


Abstract
Gene amplification is a hallmark of cancer with chromosomal instability although the underlying mechanism by which altered copy numbers are maintained is largely unclear. Cohesin, involved in sister chromatid cohesion, DNA repair, cell cycle progression and transcriptional regulation of key developmental genes, is frequently overexpressed in human cancer. Here we show that cohesin-dependent change in DNA replication controls the copy numbers of amplified genes in cancer cells with chromosomal instability. We found that the down-regulation of elevated cohesin leads to copy number-associated gene expression changes without disturbing chromosomal segregation. Highly amplified genes form typical long-range chromatin interactions, which are stabilized by enriched cohesin. The spatial proximities among cohesin binding sites within amplified genes are decreased by RAD21-knockdown, resulting in the rapid decline of amplified gene expression. After several passages, cohesin depletion inhibits DNA replication initiation by reducing the recruitment of pre-replication complexes such as minichromosome maintenance subunits 7 (MCM7), DNA polymerase α, and CDC45 at replication origins near the amplified regions, and as a result, decreases the DNA copy numbers of highly amplified genes. Collectively, our data demonstrate that cohesin-mediated chromatin organization and DNA replication are important for stabilizing gene amplification in cancer cells with chromosomal instability.
INTRODUCTION
In order to maintain genome integrity, genetic and epigenetic alteration is strictly regulated during mammalian development (1). However, genomic instability, including deletion/insertion, alternations in chromosome number, chromosome translocations and gene amplifications, is frequently observed in human cancer (2). The continuous accumulation of genomic instability leads to an imbalance in aneuploidy, an increased rate of loss-of-heterozygosity and gains or losses of whole or partial chromosomes that is called chromosomal instability (CIN) (2,3).
Gene amplification, which is observed in many types of cancer with CIN (4), drives tumor progression by increasing the expression of oncogenes such as c-Myc, HER2 and EGFR (5). The extra copies of amplified DNA in human cancers can be organized as cytologically visible homogeneously staining regions (HSRs) and extrachromosomal double minutes (DMs) (4). DMs, an autonomously replicating extrachromosomal circular DNA, can be initiated by somatic genome rearrangement through DNA breakage and repair processes (called the breakage-fusion-bridge (BFB) cycle) in human cancers (6,7). The creation of DNA double strand breaks (DSBs) followed by replication stress and fusion of chromosome ends results in an unstable dicentric chromosome, which leads to the accumulation of additional DNA breaks (7,8). Thus, continuous DSBs formation and subsequent inaccurate DNA repair may provoke the amplification of DMs near the breakage sites (4,9). DMs are delivered to the daughter cell by attaching to the mitotic chromosome during mitosis (7). In addition, DMs can be integrated into the chromosome arm, followed by repeated initiation of BFB cycle triggered by site-specific DSBs, finally leading to HSRs formation (7,10). However, the molecular mechanisms responsible for maintaining gene amplification in human cancers are not completely understood yet. Since gene amplification not only confers a selective advantage during tumor development but also minimizes sensitivity to anti-cancer drugs (11), therefore, understanding the maintenance processes operating for amplified genes may provide an opportunity to overcome drug resistance caused by oncogene amplification (9,11).
Cohesin is composed of four major core subunits: SMC1, SMC3, RAD21 and SCC3 (12). This complex was originally found to be involved in sister chromatid cohesion, DNA repair and cell cycle progression (13,14). Thus, mutational inactivation of the cohesin complex causes CIN and aneuploidy in human cancer cells due to improper chromosome segregation fidelity (15,16). In addition to its major influence on sister-chromatid cohesion and DNA repair, the cohesin complex affects gene transcription by facilitating long-range interactions among members of many developmentally regulated gene families (17–22). Interestingly, aberrant expression of cohesin components is also present in many human cancers (23). The recent discovery that the overexpression of cohesin components confers poor prognosis and resistance to chemotherapy in breast and colorectal cancers (24,25) raises the possibility that the elevated cohesin level is essential for tumorigenesis (23). However, it is not yet clear if enhanced expression of cohesin can contribute to the gene amplification process.
In the present investigation, we comprehensively evaluated the effects of cohesin reduction on gene amplification. We found that the down-regulation of elevated cohesin abolishes long-range chromatin interactions of highly amplified genes with a concurrent reduction of transcription in human gastric cancer cells. Moreover, reduction of cohesin appears to de-stabilize high-level gene amplifications by disrupting the recruitment of pre-replication complex to the near amplified genes in chromosomally unstable cancer cells, thereby reducing DNA copy-number of amplified genes.
MATERIALS AND METHODS
Patient tissues, cell culture, virus production, transduction and cell-growth inhibition assay
Twenty four human gastric tumor tissues and the matched normal tissues were obtained from the Tissue Bank of Seoul National University Hospital. The study protocol was reviewed and approved by the institutional review board of Seoul National University Hospital. Four CIN+ cell types (SNU16, N87, COLO 320-HSR and COLO 320-DM) with multiple chromosomal structure changes and three CIN− cancer-cell types (HCT116, LoVo and HepG2 cells; diploid/near-diploid karyotype with a few structural alterations) were obtained from American Tissue Culture Collection or the Korean Cell Line Bank and have not been cultured for longer than 6 months. Cells were cultured in DMEM or RPMI 1640 supplemented with 10% fetal bovine serum and gentamicin (10 μg/ml) at 37 °C in a 5% CO2-humidified atmosphere (7,15,26–29). Control and RAD21-directed TRC lentiviral shRNAs were purchased from Open Biosystems. Lentiviruses were produced by transducing 293FT cells with shRNA using a Virapower packaging mix (Invitrogen) as previously described (30). The viruses were harvested from the media on day 3 by centrifugation, and cells were then incubated with viral supernatant in the presence of 6 μg/ml polybrene (Sigma). After 2 days of incubation, the transduced cells were cultured in the presence of 1 μg/ml puromycin (Sigma) for another 3 days before collection as previously described (17). Silencing was confirmed by western blot analysis and qPCR. To generate stably transfected cells, several single colonies were isolated and independently expanded in the presence of puromycin as previously described (30). Flow cytometry was performed as previously described (17). Cell-growth inhibition was measured by an MTT assay as previously described (31).
Reverse transcription and western blot analysis
Two μg total RNA was reverse transcribed with random hexamers as previously described (22,32). Whole cell extracts were prepared, and western blot analysis was performed as previously described (31).
Array-comparative genome hybridization (array-CGH) analysis
Genomic DNA from cells was analyzed by array-CGH (33) using a 2 × 400K oligonucleotide microarray (Agilent Technologies) according to the manufacturer's recommendations as previously described (34). Test DNA (2 μg) and reference DNA (2 μg) was digested with AluI and RsaI (Promega). The digested test DNA and reference DNA were labeled with cyanine (Cy) 3-deoxyuridine triphosphate (dUTP) or Cy5-dUTP, respectively, using an Agilent Genomic DNA Labeling Kit PLUS (Agilent). The individually labeled test and reference samples were then purified using Microcon YM-30 filters (Millipore, Billerica, MA). Following purification, the Cy3-labeled test DNA and Cy5-labeled reference DNA were mixed together and combined with 2× hybridization buffer (Agilent), 10× blocking agent (Agilent) and human Cot-1 DNA (Invitrogen). The hybridization mixture was slowly dispensed onto a microarray chip and assembled with an Agilent SureHyb chamber (Agilent). The assembled slide chamber (Agilent) was placed in the rotator rack in a hybridization oven for 40 h at 65°C with suitable rotation. Hybridization was followed by two washes with Washing Buffer 1 and Washing Buffer 2 (Agilent) according to the manufacturer's instructions. After washing, all microarray slides were scanned with an Agilent Microarray Scanner G2505C at a 5-μm resolution. Captured images were transformed into data with Feature Extraction Software, version 10.7 (Agilent), and then imported into Agilent CGH Analytics 5.0.14 software for evaluation. From the array-CGH data, we subtracted the background intensity from the total spot intensity. To remove systematic bias of the chip, within-slide normalization was performed using Lowess normalization for log2 transformed data. Copy number data for the target and control samples were manually inspected. Probe-level data were segmented using the circular binary segmentation method (35) to detect statistically significant CNA values. Copy number gain or loss beyond log2 (RAD21-shRNA/control-shRNA) ± 0.3 was cataloged for each sample. All calculations were performed using R.
