


Abstract
Cellular RNA labeling strategies based on bioorthogonal chemical reactions are much less developed in comparison to glycan, protein and DNA due to its inherent instability and lack of effective methods to introduce bioorthogonal reactive functionalities (e.g. azide) into RNA. Here we report the development of a simple and modular posttranscriptional chemical labeling and imaging technique for RNA by using a novel toolbox comprised of azide-modified UTP analogs. These analogs facilitate the enzymatic incorporation of azide groups into RNA, which can be posttranscriptionally labeled with a variety of probes by click and Staudinger reactions. Importantly, we show for the first time the specific incorporation of azide groups into cellular RNA by endogenous RNA polymerases, which enabled the imaging of newly transcribing RNA in fixed and in live cells by click reactions. This labeling method is practical and provides a new platform to study RNA in vitro and in cells.
INTRODUCTION
Chemical modification of RNA has become indispensable in the study of its structure and function and in the development of nucleic acid-based diagnostic and therapeutic tools (1,2). Typically, RNA labeling strategies based on solid-phase chemical synthesis and enzymatic methods are sufficient for most in vitro applications. However, analogous labeling strategies for cellular RNA are much less developed. In particular, paucity of efficient RNA imaging tools has been a major impediment in the study of cellular RNA biogenesis, localization and degradation, a combination of processes that tightly regulates gene expression (3). Methods to visualize RNA commonly rely on metabolic labeling of RNA with ribonucleoside or ribonucleotide analogs such as BrU or BrUTP followed by immunostaining with fluorescent antibody for BrU (4,5). However, these methods involve laborious assay setups and are not applicable to all cell types and tissue samples due to limited permeability of the antibodies. Endogenous RNA has also been visualized by using fluorescently-modified antisense oligonucleotide (ON) probes (6,7), molecular beacons (8), nucleic acid-templated reactions (9,10) and more recently, aptamer-binding fluorophores (11). Apart from synthetic challenges in preparing the ON probes, these methods also suffer from poor membrane permeability and short half-life of the probes and background fluorescence due to non-specific interactions (12).
Alternatively, postsynthetic functionalization by using chemoselective reactions (e.g. azide-alkyne cycloaddition, Staudinger ligation) has recently emerged as a valuable method to label glycans, proteins, lipids and nucleic acids for a variety of applications (13–18). In this methodology, a nucleoside containing an unnatural reactive group is incorporated into an ON sequence by chemical or enzymatic method. Further functionalization is achieved postsynthetically by performing a chemoselective reaction between the labeled ON and a probe containing the cognate reactive group. While DNA labeling and imaging techniques based on this strategy are well documented (19–27), postsynthetic RNA manipulations are less prevalent (28–30) as methods developed for DNA often do not work for RNA due to its inherent instability. Moreover, the azide group, which participates in a wide range of bioorthogonal reactions in comparison to alkyne functionality, cannot be easily incorporated into nucleic acids by solid-phase ON synthesis protocols because most azide substrates are unstable in solution and undergo Staudinger-type reaction with phosphoramidite substrates (31–33). Hence, except for a very few examples wherein the azide group has been incorporated into DNA (34,35), these procedures mostly use easily accessible alkyne-modified nucleic acids, thereby making this postsynthetic modification method one-dimensional (36–41).
Owing to these practical problems in current labeling procedures and paucity of efficient RNA imaging tools, we sought to develop a robust and modular labeling tool that would enable the study of RNA in vitro as well as in cells. Towards this endeavour, we have recently reported the effective incorporation of an azide group into short RNA ONs by in vitro transcription reactions using 5-azidopropyl-modified UTP analog (42). The azide-modified RNA ONs were suitable for posttranscriptional chemical modification by copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) and Staudinger reduction reactions (43). Encouraged by these results we wished to develop a small series of azide-modified nucleotide analogs, which would enable detailed investigation of the utility of our azide labeling technique to functionalize RNA with biophysical probes by CuAAC, copper-free strain-promoted azide-alkyne cycloaddition (SPAAC) and azide-phosphine Staudinger ligation reactions in vitro and in cells. Here, we describe the development of a versatile toolbox composed of azide-modified uridine triphosphates, which facilitates the direct incorporation of azide functionality into RNA transcripts by transcription reaction (Figure 1). The azide-modified RNA is readily functionalized with biophysical probes in a modular fashion by CuAAC, copper-free strain-promoted azide-alkyne cycloaddition (SPAAC) and azide-phosphine Staudinger ligation reactions. Importantly, we show for the first time the specific incorporation of azide groups into cellular RNA transcripts by endogenous RNA polymerases. The azide-labeled cellular RNA transcripts are conveniently visualized in fixed cells and live cells by fluorescence microscopy upon click reaction with fluorescent alkynes in the presence and absence of a copper catalyst.
Figure 1.
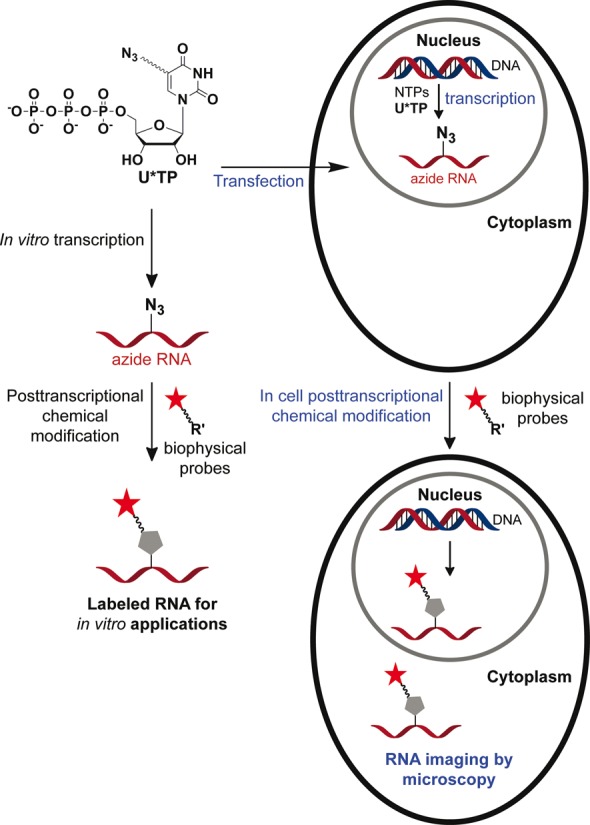
Schematic diagram illustrating the posttranscriptional chemical labeling of RNA transcripts in vitro and in cells by using azide-modified UTP analogs.
MATERIALS AND METHODS
Enzymatic incorporation of azide-modified UTP 4 and 6
Transcription reactions with α-32P ATP
The promoter-template duplexes (5 μM) were assembled by heating T7 RNA polymerase consensus promoter DNA sequence and DNA template (T1–T5) in TE buffer (10 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, pH 7.8) at 90°C for 3 min. The solution was allowed to come to room temperature slowly and then placed on an ice bath for 20 min, and stored at −40°C. The transcription reactions were carried out at 37°C in 40 mM Tris-HCl buffer (pH 7.9) containing 250 nM annealed promoter-template duplexes, 10 mM MgCl2, 10 mM NaCl, 10 mM of dithiothreitol (DTT), 2 mM spermidine, 1 U/μl RNase inhibitor (Riboblock), 1 mM GTP, CTP, UTP and or modified UTP 4/6, 20 μM ATP, 5 μCi α-32P ATP and 3 U/μl T7 RNA in a total volume of 20 μl. The reaction was quenched after 3.5 h by adding 20 μl of loading buffer (7 M urea in 10 mM Tris-HCl, 100 mM EDTA, 0.05% bromophenol blue, pH 8), heated for 3 min at 75°C followed by cooling the samples on an ice bath. The samples (4 μl) were loaded on a sequencing 18% denaturing polyacrylamide gel and electrophoresed. The radioactive bands were phosphorimaged and then quantified using the GeneTools software from Syngene to determine the relative transcription efficiencies. Relative percentage incorporation of azide-modified ribonucleoside triphosphates 4and6 into full-length transcripts has been reported with respect to the amount of full-length transcript formed in the presence of natural NTPs. All reactions were performed in duplicate and the errors in yields were ≤ 4%.
