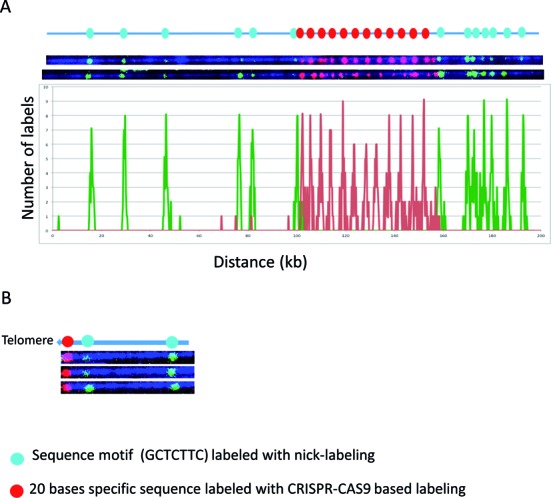
The combination of Cas9n sequence specific and nicking motif labeling. The Cas9n fluorescent nick-labeling system is effective at labeling specific repetitive sequences and locating them relative to flanking large DNA segments labeled using conventional sequence-motif labeling. The expected labeling pattern and distances between labels is shown above corresponding fluorescent microscopy images. (A) The Cas9n fluorescent nick-labeling method was used to label the repetitive sequences the DUF1220 triplet and is shown with red labels. These molecules were then labeled with the conventional sequence-motif labeling method with Nt.BspQI (green labels). The histogram of the labels is shown below the molecules. The red peaks represent the DUF gRNA labels and the green the Nt.BspQI labeling sites. A total of 43 molecules were evaluated for the labeling efficiency. (B) The Cas9n fluorescent nick-labeling method was used to label the repetitive telomere sequences at the left end of these telomere-terminal DNA fragments (red labels). The sequence-motif labeling method with Nt.BspQI was performed after Cas9n/gRNA system labeling of telomeres, and is displayed with green labels.
The same approach was also applied to measure the telomere repeat length of a telomere-terminal fragment of chromosome 8q cloned in a fosmid. This fosmid carries 800 bp of the repetitive (TTAGGG)n sequence, which lacks motif nicking sites recognized by currently available nicking endonucleases and therefore cannot be labeled with current sequence-motif based methods. A gRNA specific for the telomere was designed. In Figure 4B, the telomere was first labeled with Cas9n fluorescent nick-labeling of red fluorescent nucleotides. The labeled DNA molecules were then labeled with green nucleotides by Nt.BspQI nick-labeling to target the GCTCTTC motif. The telomere was correctly labeled on the end of the sequence. The length of this telomere region can be determined by measuring the length of the red fluorophore region and the intensity of its fluorescence relative to known controls after imaging. Sequence-motif labeling over extended subtelomere regions linked on single large DNA molecules to the Cas9n fluorescent nick-labeling (TTAGGG)n can be used to identify the specific subtelomere by comparison with genome-wide maps.
Cas9n fluorescent nick-labeling can be used to create locus-specific and variant-specific barcodes. We designed gRNAs to create barcodes to distinguish individual subtelomeres linked on single molecules to (TTAGGG)n tracts (Figure 5A, Chr 1q and Chr 11q) and to distinguish variant copies of highly similar subtelomeric segmental duplications (Chr 15q and Chr 12p) using the Cas9n sequence specific labeling methods. It has been suggested that the shortest telomere or a small subset of the shortest telomeres in a cell determines the onset of senescence, apoptosis or genome instability (26,27), the ability to systematically measure individual dysfunctionally short (TTAGGG)n tracts (rather than average telomere tract lengths in a sample as is typically measured currently) would provide important new high-resolution information on specific telomere functional status and identity at the single-molecule level. On the telomeric fosmids from Chr 15q and Chr 12p (Figure 5A), Cas9n-gRNA directed nicks at two loci in a segmentally duplicated subtelomere region containing the WASH gene family (Linnardopoulou et al., 2007) were labeled in red. The same pair of gRNAs generated signals separated by 5.87 kb on the 15q fosmid and 7.5 kb on Chr 12p fosmid, as predicted by the reference sequences of these two highly similar but structurally non-identical regions. The patterns generated by three target-specific gRNAs on Chr1q and two gRNAs on 11q are unique, easily distinguishable from each other and from both of the WASH gene-related patterns, and correspond exactly to what is expected from their respective reference sequences. These initial experiments indicate that specific gRNAs can be pooled to generate multiple specific nicks that, when labeled, can create custom barcodes predicted precisely by the respective reference sequences.