Paired-end transcriptome analysis
Sequencing libraries were generated according to the standard protocol of Illumina Inc. for high-throughput sequencing. The transcriptome was then sequenced using a Genome Analyzer IIx (Illumina Inc.) as previously described (34). We mapped 101-bp sequenced fragments to the human genome using TopHat2.0.8, which can allow up to two mismatches with the references. Sequenced reads were aligned to human transcript reference sequences from the UCSC database (Homo_sapiens.GRCh37/hg19) for expression analysis at the gene/transcript levels (36). We use the DEGseq R package program to detect differential expression mainly because this software supports an experimental design between two samples without multiple technical replicates (37). We identified differentially expressed genes (DEGs) according to the overall differential expression from the DEGseq analysis with FDR < 0.001. The genes with FDRs of less than 0.05 and normalized fold change values greater than 2 were shown in Supplementary Figure S6B.
CNV assay
Cells and human primary gastric cancer tissues and matched normal samples were assayed for gene copy number using TaqMan Gene Copy Number Assays (38). Each probe was designed based on a genomic sequence (Homo_sapiens.GRCh37/hg19) using Applied Biosystems proprietary software. Each assay was run as a TaqMan real-time PCR reaction in triplicate, using an FAM dye-based assay targeted to 11p13 and a VIC dye-based assay for the reference gene, RNase P (PN 4316844 from Applied Biosystems). Each 20 μl PCR reaction contained 20 ng gDNA and TaqMan probe/primer mix in TaqMan Universal Master Mix, and was amplified using StepOnePlus (Applied Biosystems). Cycling conditions were 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 15 s at 92°C and 60 s at 60°C. Real-time data were collected by CopyCaller v2.0 software. This method involved the relative quantification of the test sequence versus a reference gene known to have two copies per diploid genome. Relative quantity was determined using the ΔΔCt [(FAM Ct – VIC Ct) sample – (FAM Ct – VIC Ct) calibrator] method (17) in which a reference sample or calibrator known to have two copies of the test sequence is used as the basis for comparison.
Fluorescent in situ hybridization (FISH)
Cells were fixed with Carnoy's solution on slides and then dried. The cells were then covered with 10 μl dual hybridization mixture containing a pair of painting probes and labeled with two different fluorochromes. The painting probes were labeled with MacProbe™ solution (Macrogen). The slides and probes were denatured at 75°C for 2 min and hybridized overnight at 37°C. Post-hybridization washes were conducted according to the manufacturer's protocol.
Chromatin immunoprecipitation (ChIP) assay and quantitative real-time PCR (qPCR)
ChIP assays were performed as previously described (22,30). qPCR using SYBR Green (Molecular Probes) was performed to observe enriched DNA or cDNA using StepOnePlus (Applied Biosystems) as previously described (32). The enrichment of target DNA over the input was calculated using the ΔΔCt method, and the results were presented as the mean ± SEM (17,30,39). The PCR primers used for the ChIP and qPCR assays are available upon request.
Chromosome conformation capture (3C) assay
A 3C assay was performed as previously described (22,40,41) with minor modification. Briefly, chromatin crosslinked in 1% formaldehyde was digested with 1000 U of EcoRI (NEB) overnight followed by ligation with 2000 U of T4 DNA ligase (NEB) at 16°C for 4 h. Crosslinking was reversed, and the DNA was then purified by phenol extraction and ethanol precipitation as previously described (30). To generate control templates for the positive controls, equimolar amounts of the different BAC clones were mixed and digested with 200 U of EcoRI overnight at 37°C as previously described (42). After phenol extraction and ethanol precipitation, DNA fragments (200 ng/μl) were ligated with T4 DNA ligase. Digestion efficiency was calculated as previously described (43) and samples with efficiencies greater than 90% were used for the 3C assays. Crosslinking frequency and ligation efficiencies between different samples were normalized relative to the ligation frequency of two adjacent EcoRI fragments in the CalR gene (41). Quantitation of the data was performed by qPCR using SYBR Green (Molecular Probe).
Antibodies
Antibodies specific for the following factors were used in this study: c-Myc (SC-40), normal rabbit IgG (SC-2027), CDC45 (SC-20685) and RNA pol II (SC-899) from Santa Cruz Biotechnology; RAD21 (ab992) and DNA Pol α (ab31777) from AbCam, and CTCF (07–729) from Millipore. Anti-CD44 antibody (5640S) and anti-MCM7 (3735S) was purchased from Cell Signaling Technology. Anti-SMC1 antibody (A300–055A) was from Bethyl Lab.
Immunofluorecsence analysis
Cells were seeded on 0.01% poly-L-lysine (Sigma-Aldrich)-coated coverslips. Next day, the coverslips were rinsed once in PBS (37°C), fixed in 4% formaldehyde for 15 min, permeabilized with 0.5% Triton X-100 for 5 min and then incubated with primary antibody for 1 h at RT. The primary antibodies used in this study were rabbit polyclonal anti-MCM7 and anti-RAD21 at a dilution of 1:100. The coverslips were rinsed three times with PBS, followed by incubation with the appropriate fluorophore-conjugated secondary antibody (Invitrogen) for 1 h at RT. The cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 300 nmol/L; Invitrogen) and the coverslips were mounted on slides using Faramount aqueous mounting medium (DAKO).
RESULTS
Reduction of cohesin induces neither mitotic arrest nor defective chromosomal segregation in SNU16 cells
First, to explore if elevated cohesin level is required to maintain gene amplification, we stably reduced the expression of cohesin by RAD21-knockdown (KD) in SNU16 human gastric cancer cells with multiple chromosomal instability (CIN+) (Supplementary Figure S1A) (27,29). RAD21 is a core subunits of the cohesin complex (12). Two different RAD21-directed shRNAs reduced RAD21 expression with a similar efficiency, thus decreasing RAD21 mRNA and protein levels by approximately 90% in SNU16 cells (Figure 1A and B). In contrast, the levels of CTCF, which position cohesin on its site (44), were not affected in RAD21-KD SNU16 cells (Figure 1A and B). Using FACS analysis we observed that stable RAD21-KD SNU16 cells seemed to exit mitosis, and then divided and survived well without pronounced cell death (Figure 1C). Moreover, following treatment with nocodazol, a microtubule polymerization inhibitor (16), we also found that stable RAD21-KD does not induce aberrant sister chromatid separation (Figure 1D and E). Collectively, we conclude that the stable down-regulation of cohesin by RAD21-KD induces neither mitotic arrest nor defective chromosomal segregation during mitosis and meiosis in SNU16 cells with CIN+. We obtained similar results in three CIN+ (N87, COLO 320-HSR and COLO 320-DM) (7) and three cancer cells with stable chromosomes (CIN−) (HCT116, LoVo and HepG2) (Supplementary Figure S2 and S3) (15,16,26).