Large-scale transcription reactions
Large-scale transcription reactions were performed using DNA template T1. Each reaction (250 μl) was performed in the presence of 2 mM GTP, CTP, ATP, and modified UTP 4/5/6, 20 mM MgCl2, 0.4 U/μl RNase inhibitor (Riboblock), 300 nM annealed template and 800 units T7 RNA polymerase. After incubating for 12 h at 37°C, the reaction volume was reduced to 1/3 by speed vac. 50 μl of the loading buffer was added and the sample was loaded on a preparative 20% denaturing polyacrylamide gel. The gel was UV shadowed, appropriate band was cut out, extracted with 0.3 M sodium acetate and desalted using Sep-Pak classic C18 cartridge.
Enzymatic digestion of RNA ONs 7 and 9
Approximately 4 nmol of the modified oligoribonucleotide transcripts 7 and 9 were treated with snake venom phosphodiesterase I (0.01 U), calf intestinal alkaline phosphatase (10 μl, 1 U/μl) and RNase A (0.25 μg) in a total volume of 100 μl in 50 mM Tris-HCl buffer (pH 8.5, 40 mM MgCl2, 0.1 mM EDTA) at 37°C. ON 7 was incubated for 5 h and ONs 9 was incubated for 12 h. After this period, RNase T1 (0.2 U/μl) was added, and the samples were incubated for another 1 h (7) and 4 h (9) at 37°C (44). The ribonucleoside mixture obtained from the digest was analyzed by reversed-phase HPLC using Phenomenex-Luna C18 column (250 × 4.6 mm, 5 μm) at 260 nm. See Reference 43 for enzymatic digestion of RNA ON 8.
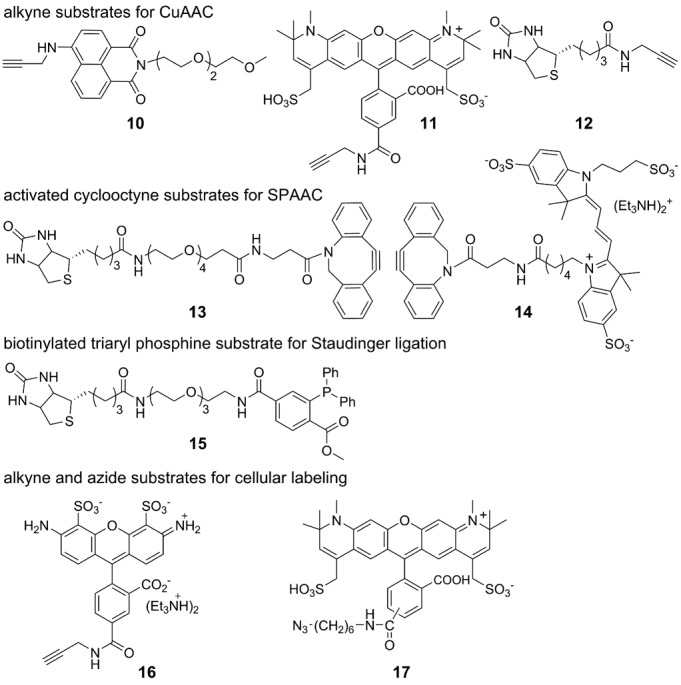
Posttranscriptional modification of azide-modified RNA ONs 7–9 by CuAAC reaction
A solution of THPTA (3.3 μl, 90 mM), CuSO4 (3.3 μl, 45 mM) and sodium ascorbate (3.3 μl, 90 mM) was added to an aqueous solution of azide-modified RNA ONs 7/8/9 in water (27 μl, 0.55 mM). Stock solutions (7.5 mM) of alkyne substrates 10, 11 and 12 were prepared in DMSO. Alkyne substrates (10 μl, 7.5 mM) were added to individual reaction mix and the reaction volume was adjusted to 50 μl by adding water. The final concentration of reaction components was the following: THPTA (6.0 mM), CuSO4 (3.0 mM), sodium ascorbate (6.0 mM), RNA ON (0.30 mM, 15 nmol), alkyne substrate (1.5 mM) and DMSO (20%). The reaction mixtures were incubated at 37°C for 30 min and purified by PAGE under denaturing conditions. The bands corresponding to clicked products were cut and extracted in 0.5 M ammonium acetate and desalted using Sep-Pak classic C18 cartridge. For structure of clicked products see Supplementary Scheme S4, and for yields and mass data see Supplementary Table S2.
Posttranscriptional modification of azide-modified RNA ONs 7–9 by copper-free SPAAC reaction with cyclooctyne building blocks of biotin (13) and Cy3 (14)
A solution of azide-modified RNA ONs 7/8/9 in water (23 μl, 0.65 mM) was mixed with alkyne substrate 13 or 14 (4.5 μl, 10 mM) dissolved in DMSO. Total volume of the reaction was adjusted to 37.5 μl by adding 10 μl of water such that the final concentration of ONs was 0.40 mM (15 nmol), alkyne substrates was 1.2 mM and percentage of DMSO was 12. The individual reaction mixture was incubated at 37°C for 1 h and the clicked product was purified by PAGE under denaturing conditions. The bands corresponding to clicked product was cut and extracted in 0.5 M ammonium acetate and desalted using Sep-Pak classic C18 cartridge. For structure of clicked products see Supplementary Scheme S5, and for yields and mass data see Supplementary Table S2.
Posttranscriptional modification of azide-modified RNA ONs 7–9 by Staudinger ligation reaction with biotinylated triaryl phosphine substrate 15
A solution of azide-modified RNA ONs 7/8/9 in water (27 μl, 0.55 mM) was mixed with PBS buffer (10 μl, 100 mM). The biotinylated phosphine substrate 15 (10 μl, 15 mM) in DMSO was then added to the above solution and mixed well (45). The reaction volume was adjusted to 50 μl by adding water. The final concentration of reaction components was the following: RNA ONs (0.30 mM, 15 nmol), 15 (3.0 mM) and DMSO (20%). The reaction mixture was incubated at 60°C for 15 h and was purified by PAGE under denaturing conditions. Bands corresponding to the ligated products were cut and extracted in 0.5 M ammonium acetate and desalted using Sep-Pak classic C18 cartridge. For structure of products see Supplementary Scheme S6, and for yields and mass data see Supplementary Table S2.
AMU labeling of RNA transcripts in HeLa cells using AMUTP (4)
Mammalian (HeLa) cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). 0.3–0.5 million HeLa cells were plated in 6 well plates on glass cover slips for nearly 24 h before the experiment. Transfection solution (150 μl) containing varying concentrations of AMUTP 4 (50 μM–4.0 mM) and DOTAP (N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate, 30 μl, 1 μg/μl) were prepared in HEPES buffered saline (HBS, 20 mM HEPES, 150 mM NaCl, pH 7.4). Each solution was incubated for 15 min at RT, and was mixed with 450 μl of Opti-MEM. The cells were treated with above transfection solution (600 μl) for 1 h at 37°C. The cells were then washed with PBS (3 × 1 ml), and subsequently fixed and permeabilized with 3.7% formaldehyde and 0.5% Triton X-100 in PBS, respectively, for 30 min at RT. The cells were washed with PBS (3 × 5 min) before click reaction. AMU-labeled RNA transcripts were detected by performing CuAAC reaction with Alexa594-alkyne 11 under the following conditions.
Fixed cells were treated with click reaction mix (500 μl) containing CuSO4 (4 mM), 11 (15 μM) and sodium ascorbate (10 mM) in TBS (50 mM Tris, 150 mM NaCl, pH 7.4) for 30 min at RT. After this period, the cells were washed with PBS containing sodium azide (2 mM) for 20 min and rinsed again with PBS. DNA was stained with DAPI (500 μl, 2 μg/ml) in dark for 2 min at RT. The coverslip was then washed with PBS (1 ml) and glued upside-down on microscopy slide using 10 μl of slowfade antifade gold. The cells were imaged using a Confocal Laser Scanning Microscope with an oil immersion using 40X and 63X lenses.