Figure 5.
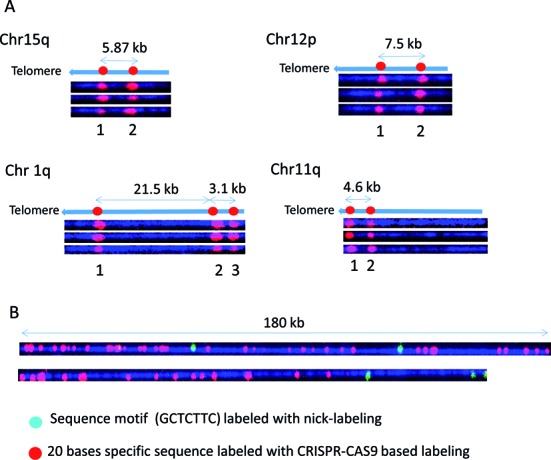
Locus-specific and variant-specific barcodes based on Cas9n labeling. Cas9n fluorescent nick-labeling creates locus-specific and variant-specific barcodes. The expected labeling pattern and distances between labels is shown above corresponding fluorescent microscopy images. (A) For each of the tested subtelomere regions specific gRNAs were designed and pooled in the in vitro nicking reaction mix in order to produce a unique pattern. In each instance, the expected pattern of the red fluorophores in the Cas9n fluorescent nick-labeling system was obtained. (B) Human genomic DNA was labeled with Alu gRNAs, followed by conventional sequence-motif labeling with Nt.BspQI. Conventional sequence-motif dependent labeling sites (green labels) are found infrequently within these genomic DNA regions. However, Alu-specific gRNAs direct Cas9n D10A to generate information-rich long-range barcoding patterns (red labels).
Figure 5B shows the results of experiments combining the Cas9n fluorescent nick-labeling and nicking endonuclease sequence motif labeling to map Alu elements in the human genome. Alu sequences, with about one million copies, are the most abundant retroelements in humans, and account for up 10% of the human genome. These SINE (Short Interspersed Nuclear Elements) sequences, exclusively found in primate genomes, have been particularly active in the human lineage even after human–chimpanzee divergence, where they are theorized to have contributed to some of the human-specific characteristics such as brain size (28). A gRNA sequence was designed to target 280 000 Alu sites out of one million copies. Typical genomic DNA molecules are imaged and shown in Figure 5B. One 180 kb molecule displays dense Alu elements with only two GCTCTTC motifs. Another DNA molecule shows dense Alu elements with two GCTCTTC motifs. The combined information can be used to map the DNA molecules to the reference genome and profile the distribution of Alu elements on the whole genome scale.
CONCLUSION
We have demonstrated the capabilities of our integrated approach of sequence motif fluorescent tagging and sequence specific labeling with Cas9n fluorescent nick-labeling system. The flexible and efficient fluorescent tagging of specific sequences allow us to obtain context specific sequence information along the long linear DNA molecules in the BioNano Genomics nanochannel. Our global nick-labeling scheme tags short recognition sequences, whose spatial relation can be translated into a genomic map. Not only can this integrated fluorescent DNA double strand labeling make the whole genome mapping more accurate, and provide more information, but it can also specifically target certain loci for clinical testing. Importantly, it renders the labeled double-stranded DNA available in long intact stretches for high-throughput analysis in nanochannel arrays as well as for lower throughput targeted analysis of labeled DNA regions using alternative methods for stretching and imaging the labeled large DNA molecules. Thus, this method will dramatically improve both automated high-throughput genome-wide mapping as well as targeted analyses of complex regions containing repetitive and structurally variant DNA.
Acknowledgments
CH17–353B19 BAC clone is a gift from Dr Pui-Yan Kwok of University of California at San Francisco. The gRNA targeting the Alu elements was designed by Dr Michal Sakin at University of California at San Francisco.