Figure 1.
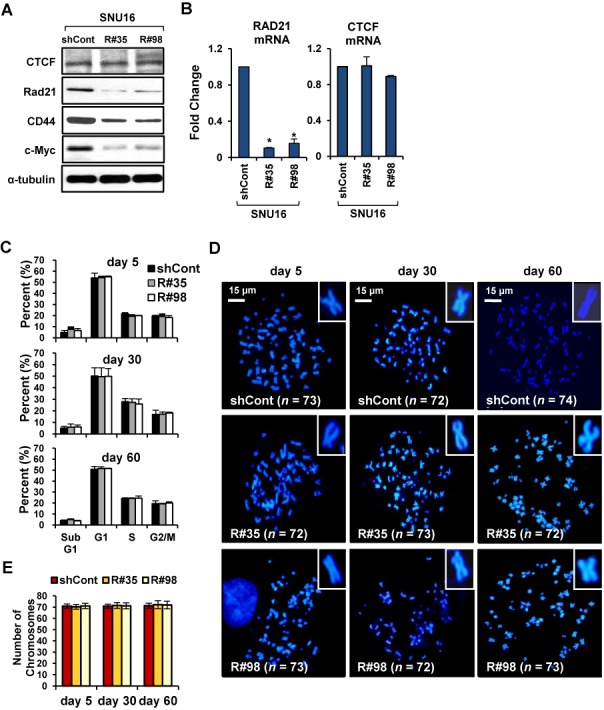
Stable down-regulation of cohesin does not led to mitotic arrest or the development of chromosomal segregation defect in SNU16 cells. (A, B) SNU16 cells were stably transduced with control GFP-shRNA or two different RAD21-shRNAs (R#35 and R#98). (A) Western blot analysis was performed with the indicated antibodies on day 60 after RAD21-KD in SNU16 cells. α-tubulin served as a loading control. (B) RAD21 or CTCF mRNA expression was analyzed by qRT-PCR on day 60 after RAD21-KD in SNU16 cells. Each value was normalized to that of 18S ribosomal RNA relative to the control GFP-shRNA expressing SNU16 cells. Error bars represent the SD, n = 3 biological replicates from independent viral transduction. *P < 0.01; Student's t test. (C) SNU16 cells were stained with propidium iodide at the day 5, 30 and 60 after RAD21-KD, and then subjected to FACS analysis. The percentage of cells in the Sub G1, G1, S and G2/M phases are shown. (Mean ± SD, n = 3 biological replicates). (D) A metaphase chromosome spread was prepared by treatment with nocodazol, a microtubule polymerization inhibitor (16), at the day 5, 30 and 60 after RAD21-KD in SNU16 cells and then cells were stained with DAPI. Scale bar = 15 μm. (E) The total chromosome numbers of the control GFP-KD or RAD21-KD SNU16 cells populations were counted at the day 5, 30 and 60 after three independent viral transduction procedures. Fifty mitotic spreads were evaluated for each sample (± SD). Representative FISH images are shown in (D).
Reduction of cohesin coordinately leads to copy number-associated gene expression changes in SNU16 cells
To directly assess the influence of cohesin depletion on gene amplification, we systematically identified copy-number changes in RAD21-KD SNU16 cells. Using array-CGH analysis (34), we found that genome-wide copy-number alterations were observed in RAD21-KD SNU16 cells compared to the control GFP-shRNA expressing SNU16 cells (Supplementary Figure S1B). To further determine whether DNA copy-number alterations produce corresponding changes in gene expression, copy number-associated gene expression alterations were monitored in RAD21-KD SNU16 cells. First, to assess the transcriptional impact of cohesin reduction, we carried out paired-end transcriptome analysis (34) from the control GFP-KD or RAD21-KD SNU16 cells and identified global gene expression changes in RAD21-KD SNU16 cells (Supplementary Figure S4). Next, genome-wide array-CGH data and transcriptional profiles were integrated to search for candidate target genes with concomitantly altered DNA copy numbers and gene expression levels following cohesin reduction (data not shown). By matching differentially expressed genes to the corresponding copy-number, we identified six segment regions, including the WDR11 and APIP/PDHX/CD44 locus (see below), in which altered expression significantly correlated with changes in DNA copy-number (Supplementary Table S1). Taken together, our results suggest that reduction of cohesin by RAD21-KD coordinately lead to copy number-associated gene expression alterations in SNU16 cells.
Cohesin-mediated long-range chromatin interactions is required for transcriptional efficiency of the APIP/PDHX/CD44 locus in SNU16 cells
In order to investigate whether cohesin can directly regulate the expression of amplified genes, we evaluated the highly amplified APIP/PDHX/CD44 locus in SNU16 cells. APIP/PDHX/CD44 locus, whose expression was responsive to RAD21-KD, are localized within 500 kb in chromosome 11 (Figure 2A and B). Since CTCF plays important role in cohesin positioning on chromatin (44), we determined whether CTCF is present at the APIP/PDHX/CD44 locus. A UCSC Genome Bioinformatics database (http://genome.ucsc.edu/) search for CTCF binding motifs across the APIP/PDHX/CD44 locus revealed numerous putative candidate sites (Figure 2A). Chromatin immunoprecipitation (ChIP) assays showed that CTCF binds within the APIP/PDHX/CD44 locus in SNU16 cells (Figure 2C). Each value was normalized with gene copy number to determine the correct signal per region (data not shown). Prominent bindings of RAD21 and SMC1, two members of the cohesin complex (12), were co-localized with CTCF in SNU16 cells (Figure 2D and E), suggesting that APIP/PDHX/CD44 locus is a natural target of the CTCF and cohesin complex.
Figure 2.
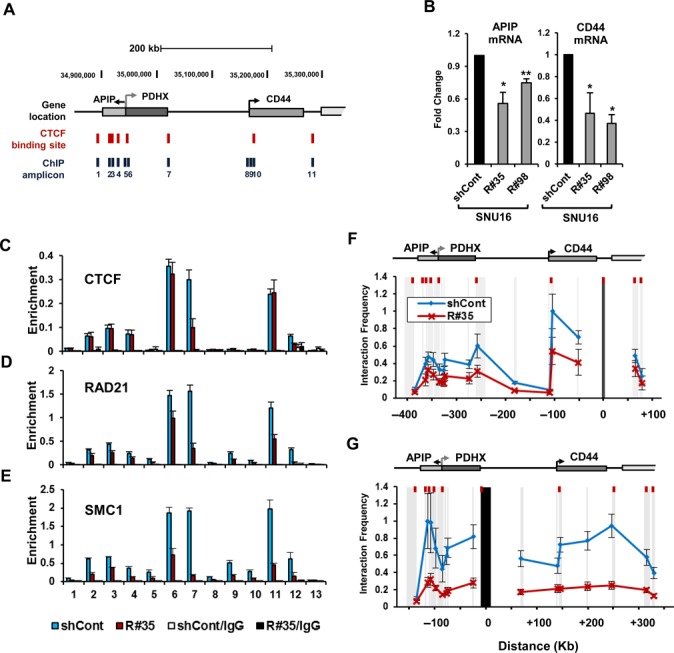
Reduction of cohesin leads to transcriptional inefficiency of the APIP/PDHX/CD44 locus in SNU16 cells. (A) The APIP/PDHX/CD44 locus at chromosome 11p13 is illustrated to scale. The location of putative CTCF/cohesin binding sites and the primer pairs used for qPCR are shown with names below. (B) APIP and CD44 expression was measured by qRT-PCR on day 60 after RAD21-KD in SNU16 cells. Each value was normalized to that of 18S ribosomal RNA relative to the control GFP-KD cells. (Mean ± SD, n = 3 biological replicates. *P < 0.01; **P < 0.05). (C–E) A ChIP assay was carried out with the control GFP-KD (blue bar) or RAD21-KD SNU16 cells (R#35; red bar) on day 60 after lentivial transduction using antibodies against to (C) CTCF, (D) RAD21, and (E) SMC1. The enrichment of target DNA over the input was calculated using the ΔΔCt method (17). 3′HS1 (amplicon 12) of the human β-globin locus and Necdin (amplicon 13) served as positive and negative controls, respectively, for CTCF/RAD21 binding (17). (Mean ± SEM, n = 3 biological replicates). (F, G) Relative crosslinking frequencies among CTCF/RAD21 binding sites within the APIP/PDHX/CD44 locus were measured with a 3C assay in the control GFP-KD (blue line) or RAD21-KD SNU16 cells (red line) on day 60 after lentivial transduction. The EcoRI restriction fragments in the APIP/PDHX/CD44 locus appear as gray shaded bars. Black shading indicates the anchor fragment of (F) the 3′ end of CD44 and (G) the 3′ end of PDHX. Each value was normalized to the crosslinking frequency at the CalR gene and the total DNA copy numbers to determine the correct signal per region (41). The maximum crosslinking frequency was set to 1. (Mean ± SEM, n = 3 biological replicates).