Hydroxyurea treatment
Cells were incubated in Opti-MEM (600 μl) containing 20 mM hydroxyurea for 5 h after which 150 μl of the media was removed. The cells were then supplemented with a freshly prepared solution of transfection mix (150 μl, mentioned above) containing 4 and incubated for another 1 h. The final concentration of hydroxyurea and 4 in the media was 15 mM and 1 mM, respectively. The cells were washed, fixed and stained by click reaction using Alexa594-alkyne 11 as mentioned above.
Actinomycin D treatment
Cells were incubated in Opti-MEM (600 μl) containing actinomycin D (150 nM or 2.5 μM) for 5 h after which 150 μl of the media was removed. The cells were then supplemented with a freshly prepared solution of transfection mix (150 μl) containing 4, and incubated for another 1 h. The final concentration of actinomycin D and 4 in the media was 100 nM/2.0 μM and 1 mM, respectively. The cells were washed, fixed and stained by click reaction using Alexa594-alkyne 11.
RNase A treatment
Cells were incubated in Opti-MEM (600 μl) with AMUTP 4 (1 mM) and DOTAP (30 μg) for 1 h. The cells were washed, fixed and rinsed as before. The cells were treated with RNase A (single-stranded RNA cleaving enzyme, 200 μg/ml) for 45 min at RT. The cells were washed with PBS and stained in the presence of Alexa594-alkyne 11 as before.
Detection of AMU labeling in cellular RNA by copper-free SPAAC reaction
HeLa cells were incubated with Opti-MEM (600 μl) supplemented with AMUTP 4 (1 mM) and DOTAP (30 μg). After incubating for 1 h, the cells were washed, fixed and permeabilized as before. For staining under SPAAC condition, fixed cells were first treated with 500 μl of Triton X (0.01% in PBS) solution containing (10 μM) cyclooctyne 14 for 2 h at RT. In a parallel experiment, cells transfected with AMUTP were stained by first SPAAC reaction using 14 (red) as mentioned above. After copper-free click reaction, cells were washed once with 1 ml Triton X (0.01% in PBS) followed by washing with PBS (3 × 5 min). Subsequently, CuAAC reaction was performed using Alexa488-alkyne 16 as mentioned before.
Simultaneous imaging of replicating DNA and newly transcribing RNA in cells
HeLa cells were incubated in DMEM supplemented with 10% FBS and containing EdU (10 μM) for 4 h. The media was removed and immediately supplemented with Opti-MEM (600 μl) containing AMUTP 4 (1 mM) and DOTAP (30 μg). After incubating for 1 h, the cells were washed, fixed and stained under CuAAC conditions as before. The fixed cells were first stained with Alexa-594 azide 17 to detect EdU labeling (red) in DNA. The cells were washed and then stained with Alexa488-alkyne 16 to detect AMU labeling (green) in RNA.
RESULTS AND DISCUSSION
Synthesis and incorporation of azide-modified UTP analogs
5-Modified pyrimidine ribonucleotides are known to serve as good substrates in in vitro transcription reaction catalyzed by T7 RNA polymerase (44,46). Therefore, we assembled a toolbox containing 5-azido-modified UTP analogs (4–6) to perform posttranscriptional chemical labeling and imaging of RNA (Scheme 1). The ribonucleosides (1–3) were synthesized according to analogous procedures reported in the literature (Supplementary Data). The corresponding modified triphosphate substrates (4–6) necessary for transcription reactions were obtained by first phosphorylating the nucleoside with POCl3 followed by a reaction with bis-tributylammonium pyrophosphate (Scheme 1).
Scheme 1.
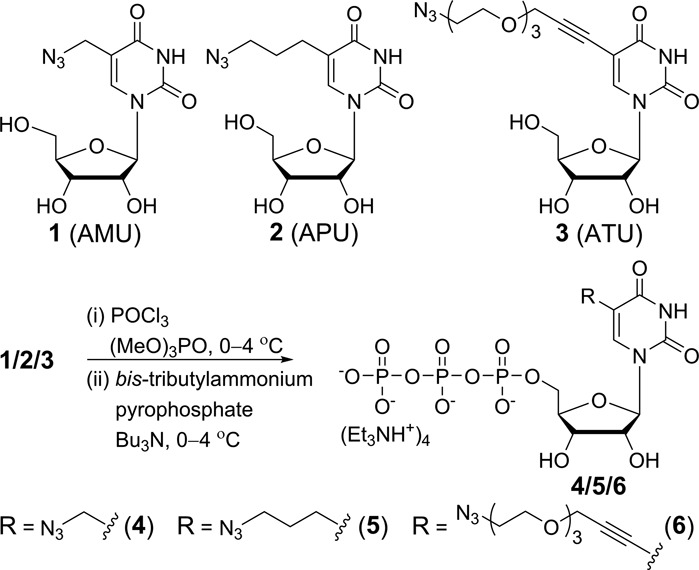
General scheme for the synthesis of azide-modified UTP analogs 4–6 from ribonucleosides 1–3, respectively (Supplementary Data).
The suitability of transcription reaction in producing azide-modified RNA was first evaluated in vitro by using a series of short promoter-template DNA duplexes and T7 RNA polymerase (Figure 2). Deoxyadenosine (dA) residue was placed in the coding region of the templates at one or two sites to direct the incorporation of azide-modified UTP into RNA transcripts. The templates were also designed to contain a dT residue at the 5′-end to direct the incorporation of a unique A residue at the 3′-end of each transcript. Transcription reactions were performed with templates T1–T5 in the presence of GTP, CTP, UTP/modified UTP and α-32P ATP. The products were then resolved by polyacrylamide gel electrophoresis (PAGE) and phosphorimaged. In this experimental setup successful transcription reactions producing 3′-end radiolabeled full-length RNA transcripts will be detected and all failed reactions resulting in the formation of truncated transcripts will remain undetected.
Figure 2.
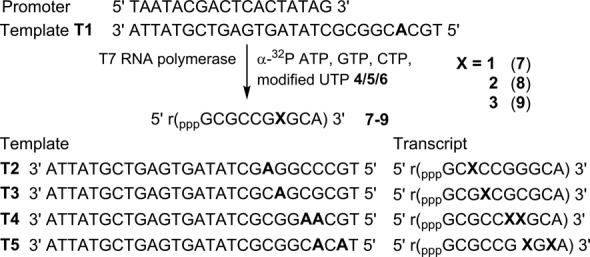
Incorporation of azide-modified UTP analogs (4–6) into RNA ONs by in vitro transcription reactions. Incorporation of 4–6 in the presence of DNA template T1 will produce RNA transcripts 7–9, respectively. Transcripts resulting from T2–T5 are also shown.
Transcription reactions performed in the presence of template T1 and modified UTPs 4–6 resulted in very high yields of azide-modified full-length RNA transcripts 7–9, respectively, in comparison to a control reaction in the presence of natural UTP (Figure 3, lane 2). The retarded mobility of modified transcripts as compared to control unmodified transcript indicated the incorporation of heavier nucleotides into transcripts (Figure 3, compare lanes 1 and 2). In addition to full-length product, non-templated random incorporation of nucleotides resulted in the formation of trace amounts of longer transcripts. This observation is not unusual as in vitro transcription reactions with short templates are known to produce N+1 and N+2 products (47). Furthermore, absence of a band corresponding to full-length transcript in a control reaction performed with no UTP or modified UTP ruled out any misincorporation during the transcription reaction (Figure 3, lane 3). To test the preference of RNA polymerase, reactions were performed in the presence of 1:1 concentration of natural UTP and modified UTP. Rewardingly, the enzyme incorporated both natural as well as modified UTPs reasonably well, indicating that the azide group could be potentially incorporated into cellular RNA transcripts by using cell's biosynthetic machinery (Figure 3, lane 4). Furthermore, reactions with templates T2–T5 revealed that the modified uridine analogs could be incorporated into RNA transcripts near the transcription initiation site (TATA box) and at more than one site with moderate to high efficiency (Figure 3, lanes 6, 8, 10 and 12).