FUNDING
National Institutes of Health [HG005946 to Pui-Yan Kwok and M.X, CA177395 to H.R and M.X]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Siegel A.F., van den Engh G., Hood L., Trask B., Roach J.C. Modeling the feasibility of whole genome shotgun sequencing using a pairwise end strategy. Genomics. 2000;68:237–246. doi: 10.1006/geno.2000.6303. [DOI] [PubMed] [Google Scholar]
- 2.Gnerre S., MacCallum I., Przybylski D., Ribeiro F.J., Burton J.N., Walker B.J., Sharpe T., Hall G., Shea T.P., Sykes S., et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton J.N., Adey A., Patwardhan R.P., Qiu R., Kitzman J.O., Shendure J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adey A., Kitzman J.O., Burton J.N., Daza R., Kumar A., Christiansen L., Ronaghi M., Amini S., Gunderson K.L., Steemers F.J., et al. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 2014;24:2041–2049. doi: 10.1101/gr.178319.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaper F., Swamy S., Klotzle B., Munchel S., Cottrell J., Bibikova M., Chuang H.-Y., Kruglyak S., Ronaghi M., Eberle M.A., et al. Whole-genome haplotyping by dilution, amplification, and sequencing. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5552–5557. doi: 10.1073/pnas.1218696110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuleshov V., Xie D., Chen R., Pushkarev D., Ma Z., Blauwkamp T., Kertesz M., Snyder M. Whole-genome haplotyping using long reads and statistical methods. Nat. Biotechnol. 2014;32:261–266. doi: 10.1038/nbt.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters B.A., Kermani B.G., Sparks A.B., Alferov O., Hong P., Alexeev A., Jiang Y., Dahl F., Tang Y.T., Haas J., et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487:190–195. doi: 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voskoboynik A., Neff N.F., Sahoo D., Newman A.M., Pushkarev D., Koh W., Passarelli B., Fan H.C., Mantalas G.L., Palmeri K.J., et al. The genome sequence of the colonial chordate, Botryllus schlosseri. Elife. 2013;2:e00569. doi: 10.7554/eLife.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaisson M.J.P., Huddleston J., Dennis M.Y., Sudmant P.H., Malig M., Hormozdiari F., Antonacci F., Surti U., Sandstrom R., Boitano M., et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015;517:608–611. doi: 10.1038/nature13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samad A., Huff E.J., Cai W.W., Schwartz D.C. Optical mapping - a novel, single-molecule approach to genomic analysis. Genome Res. 1995;5:1–4. doi: 10.1101/gr.5.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Teague B., Waterman M.S., Goldstein S., Potamousis K., Zhou S., Reslewic S., Sarkar D., Valouev A., Churas C., Kidd J.M., et al. High-resolution human genome structure by single-molecule analysis. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10848–10853. doi: 10.1073/pnas.0914638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hastie A.R., Dong L., Smith A., Finklestein J., Lam E.T., Huo N., Cao H., Kwok P.-Y., Deal K.R., Dvorak J., et al. Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLos One. 2013;8:e55864. doi: 10.1371/journal.pone.0055864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam E.T., Hastie A., Lin C., Ehrlich D., Das S.K., Austin M.D., Deshpande P., Cao H., Nagarajan N., Xiao M., et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012;30:771–776. doi: 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feuk L., Carson A.R., Scherer S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 15.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.-W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anton T., Bultmann S., Leonhardt H., Markaki Y. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus. 2014;5:163–172. doi: 10.4161/nucl.28488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koike-Yusa H., Li Y., Tan E.P., Velasco-Herrera Mdel C., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 22.Dumas L.J., O'Bleness M.S., Davis J.M., Dickens C.M., Anderson N., Keeney J.G., Jackson J., Sikela M., Raznahan A., Giedd J., et al. DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am. J. Hum. Genet. 2012;91:444–454. doi: 10.1016/j.ajhg.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroder A.R.W., Shinn P., Chen H.M., Berry C., Ecker J.R., Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 24.Cohn L.B., Silva I.T., Oliveira T.Y., Rosales R.A., Parrish E.H., Learn G.H., Hahn B.H., Czartoski J.L., McElrath M.J., Lehmann C., et al. HIV-1 integration landscape during latent and active infection. Cell. 2015;160:420–432. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desfarges S., Ciuffi A. Retroviral integration site selection. Viruses. 2010;2:111–130. doi: 10.3390/v2010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaul Z., Cesare A.J., Huschtscha L.I., Neumann A.A., Reddel R.R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012;13:52–59. doi: 10.1038/embor.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou Y., Sfeir A., Gryaznov S.M., Shay J.W., Wright W.E. Does a sentinel or a subset of short telomeres determine replicative senescence. Mol. Biol. Cell. 2004;15:3709–3718. doi: 10.1091/mbc.E04-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Britten R.J. Transposable element insertions have strongly affected human evolution. Proc. Natl. Acad. Sci. U.S.A. 2010;107:19945–19948. doi: 10.1073/pnas.1014330107. [DOI] [PMC free article] [PubMed] [Google Scholar]