Increasing evidence has recently indicated that cohesin, along with CTCF, mediates high-order chromatin structures among CTCF/cohesin binding sites for the transcriptional regulation of many developmentally regulated gene families (17–20,22). To test whether the APIP/PDHX/CD44 locus forms typical long-range chromatin interactions through cohesin binding, we carried out a chromosome conformation capture (3C) analysis (40) to assess the proximity of chromatin across the APIP/PDHX/CD44 locus. Because high levels of cohesin occupancy is apparent in the CTCF/cohesin binding site at the 3′ end of CD44 in SNU16 cells (amplicon 11 in Figure 2D), the EcoRI restriction fragment containing this region was used as an anchor primer. The 3′ end of CD44 was found to strongly interact with the CTCF/cohesin binding sites within the coding region of APIP, the 3′ end of PDHX and the promoter region of CD44 in SNU16 cells (blue line, Figure 2F). When we used primers complementary to the 3′ end of PDHX as anchors (amplicon 7 in Figure 2D), we also found strong interactions between PDHX and both APIP and CD44 in SNU16 cells (blue line, Figure 2G), suggesting that the APIP/PDHX/CD44 locus has a high-order chromosome architecture in SNU16 cells.
Next, we examined whether cohesin was crucial for the observed long-range chromatin interactions and gene expression. First, we confirmed that high level of APIP and CD44 expression was reduced by RAD21-KD in SNU16 cells (Figure 2B). Second, our ChIP experiments showed that RAD21 and SMC1 enrichments at the APIP/PDHX/CD44 locus in RAD21-KD SNU16 cells were reduced twofold on average compared with those in the control GFP-shRNA expressing cells (Figure 2D and E). Consistent with the decreased enrichment of cohesin, the physical proximity in the RAD21-KD SNU16 cells (red line, Figure 2F and G) was significantly lower (twofold) than that in the control GFP-KD cells (blue line, Figure 2F and G). We observed the down-regulation of cohesin enrichment and a reduction of long-range chromatin interactions between the CTCF/cohesin binding sites within these amplified regions as early as 5 days after RAD21-KD in SNU16 cells (Supplementary Figure S5 and S6). Thereafter, gene expression of highly amplified APIP/PDHX/CD44 was significantly reduced in RAD21-KD SNU16 cells (Supplementary Figure S7C). Collectively, these results suggest that cohesin-mediated high-order chromosome architecture is required for transcriptional efficiency of the APIP/PDHX/CD44 locus in SNU16 cells.
Copy-numbers of amplified APIP/PDHX/CD44 locus are decreased by cohesin reduction in SNU16 cells
Not only the gene transcription but also the amplified copy-number of the APIP/PDHX/CD44 locus in SNU16 cells was notably reduced by RAD21-KD (Figure 3A). Interestingly, the amplified APIP/PDHX/CD44 locus appeared as DMs (indicated as  in Figure 3B), HSRs (indicated as
in Figure 3B), HSRs (indicated as  in Figure 3B) and distributed insertions (indicated as
in Figure 3B) and distributed insertions (indicated as  in Figure 3B) in parental SNU16 cells (4,7). Thus, to elucidate the copy-number changes induced by cohesin reduction with greater accuracy, a TaqMan-quantitative PCR-based CNV assay (38) targeting APIP/PDHX/CD44 locus was performed. Compared with the two copies of APIP and CD44 in the HCT116 cells, more than 100 copies of APIP and CD44 were found in SNU16 cells (Figure 3C). Notably, the copy-number of APIP and CD44 in the RAD21-KD SNU16 cells was significantly reduced relative to that in the control GFP-KD cells (Figure 3C). Next, FISH analysis of a metaphase spread of RAD21-KD SNU16 cells revealed that the amplified APIP/PDHX/CD44 locus located in the HSRs was slightly reduced by RAD21-KD (Figure 3D). In addition, RAD21-KD in SNU16 cells also led to a decrease in the total number of DMs bearing the APIP/PDHX/CD44 locus (Figure 3E), suggesting that cohesin reduction via RAD21-KD decreases the focal copy-number of amplified APIP/PDHX/CD44 segments, which exist in both HSRs and DMs in SNU16 cells. DNA copy-numbers of APIP and CD44 started to dwindle 30 days after viral transduction of RAD21-directed shRNAs and subsequently remained low throughout the rest of the experiment (Supplementary Figure S7A). However, the copy-number changes of the same set of genes were not detected following RAD21-KD in HCT116, LoVo and HepG2 cancer cells (Supplementary Figures S7B and S8). Collectively, these results suggest that down-regulation of cohesin induces copy-number loss of amplified APIP/PDHX/CD44 locus on HSRs and DMs only in CIN+ SNU16 cells but not in other cancer cells having stable chromosome.
in Figure 3B) in parental SNU16 cells (4,7). Thus, to elucidate the copy-number changes induced by cohesin reduction with greater accuracy, a TaqMan-quantitative PCR-based CNV assay (38) targeting APIP/PDHX/CD44 locus was performed. Compared with the two copies of APIP and CD44 in the HCT116 cells, more than 100 copies of APIP and CD44 were found in SNU16 cells (Figure 3C). Notably, the copy-number of APIP and CD44 in the RAD21-KD SNU16 cells was significantly reduced relative to that in the control GFP-KD cells (Figure 3C). Next, FISH analysis of a metaphase spread of RAD21-KD SNU16 cells revealed that the amplified APIP/PDHX/CD44 locus located in the HSRs was slightly reduced by RAD21-KD (Figure 3D). In addition, RAD21-KD in SNU16 cells also led to a decrease in the total number of DMs bearing the APIP/PDHX/CD44 locus (Figure 3E), suggesting that cohesin reduction via RAD21-KD decreases the focal copy-number of amplified APIP/PDHX/CD44 segments, which exist in both HSRs and DMs in SNU16 cells. DNA copy-numbers of APIP and CD44 started to dwindle 30 days after viral transduction of RAD21-directed shRNAs and subsequently remained low throughout the rest of the experiment (Supplementary Figure S7A). However, the copy-number changes of the same set of genes were not detected following RAD21-KD in HCT116, LoVo and HepG2 cancer cells (Supplementary Figures S7B and S8). Collectively, these results suggest that down-regulation of cohesin induces copy-number loss of amplified APIP/PDHX/CD44 locus on HSRs and DMs only in CIN+ SNU16 cells but not in other cancer cells having stable chromosome.