Figure 3.
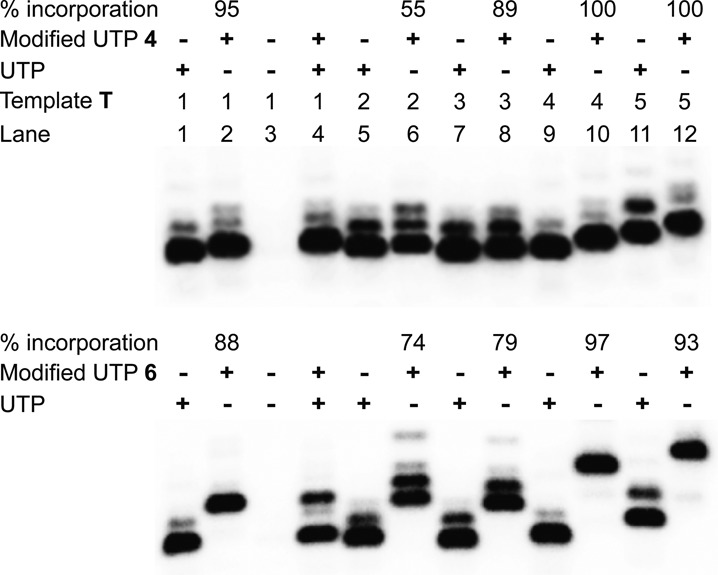
Incorporation of modified UTPs into RNA ONs by T7 RNA polymerase. Transcription reactions were performed with templates T1–T5 in the presence of UTP and or modified UTPs 4 and 6. Relative% incorporation of azide-modified UTPs into modified full-length transcript is given with respect to the amount of full-length transcript formed in the presence of natural NTPs. For complete gel image see Supplementary Figure S1. Gel image of transcription reactions with UTP 5 is also provided in Supplementary Figure S1 (43).
To unambiguously confirm the incorporation of azide functionality into RNA ONs, large-scale transcription reactions were performed in the presence of template T1 and modified UTPs 4–6. Mass analysis of transcripts isolated after PAGE purification clearly confirmed the integrity of modified full-length RNA products (see Supplementary Table S1). In addition, HPLC analysis of ribonucleoside products obtained from digestion of modified transcripts clearly ascertained the incorporation of azide-modified uridine analogs into RNA (see Supplementary Figures S2, S3).
The efficacy of RNA polymerase to incorporate azide groups at multiple sites in RNA sequences of biological relevance was evaluated by performing transcription reactions in the presence of linearized plasmid DNA that would generate nearly 1.1 kb RNA transcript (Supplementary Data). All modified UTPs (4–6) were found to be effectively incorporated by RNA polymerase to produce respective transcripts. As before, HPLC analysis of ribonucleoside products obtained from enzymatic digestion of transcripts confirmed the incorporation of modification into the transcripts (Supplementary Figure S4). The fidelity of transcription reaction in the presence of modified UTPs (e.g. 4) was further evaluated by reverse transcribing modified transcripts into cDNA, which was then PCR amplified and sequenced. The sequencing data of PCR product though showed 100% sequence identity with the template DNA used for in vitro transcription reactions (Supplementary Figures S5–S7), we performed additional cloning and sequencing experiments. The PCR amplified fragment was further subjected to A-tailing using Taq polymerase, which was then cloned using TA cloning kit. The individual colonies were sequenced and sequence reads were then aligned to the DNA template sequence. Out of the eight clones that yielded full-length sequences, five clones exhibited 100% identity and in the remaining three only one base differed at three different locations (Supplementary Figure S8). Together these results reveal that the azide-modified UTPs are effectively incorporated into RNA transcripts and the transcription products are recognized by reverse transcriptase to generate respective cDNA with reasonable accuracy. This feature of nucleoside analogs could be beneficial in expanding the structural and functional diversity of RNA library used in the in vitro selection protocols (48,49).
Posttranscriptional chemical functionalization of azide-modified RNA ONs
The compatibility of azide-modified RNA ONs 7–9 to posttranscriptional chemical functionalization was evaluated by performing click and Staudinger ligation reactions in the presence of a variety of biophysical probes (Figure 4). First, the azide-modified RNA ONs 7–9 were subjected to CuAAC reaction with alkyne substrates in the presence of a water-soluble Cu(I) stabilizing ligand, tris-(3-hydroxypropyltriazolylmethyl)amine (THPTA) (36,50), CuSO4 and sodium ascorbate at 37°C. Typically, reaction mixture after 30 min was resolved by PAGE under denaturing conditions, and the band corresponding to the product was isolated. The cycloaddition reaction with naphthalimide-alkyne (10), Alexa594-alkyne (11) and biotin-alkyne (12) was completed in 30 min to afford the desired fluorescent- and biotin-labeled RNA ONs in moderate to good yields (Figure 5, Supplementary Figure S9, Supplementary Table S2). Reactions between ON 7–9 and biotin-alkyne substrate gave the expected clicked products and also an unknown slower migrating side–product in trace amounts (Supplementary Figure S9). On the other hand, RNA ONs 7–9 underwent facile copper-free SPAAC reaction with commercially available cyclooctyne building blocks of biotin (13) and Cy3 (14) in 60 min at 37°C (Figure 5, Supplementary Figure S9, Supplementary Table S2) to produce the clicked products in moderate to very high yields. In SPAAC reaction conditions, we did not detect any side–products. Next, the azide-modified ONs were subjected to Staudinger ligation reaction with biotinylated triaryl phosphine substrate 15. Since the kinetics of ligation reaction is slow (13), the desired biotinylated RNA products (15a–15c) were formed in isolable amounts when the reaction was performed for 15 h at elevated temperature (60°C, Supplementary Table S2). Some amount of unreacted azide-modified RNA transcript was noticed as the reaction did not proceed to completion (Supplementary Figure S9).
Figure 4.
Building blocks for posttranscriptional chemical modification (Supplementary Data).
Figure 5.
Posttranscriptional chemical functionalization of azide-modified RNA ONs 7–9 by CuAAC, SPAAC and Staudinger ligation reactions. For chemical structure of the products see Supplementary Scheme S4–S6.
Notably, ATU (3) labeled RNA ON 9 containing a longer tetraethyleneglycol spacer gave the best yield with all substrates as compared to ONs 7 and 8 containing shorter linkers. Under these posttranscriptional modification conditions we observed minimum degradation of RNA (Supplementary Figure S9). Typically, 15 nmol reaction scale yielded 4–12 nmol of the labeled product after gel electrophoretic purification. The integrity of all clicked and ligated RNA products was confirmed by mass analysis (Supplementary Table S2). Taken together, these results clearly demonstrate that this posttranscriptional chemical labeling methodology is simple, modular, and generates labeled RNA ONs in sufficient amounts for biophysical analysis.
Incorporation of azide-modified UTPs into cellular RNA transcripts by endogenous RNA polymerases
Encouraged by these results, we sought to devise a cellular RNA labeling and imaging method using our ribonucleoside toolbox. We hypothesized that azide groups could be metabolically introduced into cellular RNA transcripts by using azide-modified ribonucleosides (1–3) or ribonucleotides (4–6, Figure 1). The labeled RNA could then be chemically functionalized by click reactions with a variety of probes that could be utilized in the study of cellular RNA by various biophysical techniques. The success of this strategy will depend on (i) the cell permeability of modified nucleoside or nucleotides, (ii) the ability of cell's biosynthetic machinery to incorporate the modifications specifically into RNA transcripts and (iii) the compatibility of modified cellular RNA to posttranscriptional chemical modification under bioorthogonal reaction conditions.
The incorporation of azide functionalities into cellular RNA transcripts was first tested by incubating cultured human cervical cancer cells (HeLa) with ribonucleoside analogs 1–3. The cells were fixed, permeabilized, stained with Alexa594-alkyne (11) under CuAAC conditions and imaged by fluorescence microscopy. Unfortunately, the cells did not show appreciable fluorescence signal indicating that the ribonucleoside analogs are probably cell-impermeable and or not good substrates for metabolic labeling by the ribonucleoside salvage pathway (data not shown). As a result, we decided to bypass the phosphorylation steps involved in the salvage pathway by transfecting the cells with modified UTPs, which could now be directly incorporated into cellular RNA by RNA polymerases. Rewardingly, among the nucleotides, cells transfected with 5-azidomethyl UTP (AMUTP, 4) resulted in AMU-labeled RNA transcripts, which were detected by click reactions with fluorescent alkyne probes. Transfection of UTP analogs 5 and 6 in which the azide group is linked via a propyl and tetraethylene glycol linker did not result in detectable incorporation of azide groups into cellular RNA (Supplementary Figure S10). This observation indicates that the endogenous RNA polymerases efficiently incorporate the azide-modified UTP 4 containing a shorter linker into RNA as compared to 5 and 6.
Imaging cellular RNA transcription using AMUTP 4
The AMU labeling of RNA by endogenous polymerases was evaluated by transfecting the HeLa cells with increasing concentrations of AMUTP (50 μM to 1 mM) for 1 h using DOTAP. Upon fixing and staining the cells with Alexa594-alkyne (11) under CuAAC conditions, nuclear and cytoplasmic staining was observed in concentration-dependent manner (Figure 6). As low as 50 μM of AMUTP and incubation time of 15 min produced significant fluorescence signal in all cells (Figure 6, Supplementary Figure S11). The nucleoli, where abundant rRNA synthesis takes place, stained more intensely indicating higher levels of AMU labeling, presumably in the pre-rRNA (Supplementary Figure S12). Apparently, longer incubation times (≥ 3 h) and concentrations of AMUTP ≥ 2 mM resulted in discernable reduction in staining due to cell death (Supplementary Figure S11, S13). Expectedly, cells cultured in the absence or presence of AMUTP alone (without DOTAP) displayed no detectable fluorescence signal upon fixing and staining under similar conditions.
Figure 6.
Imaging cellular RNA transcription using AMUTP 4. Cultured HeLa cells were transfected with 4 (50 μM to 1 mM) for 1 h using DOTAP. The cells were then fixed, permeabilized and the AMU-labeling was detected by performing CuAAC reaction with Alexa594-alkyne 11.
AMUTP specifically labels RNA
The observed fluorescence signal from the cells could be due to (i) the specific incorporation of AMU into RNA transcripts followed by a click reaction with Alexa594-alkyne, (ii) a click reaction between unincorporated AMUTP and Alexa594-alkyne and or (iii) non-specific labeling of azide moiety into DNA. To determine the specific incorporation of azide functionality into transcripts by endogenous RNA polymerases, the following tests were performed.
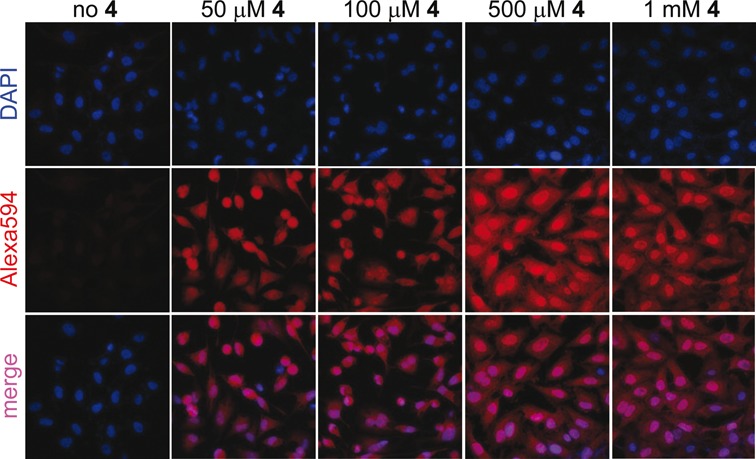
AMUTP can be potentially dephosphorylated into AMUDP by cellular phosphatases, which can serve as a substrate for ribonucleoside diphosphate reductase leading to the production of AMdUDP. AMdUDP can be phosphorylated into AMdUTP, which then could get incorporated into replicating DNA. In such a scenario the observed fluorescence staining would not be exclusively due to AMU-labeled RNA transcripts. Hydroxyurea, a known inhibitor of ribonucleoside diphosphate reductase activity, reduces the effective concentration of dNTPs in the cell thereby stalling the DNA replication process (51). Cells incubated with AMUTP in the presence of hydroxyurea did not show significant changes in the subcellular staining pattern as compared to in the absence of hydroxyurea (Figure 7A). However, control experiments performed in the presence of 5-ethynyl-2’-deoxyuridine (EdU) that incorporates efficiently into DNA, revealed that addition of hydroxyurea abolished the labeling of EdU into DNA. These observations confirm that AMUTP does not label DNA.
Figure 7.
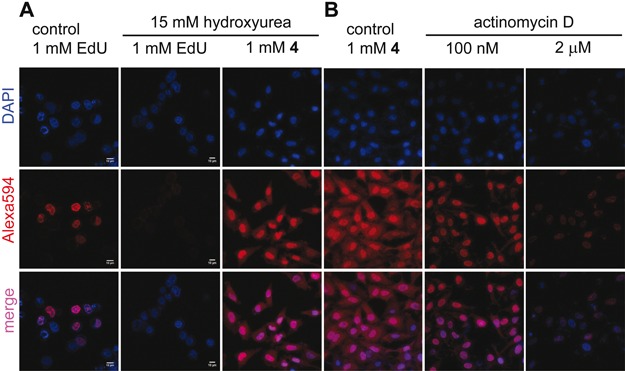
(A) AMUTP 4 is incorporated into RNA and not into DNA. HeLa cells were incubated with 15 mM hydroxyurea (ribonucleotide reductase inhibitor) and EdU/4, and were click-stained as before using Alexa594-azide (17) or Alexa594-alkyne (11). DOTAP was used when cells were incubated with 4. Inhibition of DNA synthesis by using hydroxyurea abolished the incorporation of EdU. No significant change in staining pattern in cells incubated with 4 in the presence of hydroxyurea indicates that 4 is not incorporated into DNA. (B) AMUTP 4 specifically labels transcribing RNA in cells. HeLa cells were transfected with 4 in the presence and absence of variable concentrations of actinomycin D, an RNA polymerase inhibitor. Progressive reduction in fluorescence signal in cells treated with actinomycin D confirms specific AMU labeling of cellular RNA.
Next, we studied the effect of inhibition of RNA polymerases on the AMU labeling. A lower concentration of actinomycin D (100 nM) at which RNA polymerase I is inhibited (52) a significant reduction in fluorescence signal in the nucleus was observed (Figure 7B). Further, higher concentration of actinomycin D (2.0 μM), which blocks RNA polymerase II also abolished the staining throughout the cells. In a parallel experiment, cells incubated with AMUTP for 1 h were fixed, permeabilized and treated with RNase A (single-stranded RNA cleaving enzyme) for 45 min. The cells were then stained by performing cycloaddition reaction with 11. RNase A treatment resulted in a significant reduction in fluorescence intensity (Supplementary Figure S14). Collectively, these results demonstrate that AMU is specifically incorporated into RNA transcripts by endogenous RNA polymerases.