Figure 3.
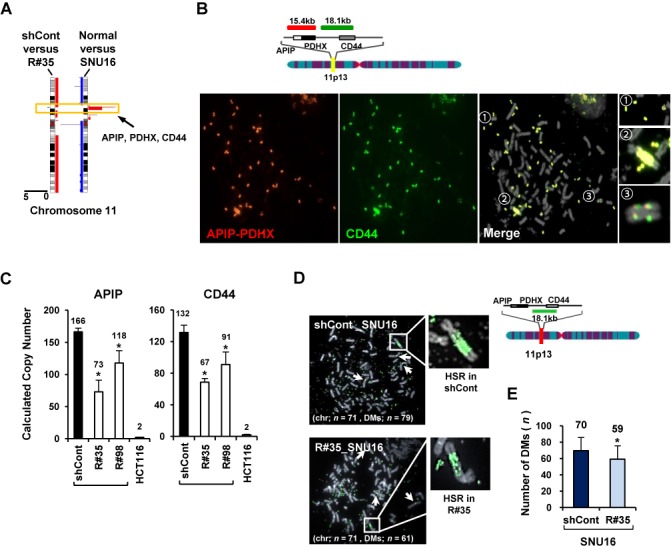
Copy-numbers of amplified APIP/PDHX/CD44 locus are decreased by cohesin reduction in SNU16 cells. (A) High-resolution array-CGH analysis was performed with the control GFP-KD or RAD21-KD SNU16 cells on day 60 after RAD21-KD. Chromosome 11 is represented by ideograms showing G-banding patterns (left ideogram, RAD21-KD SNU16 cells compared with the control GFP-KD cells; right ideogram, parental SNU16 cells compared with normal gastric cells). Gains/amplifications (red) are shown on the right side of each ideogram, while losses (blue) appear on the left side. APIP/PDHX/CD44 locus for further analyses are indicated by yellow boxes. (B) Localization of the FISH probes specific for APIP/PDHX (labeled with Cy3, red), and CD44 (labeled with FITC, green) located on chromosome 11p13 is depicted. Metaphase FISH analysis of the APIP/PDHX/CD44 locus on chromosome 11p13 in SNU16 cells revealed that two different probes co-localized on the same locus and exist in three types of amplification: DMs (), HSRs () and distributed insertions (). (C) Cohesin-mediated focal copy-number changes of APIP and CD44 were evaluated using a TaqMan-quantitative PCR-based CNV assay with control GFP-KD or RAD21-KD SNU16 cells on day 60 after viral transduction. Data are presented as averages ± SD of biological triplicate independent viral transduction experiments. *P < 0.01. (D) A metaphase FISH analysis was performed to assess CD44 on HSR or DMs in the control GFP-KD (shCont) and RAD21-KD SNU16 cells (R#35) on day 60 after lentivial transduction. Chromosome-2 centromere (labeled with Cy3 for the control; white arrows) served as control. A schematic representation of the localization of the FISH probes specific for CD44 (labeled with FITC; green) located at chromosome 11p13 is shown above. (E) The total numbers per cell of DMs containing CD44 were blindly counted in the control GFP-KD and RAD21-KD SNU16 cells on day 60 after lentivial transduction. Fifty mitotic spreads were evaluated per sample (± SD). Representative FISH images are shown in (D). *P < 0.01.
Cohesin depletion reduces the enrichment of pre-replication complex at replication origins near amplified regions
DNA replication occurs in two steps: the licensing of origin of replication and the initiation of DNA replication (45–47). Pre-replication complex (pre-RC) is responsible for proper recruitment of replication licensing machinery at replication origins (45,46). Interestingly, several pieces of evidence have suggested that perturbation of DNA replication initiation can eliminate the number of amplified genes on DMs in several human cancers (48,49). Thus, a regional DNA replication defect by improper recruitment of pre-RC may contribute to change the overall copy-number of amplified genes (4,8,47,50). Recently, cohesin has been shown to directly regulate DNA replication (39,51). Moreover, cohesin complexes are particularly enriched at replication origins during DNA replication (39,52,53). Hence, it is of interest to determine whether cohesin depletion might affect the enrichment of pre-RC at replication origins near amplified genes.
First, we compared the binding of pre-RC to the APIP/PDHX/CD44 locus in CIN+ SNU16 cells with a highly amplified APIP/PDHX/CD44 locus on HSRs and DMs and HCT116 cells having two copies of APIP/PDHX/CD44. Based on a DNA replication origin database (DeOri; http://tubic.tju.edu.cn/deori/), we identified four DNA replication origins near the APIP/PDHX/CD44 locus (Figure 4). pre-RC is composed of several proteins, such as the origin recognition complex (ORC), six minichromosome maintenance subunits (MCM 2–7), CDC45, GINS tetramer and DNA polymerases (Pol α, Pol ϵ and Pol δ) (54–56). Binding of Pol α, which is required for eukaryotic DNA replication (57), at replication origins within the APIP/PDHX/CD44 locus was strongly enriched in SNU16 cells (Figure 4A). In contrast, relatively low Pol α binding was detected in HCT116 cells. Each value was normalized with gene copy number to determine the correct signal per region (data not shown). Similarly, enrichment of MCM7, which forms a pre-RC to recruit the DNA polymerase complex to target origins and functions as a replicative DNA helicase (58,59), in SNU16 cells was higher than in HCT116 cells. We also observed strong recruitment of CDC45, a well-known MCM helicase activator (59), at replication origins within the APIP/PDHX/CD44 locus in SNU16 cells but not in HCT116 cells, suggesting a positive correlation between DNA copy number and the enrichment of pre-RC.
Figure 4.
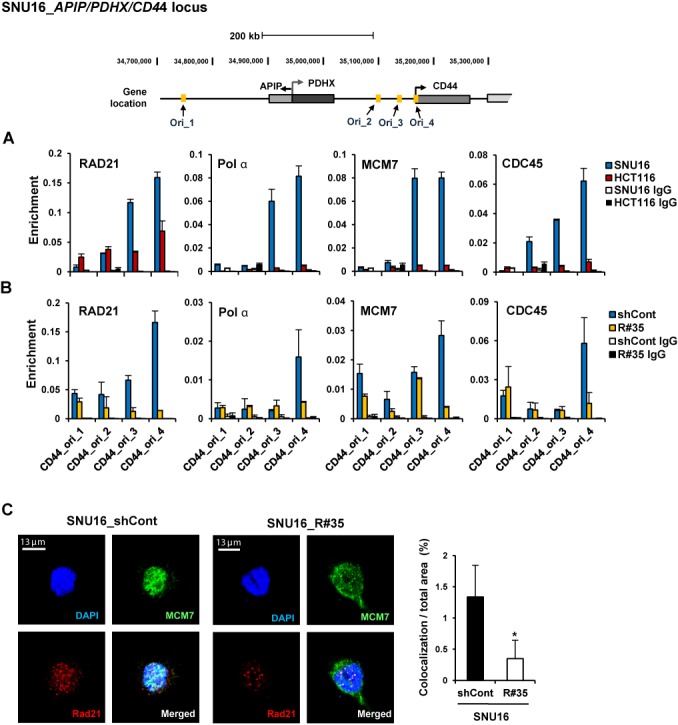
Down-regulation of cohesin impairs localization of pre-replication complex at replication origins within the amplified APIP/PDHX/CD44 locus. (A) A ChIP assay was performed with SNU16 (blue bar) and HCT116 (red bar) cells using antibodies against RAD21, DNA polymerase α (Pol α), MCM7 and CDC45. The location of previously published replication origin sequences (70) near the APIP/PDHX/CD44 locus used for qPCR are shown with names below. (Mean ± SEM, n = 3 biological replicates). (B) A ChIP assay was performed with control GFP-KD (blue bar) or RAD21-KD SNU16 cells (R#35; yellow bar) on day 60 after lentivial transduction using antibodies against RAD21, Pol α, MCM7 and CDC45. (Mean ± SEM, n = 3 biological replicates). (C) Immunofluorescence staining of DNA (DAPI, blue), MCM7 (green) and RAD21 (red) in control GFP- KD (left panel) or RAD21-KD SNU16 cells (right panel). (D) Percentage of MCM7 and RAD21 co-localization in control GFP-KD (filled bar) or RAD21-KD SNU16 cells (unfilled bar) by unbiased automatic quantification (39). Data are presented as averages ± SD of individual cells (n = 100). *P < 0.01; Student's t test.
We next found that strong binding of pre-RC was notably impaired in RAD21-KD SNU16 cells compared to control GFP-KD cells. Significantly fewer pre-RC was detected in RAD21-KD SNU16 cells at replication origins within the APIP/PDHX/CD44 locus (Figure 4B and Supplementary Figure S9D and E). Using immunofluorescence assays, we found that RAD21 depletion led to reduced co-localization of MCM7 and RAD21 (Figure 4C and D). However, reduction of cohesin by RAD21-KD did not change the basal mRNA expression of pre-RC (Supplementary Figure S9C) (39). Taken together, these results suggest that cohesin depletion reduces the enrichment of pre-RC at replication origins near amplified regions on HSRs and DMs. Thus, continuous reduction of active DNA-replication, which is essential for the maintenance of amplified genes on HSRs and DMs (7), by cohesin depletion might provoke copy-number loss of amplified oncogenes in CIN+ SNU16 cancer cells (4,8,47).
Copy-numbers of amplified gene existing in both HSRs and DMs are decreased by cohesin reduction
Next, to further confirm that two different types of gene amplification, HSRs and DMs, could be affected by cohesin reduction, a CNV assay was performed with COLO 320-HSR and COLO 320-DM cells stably transfected with control GFP-shRNA or two different RAD21-shRNAs (Supplementary Figure S3B and C). COLO 320-HSR and COLO 320-DM cells are derived from human colon carcinoma and carry multiple copies of c-Myc localized in HSR and DMs, respectively (7). By RAD21-KD in the COLO 320-HSR cells, we found that cohesin reduction induced copy-number loss of the highly amplified c-Myc without loss of HSR harboring c-Myc (Figure 5A–C). We similarly observed that RAD21-KD resulted in copy-number loss of the c-Myc localized in DMs in COLO 320-DM cells (Figure 6A–C), suggesting that down-regulation of cohesin decreases the high-level c-Myc amplifications existing in both HSRs and DMs in cancer cells.
Figure 5.
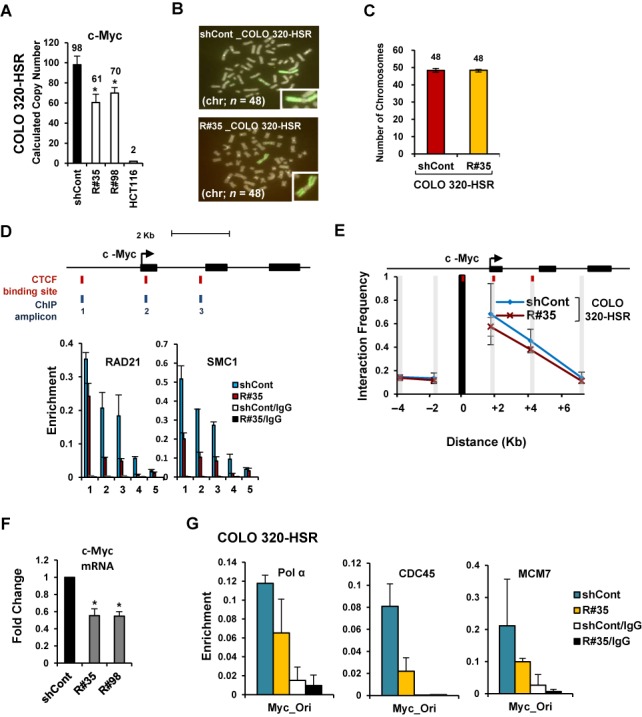
Copy-numbers of amplified gene existing in HSRs are decreased by cohesin reduction. (A) COLO 320-HSR cells were stably transduced with control GFP-shRNA or two different Rad21-shRNAs (R#35 and R#98). Changes in copy-numbers of the c-Myc were quantified using a TaqMan quantitative PCR-based CNV assay. The average of three viral transduction experiments ± SD is presented. *P < 0.01. (B) Metaphase FISH analysis with c-Myc (labeled with FITC; green) and chromosome 2 centromere (labeled with Cy3 for the control; red) revealed a reduction of c-Myc located within HSR in RAD21-KD COLO 320-HSR cells. Notably, amplified c-Myc exists as HSR and inverted repeats within the chromosome of the COLO 320-HSR cells. (C) Total chromosome numbers of the control GFP-KD or RAD21-KD COLO 320-HSR cells were counted after treatment with nocodazol. Fifty mitotic spreads were evaluated for each sample (± SD). (D) A ChIP was performed with antibodies to Rad21 and SMC1 in control GFP-KD (blue bar) or RAD21-KD COLO 320-HSR (red bar). (Mean ± SEM, n = 3). c-Myc at chromosome 8q24 are illustrated to scale. The location of putative CTCF/Rad21 binding sites and primer pairs used for qPCR are shown with names below. 3′HS1 of the human β-globin locus (amplicon 4) and Necdin (amplicon 5) served as controls. (E) 3C assay of c-Myc in COLO 320-HSR cells after RAD21-KD. DpnII restriction sites in c-Myc appear as gray shaded bars. Black shading indicates the anchor fragment of the 5′ CTCF binding sites of c-Myc. (Mean ± SEM, n = 3 biological replicates). (F) The expression of c-Myc was measured by qRT-PCR on day 60 after RAD21-KD in COLO 320-HSR cells. Each value was normalized to that of 18S ribosomal RNA relative to the control GFP-KD cells. The average of biological triplicate independent viral transduction experiments ± SD is presented. *P < 0.01. (G) A ChIP assay using previously published replication origin sequences near the c-Myc locus (70) was performed with antibodies against Pol α, CDC45 and MCM7 in control GFP-KD or RAD21-KD COLO 320-HSR cells on day 60 after lentivial transduction. (Mean ± SEM, n = 3 biological replicates).
Figure 6.