RNA distribution in cells has been studied by using RNA-selective binding fluorescent probes such as SYTORNASelect and styryl dyes (53,54). To draw comparison between RNA labeling method using AMUTP and RNA-binding dyes we have performed the following experiments. The cells cultured in the absence or presence of AMUTP 4 were fixed using pre-chilled methanol and were stained by using a commercially available RNA-binding dye, SYTORNASelect (green) and by CuAAC reaction using Alexa594-alkyne 11 (red), respectively. Cells cultured in the absence of AMUTP and stained using SYTO revealed intense punctuate staining of globular intra-nuclear structures (possibly nucleoli) and faint nuclear and cytoplasmic staining (column 2, Supplementary Figure S15). The observed SYTO staining pattern is consistent with earlier reports (53–56). Cells transfected with AMUTP 4 were fixed using methanol according to the SYTO manufacturer's instructions, permeabilized with Triton X-100 and click stained. The staining pattern observed here was similar to the one obtained when click-staining was performed on cells fixed using formaldehyde protocol, (uniform nuclear and cytoplasmic staining with intense punctuate staining of possibly nucleoli). Further, we evaluated the RNA labeling ability of both the methods in the same cell. The cells were treated with AMUTP and were fixed, permeabilized and sequentially stained using Alexa594-alkyne 11 and SYTO dye. In this protocol, cells permeabilized with Triton X-100 exhibited a slightly diffused SYTO staining (column 4, Supplementary Figure S15). Although the signal intensity was found to be slightly reduced, click reaction produced similar staining pattern as before. Nevertheless, this sequential staining procedure resulted in discernible colocalization of click-stained RNA and SYTO-stained RNA with less cell-to-cell variation. Further, cells transfected with AMUTP 4 were fixed, permeabilized, treated with RNase A and sequentially stained by using click reaction and SYTO dye. A discernible reduction in fluorescence signal from SYTO dye and click reaction with no apparent change in DAPI staining was observed (column 5, Supplementary Figure S15). These results indicated that both SYTO dye and AMUTP specifically labeled RNA transcripts in cells. Although the present click staining method requires the transfection of modified nucleotide into cells followed by a chemical reaction to label RNA, the results demonstrate that this posttranscriptional chemical labeling protocol using AMUTP is very specific, robust and modular (any alkyne probe could be attached to RNA).
Fluorescence staining of azide-labeled cellular RNA transcripts by copper-free cycloaddition reaction
SPAAC reaction using cyclooctyne probes has been widely used in the bioconjugation of azide-modified glycan, protein, lipid and recently, DNA in cells as it avoids copper catalyst, which is toxic to cells (35,57–59). However, its utility has not been extended to cellular RNA labeling procedures due to lack of efficient methods to label RNA with chemically and metabolically stable azide groups. Now that we have developed a simple method to label cellular RNA with azide functionality using AMUTP, we sought to explore the efficacy of SPAAC reaction in staining RNA posttranscriptionally. The cells were incubated with AMUTP for 1 h and then fixed and stained with cyclooctyne-conjugated Cy3 (14) for 2 h. Unlike staining under CuAAC conditions, SPAAC staining using cyclooctyne 14 was partial and produced intense fluorescence staining only in few cells (Figure 8). In order to determine cell-to-cell comparison of the two staining methods, cells were first stained by SPAAC reaction using 14 and then were washed and stained again by CuAAC reaction using Alexa-488-alkyne 16. Few cells, which were labeled by copper-free SPAAC reaction (red), were also labeled by CuAAC reaction (green, Figure 8). However, the intranuclear staining pattern was different. Apart from staining most cells, CuAAC reaction resulted in intense nucleoli staining as compared to under SPAAC reaction conditions (Figure 8 rightmost panel). Despite the differences in staining efficiency (possibly due to poor cell permeability and bulkiness of the cyclooctyne probe, 35,60), these results underscore the potential use of SPAAC reaction in detecting RNA in cells.
Figure 8.
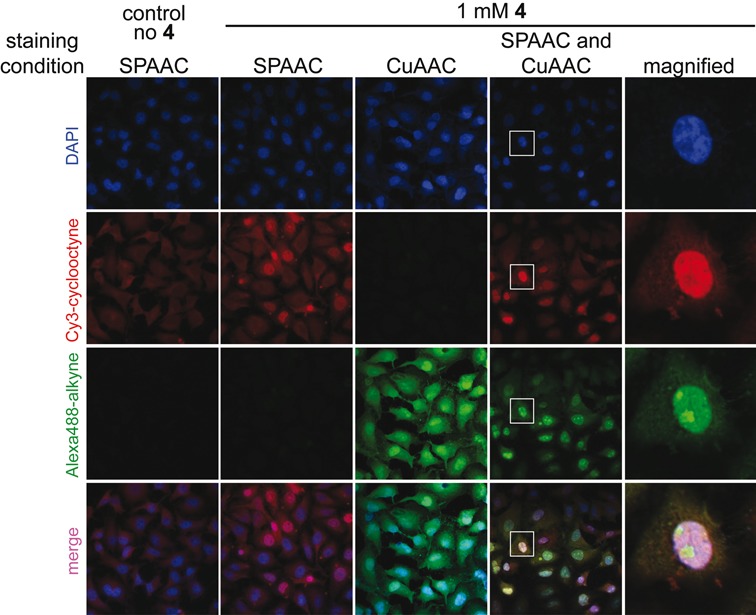
Imaging cellular RNA transcription using AMUTP 4 under copper-free SPAAC reaction conditions. HeLa cells were transfected with 1 mM of AMUTP 4 for 1 h. The cells were fixed and stained by SPAAC reaction using Cy3-cyclooctyne substrate 14 (red). In a parallel experiment, cells transfected with AMUTP were stained by first SPAAC reaction using 14 (red) followed by CuAAC reaction using Alexa488-alkyne 16 (green). Bottom panel is a merge of all three channels (blue, red and green). Boxed cell has been magnified.
The utility of copper-free SPAAC reaction to detect newly transcribing RNA in living cells was studied by first treating the cells with AMUTP for 1 h followed by transiently permeabilizing the cells without fixation using pre-cooled solution of Triton X-100 (35). Subsequently, cells were treated with cyclooctyne 14 for 30 min at 37°C, and were washed and observed by confocal microscopy. Given the poor cell permeability of currently available cyclooctyne probes (60), staining of RNA transcripts under SPAAC conditions yielded intense punctuate nucleoli staining and faint nuclear staining in few cells (Supplementary Figure S16). Further, cell viability test confirmed the low toxicity of the probe in this staining reaction conditions (Supplementary Figure S17). This RNA labeling method is advantageous as the selective incorporation of azide groups into RNA using AMUTP and SPAAC staining procedure provides an alternative route to image transcribing RNA in living cells.
Simultaneous imaging of DNA and RNA synthesis in cells
The specific incorporation of EdU into DNA and AMUTP into RNA was further exploited in designing a simple experiment to simultaneously image replicating DNA and newly transcribing RNA in cells. In this experiment, cells in culture were incubated with EdU and then transfected with AMUTP 4. The fixed cells were sequentially stained with Alexa594-azide 17 to detect EdU labeling in DNA (red) and then with Alexa488-alkyne 16 to detect AMU labeling in RNA (green) under CuAAC conditions (Figure 9). Although alkyne and azide are cognate reactive partners in cycloaddition reaction, selective incorporation of these groups into DNA and RNA by endogenous polymerases uniquely enabled the simultaneous visualization of replicating DNA and newly transcribing RNA by using click reactions.
Figure 9.
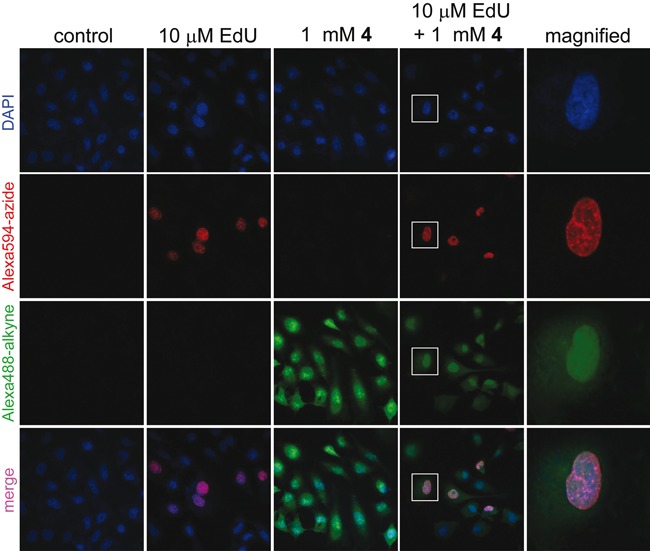
Simultaneous visualization of DNA and RNA synthesis in cells by CuAAC reactions. In separate experiments, HeLa cells were incubated with EdU/AMUTP and a combination of EdU and AMUTP. While EdU labeling in DNA was detected by a click reaction with Alexa594-azide 17 (red), AMU labeling in RNA was detected by a reaction with Alexa488-alkyne 16 (green). Bottom panel is a merge of all three channels (blue, red and green). Boxed cell has been magnified.