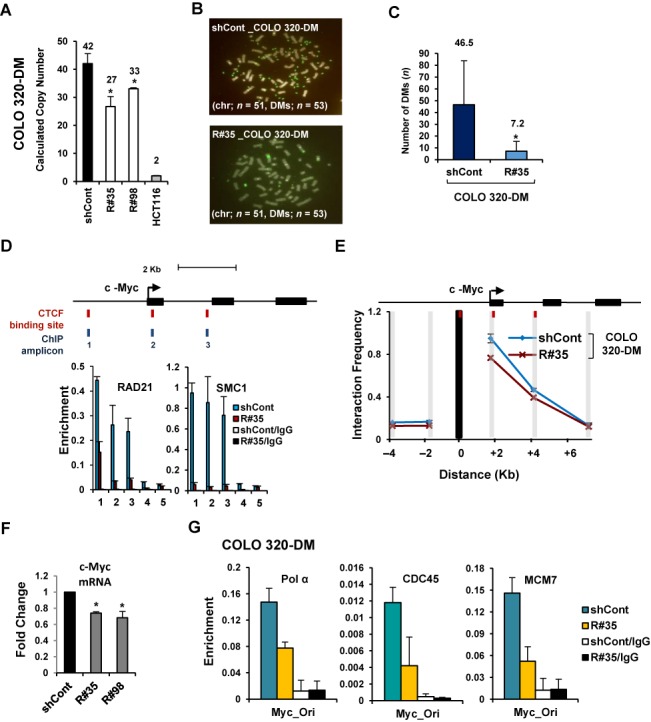
Copy-numbers of amplified gene existing in DMs are decreased by cohesin reduction. (A) COLO 320-DM cells were stably transduced with control GFP-shRNA or two different Rad21-shRNAs (R#35 and R#98). Changes in copy-numbers of the c-Myc were quantified using a TaqMan quantitative PCR-based CNV assay. The average of three viral transduction experiments ± SD is presented. *P < 0.01. (B) Metaphase FISH analysis with c-Myc (labeled with FITC; green) and chromosome 2 centromere (labeled with Cy3 for the control; red) revealed a reduction of c-Myc located within DMs in RAD21-KD COLO 320-DM cells. (C) Total numbers of DMs per cell were blindly counted in control GFP-KD and RAD21-KD COLO 320-DM cells. (Mean ± SD, n = 50). *P < 0.01. (D) A ChIP was performed with antibodies to RAD21 and SMC1 in control GFP-KD (blue bar) or RAD21-KD COLO 320-DM (red bar). (Mean ± SEM, n = 3 biological replicates). 3′HS1 of the human β-globin locus (amplicon 4) and Necdin (amplicon 5) served as controls. (E) 3C assay of c-Myc in COLO 320-DM cells after RAD21-KD. DpnII restriction sites in c-Myc appear as gray shaded bars. Black shading indicates the anchor fragment of the 5′ CTCF binding sites of c-Myc. (Mean ± SEM, n = 3 biological replicates). (F) The expression of c-Myc was measured by qRT-PCR on day 60 after RAD21-KD in COLO 320-DM cells. Each value was normalized to that of 18S ribosomal RNA relative to the control GFP-shRNA expressing cells. The average of triplicate independent viral transduction experiments ± SD is presented. *P < 0.01. (G) A ChIP assay using previously published replication origin sequences near the c-Myc locus (70) was performed with antibodies against Pol α, CDC45 and MCM7 in control GFP-KD or RAD21-KD COLO 320-DM cells on day 60 after lentiviral transduction. (Mean ± SEM, n = 3 biological replicates).
We also found that RAD21-KD blocks the formation and enrichment of cohesin complex near the c-Myc in COLO 320-HSR and COLO 320-DM cells (Figures 5D and 6D). The c-Myc segment formed long-range chromatin interactions in both COLO 320-HSR and COLO 320-DM cells (blue line, Figures 5E and 6E). Down-regulation of cohesin by RAD21-KD resulted in the release of spatial chromatin organization of the c-Myc in COLO 320-HSR and COLO 320-DM cells (red line, Figures 5E and 6E), resulting in a striking reduction in c-Myc expression (Figures 5F and 6F). We also found that the binding of Pol α, CDC45 and MCM7 at replication origins near c-Myc were diminished by RAD21-KD in COLO 320-HSR and COLO 320-DM (Figures 5G, 6G, and Supplementary Figure S9A and B). Collectively, we demonstrated that cohesin reduction via RAD21-KD decreases the expression and the focal copy-number of amplified genes existing on both HSR and DMs in CIN+ cancer cells, thereby maintaining low expression levels of previously highly amplified genes (Figure 7).
Figure 7.
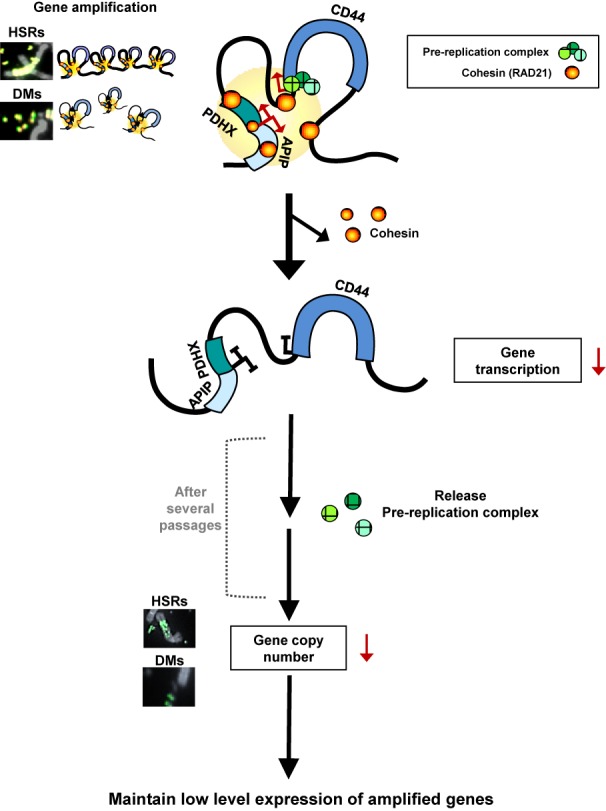
Proposed model for cohesin-mediated copy-number loss of highly amplified genes existing on DMs and HSRs in human cancer cells. APIP/PDHX/CD44 locus located in HSRs and DMs forms spatial chromatin organizations in SNU16 cells. Cohesin appeared to stabilize long-range chromatin interactions through direct interactions among CTCF/cohesin localization sites. Down-regulation of cohesin by RAD21-KD abolished the proximity between CTCF/Rad21 binding sites within these segments, thereby reducing APIP/PDHX/CD44 expression. After several passages, RAD21-KD decreases the enrichment of pre-RC at replication origins near these regions, result in reducing the copy numbers of amplified genes existing in HSRs and/or DMs in cancer cells. See text for more details.
DISCUSSION
Cohesin is required to form sister chromatid cohesion for proper chromosome segregation during mitosis and meiosis (13). Therefore, defects in chromosomal segregation caused by inactivating mutations of chromatid cohesion genes leading to increased chromosomal instability and aneuploidy have been identified in human cancer (16,60). Although a high frequency of recurrent mutations and deletions of cohesin components has been found in various types of human cancer (16,61,62), such genetic alternations in genes encoding the cohesin components seem to be very rare in our human gastric cancer samples (data not shown). Instead, aberrant overexpression of cohesin components is frequently observed in human gastric cancer (15 out of the 24 samples, 62.5%) (Supplementary Figure S10A and C). We also tested the human gastric cancer data set with 50 tumor and normal pairs from the Cancer Genome Atlas (TCGA) database and obtained similar results (Supplementary Figure S10B). This suggests the possibility that elevated cohesin level may generally affect tumorigenesis through alternative mechanisms other than the induction of sister chromatid cohesion defects in human gastric cancer.