SUMMARY AND CONCLUSION
We have developed a robust and modular posttranscriptional chemical functionalization protocol to label RNA in vitro and in cells with biophysical probes by using a novel toolbox made of azide-modified UTP analogs. These modified UTPs, synthesized in few steps, are effectively incorporated by RNA polymerases to endow RNA with azide functionality. Further, the azide-modified transcripts are conveniently labeled posttranscriptionally by CuAAC, SPAAC and Staudinger ligation reactions with a variety of substrates ranging from fluorescent probes to affinity tags. Although methods are available to metabolically incorporate the versatile azide functional group into glycan, protein and DNA, incorporation of azide groups into cellular RNA has remained elusive until now. In this context, the incorporation of azide-modified UTP analog (AMUTP) by endogenous RNA polymerases is the first example of selective labeling of cellular RNA transcripts with azide groups. The azide groups in RNA provide an easy handle to image newly transcribing RNA in fixed cells as well as live cells by CuAAC and copper-free SPAAC reactions. Further, the selective labeling of RNA with azide has enabled us to devise a simple method to simultaneously image DNA and RNA synthesis in cells by using click reactions. It is expected that this modular and practical chemical labeling methodology will provide a new platform to study RNA in vitro and in cells (e.g. RNA synthesis, localization and degradation). We are currently investigating the utility of this method in imaging RNA in live cells and animal models using fluorogenic probes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
S.G.S. thanks Council of Scientific and Industrial Research, India (02-0086/12/EMR-II) and Department of Science and Technology, India, for the research grants. Work in SG lab is supported by the Centre of Excellence in Epigenetics program of Department of Biotechnology, Government of India. Authors wish to thank the Director, NCCS for providing access to radioisotope handling facility. A.A.S. and A.A.T. thank Council of Scientific and Industrial Research, India for graduate research fellowship. A.K. acknowledges the Wellcome Trust–DBT India Alliance for early career fellowship.
FUNDING
Funding for open access charge: Indian Institute of Science Education and Research, Pune; National Centre for Cell Science, Pune.
Conflict of interest statement. None declared.
REFERENCES
- 1.Corey D.R. Chemical modification: the key to clinical application of RNA interference. J. Clin. Invest. 2007;117:3615–3622. doi: 10.1172/JCI33483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wachowius F., Höbartner C. Chemical RNA modifications for studies of RNA structure and dynamics. Chembiochem. 2010;11:469–480. doi: 10.1002/cbic.200900697. [DOI] [PubMed] [Google Scholar]
- 3.Baker M. RNA imaging in situ. Nat. Methods. 2012;9:787–790. [Google Scholar]
- 4.Haukenes G., Szilvay A.-M., Brokstad K.A., Kalland K.-H. Labeling of RNA transcripts of eukaryotic cells in culture with BrUTP using a liposome transfection reagent. Biotechniques. 1997;22:308–312. doi: 10.2144/97222st03. [DOI] [PubMed] [Google Scholar]
- 5.Cmarko D., Verschure P.J., Martin T.E., Dahmus M.E., Krause S., Fu X.-D., van Driel R., Fakan S. Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol. Biol. Cell. 1999;20:211–223. doi: 10.1091/mbc.10.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Femino A.M., Fay F.S., Fogarty K., Singer R.H. Visualization of single RNA transcripts in situ. Science. 1998;280:585–590. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 7.Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nat. Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- 9.Pianowski Z., Gorska K., Oswald L., Merten C.A., Winssinger N. Imaging of mRNA in live cells using nucleic acid-templated reduction of azidorhodamine probes. J. Am. Chem. Soc. 2009;131:6492–6497. doi: 10.1021/ja809656k. [DOI] [PubMed] [Google Scholar]
- 10.Franzini R.M., Kool E.T. Efficient nucleic acid detection by templated reductive quencher release. J. Am. Chem. Soc. 2009;131:16021–16023. doi: 10.1021/ja904138v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paige J.S., Wu K.Y., Jaffrey S.R. RNA mimics of green fluorescent protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao G., Rhee W.J., Tsourkas A. Fluorescent probes for live-cell RNA detection. Annu. Rev. Biomed. Eng. 2009;11:25–47. doi: 10.1146/annurev-bioeng-061008-124920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prescher J.A., Bertozzi C.R. Chemistry in living systems. Nat. Chem. Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 14.Neef A.G., Schultz C. Selective fluorescence labeling of lipids in living cells. Angew. Chem. Int. Ed. Engl. 2009;48:1498–1500. doi: 10.1002/anie.200805507. [DOI] [PubMed] [Google Scholar]
- 15.van Berkel S.S., van Eldijk M.B., van Hest J.C.M. Staudinger ligation as a method for bioconjugation. Angew. Chem. Int. Ed. Engl. 2011;50:8806–8827. doi: 10.1002/anie.201008102. [DOI] [PubMed] [Google Scholar]
- 16.Spicer C.D., Davis B.G. Selective chemical protein modification. Nat. Commun. 2014;5:4740. doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- 17.Gramlich P.M.E., Wirges C.T., Manetto A., Carell T. Postsynthetic DNA modification through the copper-catalyzed azide–alkyne cycloaddition reaction. Angew. Chem. Int. Ed. Engl. 2008;47:8350–8358. doi: 10.1002/anie.200802077. [DOI] [PubMed] [Google Scholar]
- 18.El-Sagheer A.H., Brown T. Click chemistry with DNA. Chem. Soc. Rev. 2010;39:1388–1405. doi: 10.1039/b901971p. [DOI] [PubMed] [Google Scholar]
- 19.Salic A., Mitchison T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gramlich P.M.E., Warncke S., Gierlich J., Carell T. Click–click–click: single to triple modification of DNA. Angew. Chem. Int. Ed. Engl. 2008;47:3442–3444. doi: 10.1002/anie.200705664. [DOI] [PubMed] [Google Scholar]
- 21.Xu Y., Suzuki Y., Komiyama M. Click chemistry for the identification of G-quadruplex structures: discovery of a DNA–RNA G-quadruplex. Angew. Chem. Int. Ed. Engl. 2009;48:3281–3284. doi: 10.1002/anie.200806306. [DOI] [PubMed] [Google Scholar]
- 22.Beyer C., Wagenknecht H.-A. In situ azide formation and ‘‘click’’ reaction of nile red with DNA as an alternative postsynthetic route. Chem. Commun. 2010;46:2230–2231. doi: 10.1039/b924471a. [DOI] [PubMed] [Google Scholar]
- 23.Qing G., Xiong H., Seela F., Sun T. Spatially controlled DNA nanopatterns by “click” chemistry using oligonucleotides with different anchoring sites. J. Am. Chem. Soc. 2010;132:15228–15232. doi: 10.1021/ja105246b. [DOI] [PubMed] [Google Scholar]
- 24.Neef A.B., Luedtke N.W. Dynamic metabolic labeling of DNA in vivo with arabinosyl nucleosides. Proc. Natl. Acad. Sci. U.S.A. 2011;108:20404–20409. doi: 10.1073/pnas.1101126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Šečkutė J., Yang J., Devaraj N.K. Rapid oligonucleotide-templated fluorogenic tetrazine ligations. Nucleic Acids Res. 2013;41:e148. doi: 10.1093/nar/gkt540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lercher L., McGouran J.F., Kessler B.M., Schofield C.J., Davis B.G. DNA modification under mild conditions by Suzuki–Miyaura cross-coupling for the generation of functional probes. Angew. Chem. Int. Ed. Engl. 2013;52:10553–10558. doi: 10.1002/anie.201304038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rieder U., Luedtke N.W. Alkene–Tetrazine ligation for imaging cellular DNA. Angew. Chem. Int. Ed. Engl. 2014;53:9168–9172. doi: 10.1002/anie.201403580. [DOI] [PubMed] [Google Scholar]
- 28.Jao C.Y., Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grammel M., Hang H., Conrad N.K. Chemical reporters for monitoring RNA synthesis and poly (A) tail dynamics. Chembiochem. 2012;13:1112–1115. doi: 10.1002/cbic.201200091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qu D., Zhou L., Wang W., Wang Z., Wang G., Chi W., Zhang B. 5-Ethynylcytidine as a new agent for detecting RNA synthesis in live cells by “click” chemistry. Anal. Biochem. 2013;434:128–135. doi: 10.1016/j.ab.2012.11.023. [DOI] [PubMed] [Google Scholar]
- 31.Wada T., Mochizuki A., Higashiya S., Tsuruoka H., Kawahara S., Ishikawa M., Sekine M. Synthesis and properties of 2-azidodeoxyadenosine and its incorporation into oligodeoxynucleotides. Tetrahedron Lett. 2001;42:9215–9219. [Google Scholar]
- 32.Pourceau G., Meyer A., Vasseur J.-J., Morvan F. Azide solid support for 3′-conjugation of oligonucleotides and their circularization by click chemistry. J. Org. Chem. 2009;74:6837–6842. doi: 10.1021/jo9014563. [DOI] [PubMed] [Google Scholar]
- 33.Santner T., Hartl M., Bister K., Micura R. Efficient access to 3′-terminal azide-modified RNA for inverse click-labeling patterns. Bioconjugate Chem. 2014;25:188–195. doi: 10.1021/bc400513z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weisbrod S.H., Marx A. A nucleoside triphosphate for site-specific labelling of DNA by the Staudinger ligation. Chem. Commun. 2007:1828–1830. doi: 10.1039/b618257g. [DOI] [PubMed] [Google Scholar]
- 35.Neef A.B., Luedtke N.W. An azide-modified nucleoside for metabolic labeling of DNA. Chembiochem. 2014;15:789–793. doi: 10.1002/cbic.201400037. [DOI] [PubMed] [Google Scholar]
- 36.El-Sagheer A.H., Brown T. New strategy for the synthesis of chemically modified RNA constructs exemplified by hairpin and hammerhead ribozymes. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15329–15334. doi: 10.1073/pnas.1006447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paredes E., Das S.R. Click chemistry for rapid labeling and ligation of RNA. Chembiochem. 2011;12:125–131. doi: 10.1002/cbic.201000466. [DOI] [PubMed] [Google Scholar]
- 38.Ishizuka T., Kimoto M., Sato A., Hirao I. Site-specific functionalization of RNA molecules by an unnatural base pair transcription system via click chemistry. Chem. Commun. 2012;48:10835–10837. doi: 10.1039/c2cc36293g. [DOI] [PubMed] [Google Scholar]
- 39.Phelps K.J., Ibarra-Soza J.M., Tran K., Fisher A.J., Beal P.A. Click modification of RNA at adenosine: structure and reactivity of 7-Ethynyl- and 7-Triazolyl-8-aza-7-deazaadenosine in RNA. ACS Chem. Biol. 2014;9:1780–1787. doi: 10.1021/cb500270x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samanta A., Krause A., Jäschke A. A modified dinucleotide for site-specific RNA-labelling by transcription priming and click chemistry. Chem. Commun. 2014;50:1313–1316. doi: 10.1039/c3cc46132g. [DOI] [PubMed] [Google Scholar]
- 41.Holstein J.M., Schulz D., Rentmeister A. Bioorthogonal site-specific labeling of the 5′-cap structure in eukaryotic mRNAs. Chem. Commun. 2014;50:4478–4481. doi: 10.1039/c4cc01549e. [DOI] [PubMed] [Google Scholar]
- 42.Rao H., Tanpure A.A., Sawant A.A., Srivatsan S.G. Enzymatic incorporation of an azide-modified UTP analog into oligoribonucleotides for posttranscriptional chemical functionalization. Nat. Protocols. 2012;7:1097–1112. doi: 10.1038/nprot.2012.046. [DOI] [PubMed] [Google Scholar]
- 43.Rao H., Sawant A.A., Tanpure A.A., Srivatsan S.G. Posttranscriptional chemical functionalization of azide-modified oligoribonucleotides by bioorthogonal click and Staudinger reactions. Chem. Commun. 2012;48:498–500. doi: 10.1039/c1cc15659d. [DOI] [PubMed] [Google Scholar]
- 44.Srivatsan S.G., Tor Y. Fluorescent pyrimidine ribonucleotide: synthesis, enzymatic incorporation, and utilization. J. Am. Chem. Soc. 2007;129:2044–2053. doi: 10.1021/ja066455r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saxon E., Bertozzi C.R. Cell surface engineering by a modified staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 46.Tanpure A.A., Srivatsan S.G. A microenvironment sensitive fluorescent pyrimidine ribonucleoside analogue: synthesis, enzymatic incorporation, and fluorescence detection of a DNA abasic site. Chem. Eur. J. 2011;17:12820–12827. doi: 10.1002/chem.201101194. [DOI] [PubMed] [Google Scholar]
- 47.Milligan J.F., Uhlenbeck O.C. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 48.Vaish N.K., Fraley A.W., Szostak J.W., McLaughlin L.W. Expanding the structural and functional diversity of RNA: analog uridine triphosphates as candidate for in vitro selection. Nucleic Acids Res. 2000;28:3316–3322. doi: 10.1093/nar/28.17.3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimito M., Yamashige R., Matsunaga K.-I., Yokoyama S., Hirao I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotech. 2013;31:453–458. doi: 10.1038/nbt.2556. [DOI] [PubMed] [Google Scholar]
- 50.Kislukhin A.A., Hong V.P., Breitenkamp K.E., Finn M.G. Relative performance of Alkynes in Copper-catalyzed azide–alkyne cycloaddition. Bioconjugate Chem. 2013;24:684–689. doi: 10.1021/bc300672b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koç A., Wheeler L.J., Mathews C.K., Merrill G.F. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem. 2003;279:223–230. doi: 10.1074/jbc.M303952200. [DOI] [PubMed] [Google Scholar]
- 52.Bensaude O. Inhibiting eukaryotic transcription. Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Q., Kim Y., Namm J., Kulkarni A., Rosania G.R., Ahn Y.-H., Chang Y.-T. RNA-Selective, Live cell imaging probes for studying nuclear structure and function. Chem. Biol. 2006;13:615–623. doi: 10.1016/j.chembiol.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 54.Wlodkowic D., Skommer J., Darzynkiewicz Z. SYTO probes in the cytometry of tumor cell death. Cytometry A. 2008;73:496–507. doi: 10.1002/cyto.a.20535. [DOI] [PubMed] [Google Scholar]
- 55.Myrdal S.E., Johnson K.C., Steyger P.S. Cytoplasmic and intra-nuclear binding of gentamicin does not require endocytosis. Hear. Res. 2005;204:156–169. doi: 10.1016/j.heares.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Couvillion M.T., Bounova G., Purdom E., Speed T.P., Collins K. A Tetrahymena Piwi bound to mature tRNA 3′ fragments activates the exonuclease Xrn2 for RNA processing in the nucleus. Mol. Cell. 2012;48:509–520. doi: 10.1016/j.molcel.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang P.V., Prescher J.A., Sletten E.M., Baskin J.M., Miller I.A., Agard N.J., Lo A., Bertozzi C.R. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. U.S.A. 2010;107:1821–1826. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Truong F., Yoo T.H., Lampo T.J., Tirrell D.A. Two-strain, cell-selective protein labeling in mixed bacterial cultures. J. Am. Chem. Soc. 2012;134:8551–8556. doi: 10.1021/ja3004667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haga Y., Ishii K., Hibino K., Sako Y., Ito Y., Taniguchi N., Suzuki T. Visualizing specific protein glycoforms by transmembrane fluorescence resonance energy transfer. Nat. Commun. 2012;3:907. doi: 10.1038/ncomms1906. [DOI] [PubMed] [Google Scholar]
- 60.Dommerholt J., Schmidt S., Temming R., Hendriks L.J.A., Rutjes F.P.J.T., van Hest J.C.M., Lefeber D.J., Friedl P., van Delf F.L. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. Engl. 2010;49:9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.