Since human gastric cancers show a high level of gene amplification (Supplementary Figure S11) (27,29,63), we therefore wondered if elevated cohesin level is required to maintain gene amplification. In the current study, we found that cohesin reduction decreases the focal copy-number of amplified APIP/PDHX/CD44 segments on HSRs and DMs in SNU16 cells. Using COLO 320-HSR and COLO 320-DM cells, we also confirmed that down-regulation of cohesin enrichment can generate the copy-number loss of amplified c-Myc existing on both HSRs and DMs. Considering the fact that our RAD21-KD cells proliferated well without developing defective sister chromatid cohesion formation or aberrant chromosome segregation during mitosis and meiosis (Figure 1 and Supplementary Figures S2 and S3), cohesin-mediated copy-number alterations might be independent of mitotic defects or chromosomal missegregation (64,65). To extend the generality of our observations demonstrating the effects of cohesin reduction, we tested the highly amplified WDR11-AS1/WDR11 locus in RAD21-KD SNU16 cells and obtained the same results (see Supplementary Text, Supplementary Table S1 and Supplementary Figure S12). In addition, we observed very similar results after RAD21-KD in N87 gastric cancer cells with HER2 amplification (see Supplementary Text and Supplementary Figure S13). However, copy-number changes of the same set of genes were not detected following RAD21-KD in near-diploid HCT116, LoVo and HepG2 cells (Supplementary Figures S7 and S8). Therefore, cohesin-mediated copy-number loss of highly amplified genes on HSRs and DMs may be not a general characteristic of cells with stable chromosomes but is instead specific to cancer cells with multiple structural chromosomal changes.
Although the nature of gene amplification in human cancer is not fully understood yet, in general, gene amplification resided on the DMs or HSR occurs and is maintained by DNA replication initiation during tumorigenesis (4,7,45,46). Since the DMs are autonomously replicating extrachromosomal genetic elements (4,7), repeated rounds of replication initiation of the DMs may be required for efficient gene amplification (7). Therefore, disruption of DNA replication initiation by treatment of DNA replication inhibitors such as hydroxyurea or ionizing radiation can eliminate the number of amplified genes on DMs in human cancers (7,48,49). Furthermore, because DMs can be generated from the repeated BFB cycle triggered by DSBs of HSR (66), failure to maintain DMs by down-regulating cohesin molecules may also reduce gene amplification resided on the HSR. In this study, we found that cohesin and pre-RC are highly enriched at replication origins near amplified genes. Cohesin depletion resulted in a reduced recruitment of pre-RC such as Pol α, CDC45 and MCM7 (Figure 4). This result suggests that deregulating DNA replication initiation by cohesin reduction might generate a regional replication defect by perturbing proper recruitment of replication licensing machinery, subsequently changing the overall copy number of amplified genes resided on the DMs or HSR (4,8,47,50).
Highly amplified APIP/PDHX/CD44, WDR11-AS1/WDR11 and c-Myc in SNU16 cells are either directly or indirectly linked to tumor progression and chemosensitivity (67–69). Down-regulation of cohesin by RAD21-KD enhanced the anti-proliferative effects of DNA-damaging agents, cisplatin or PARP inhibitor, in SNU16 and N87 cancer cells (Supplementary Figure S14), consistent with earlier data (24,25). In the case of HCT116, LoVo and HepG2 cancer cells with stable chromosome, however, no significant effect was observed following treatment with DNA-damaging agents (Supplementary Figure S14). Thus, the copy-number loss of oncogenic genes might sensitize cancer cells to DNA-damaging anti-cancer drugs (67). Given that gene amplification is associated with drug resistance and an unfavorable prognosis (4), the regulation of gene amplification by down-regulation of elevated cohesin component holds promise as a therapeutic strategy for treating cancer with genomic instability.
In summary, we have shown that down-regulation of elevated cohesin by RAD21-KD disrupts the cohesin-mediated chromatin structure and the enrichment of pre-RC at replication origins near the amplified regions existing in both HSRs and DMs, resulting in reducing the copy numbers of amplified genes in human cancer cells. Thus, cohesin is essential to stabilize high-level gene amplification in cancer cells with genomic instability. These results highlighted the importance of the elevated cohesin level in the formation of genomic instability in human cancer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors are grateful to Drs Ann Dean, Hyunsook Lee, Young-Joon Kim, Dongsoon Lee and Jong-Wan Park for critical reading and helpful comments.
FUNDING
National R&D Program for Cancer Control [Ministry of Health and Welfare, 0720540]; Basic Science Research Program [Ministry of Education, NRF-2011-0021123]; Mid-career Researcher Program [Ministry of Science, ICT and Future Planning, NRF-2013R1A2A2A01009297]; Korea Health Technology R&D Project through the Korea Health Industry Development Institute [Ministry of Health and Welfare, HI14C1277] Republic of Korea. Funding for open access charge: Mid-career Researcher Program [MSIP, NRF-2013R1A2A2A01009297].
Conflict of interest statement. None declared.
REFERENCES
- 1.Surani M.A., Hayashi K., Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Lengauer C., Kinzler K.W., Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 3.Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 4.Albertson D.G. Gene amplification in cancer. Trends Genet. 2006;22:447–455. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Santarius T., Shipley J., Brewer D., Stratton M.R., Cooper C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka H., Yao M.C. Palindromic gene amplification–an evolutionarily conserved role for DNA inverted repeats in the genome. Nat. Rev. Cancer. 2009;9:216–224. doi: 10.1038/nrc2591. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu N. Extrachromosomal double minutes and chromosomal homogeneously staining regions as probes for chromosome research. Cytogenet. Genome Res. 2009;124:312–326. doi: 10.1159/000218135. [DOI] [PubMed] [Google Scholar]
- 8.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kondratova A., Watanabe T., Marotta M., Cannon M., Segall A.M., Serre D., Tanaka H. Replication fork integrity and intra-S phase checkpoint suppress gene amplification. Nucleic Acids Res. 2015;43:2678–2690. doi: 10.1093/nar/gkv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coquelle A., Pipiras E., Toledo F., Buttin G., Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 11.Stuart D., Sellers W.R. Linking somatic genetic alterations in cancer to therapeutics. Curr. Opin. Cell Biol. 2009;21:304–310. doi: 10.1016/j.ceb.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Dorsett D., Strom L. The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr. Biol. 2012;22:R240–R250. doi: 10.1016/j.cub.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nasmyth K., Haering C.H. Cohesin: its roles and mechanisms. Annu. Rev. Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 14.Watrin E., Peters J.M. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 2009;28:2625–2635. doi: 10.1038/emboj.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber T.D., McManus K., Yuen K.W., Reis M., Parmigiani G., Shen D., Barrett I., Nouhi Y., Spencer F., Markowitz S., et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solomon D.A., Kim T., Diaz-Martinez L.A., Fair J., Elkahloun A.G., Harris B.T., Toretsky J.A., Rosenberg S.A., Shukla N., Ladanyi M., et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–1043. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou C., Dale R., Dean A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. U.S.A. 2010;107:3651–3656. doi: 10.1073/pnas.0912087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nativio R., Wendt K.S., Ito Y., Huddleston J.E., Uribe-Lewis S., Woodfine K., Krueger C., Reik W., Peters J.M., Murrell A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009;5:e1000739. doi: 10.1371/journal.pgen.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadjur S., Williams L.M., Ryan N.K., Cobb B.S., Sexton T., Fraser P., Fisher A.G., Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gosalia N., Neems D., Kerschner J.L., Kosak S.T., Harris A. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 2014;42:9612–9622. doi: 10.1093/nar/gku648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Losada A. Cohesin in cancer: chromosome segregation and beyond. Nat. Rev. Cancer. 2014;14:389–393. doi: 10.1038/nrc3743. [DOI] [PubMed] [Google Scholar]
- 22.Yun W.J., Kim Y.W., Kang Y., Lee J., Dean A., Kim A. The hematopoietic regulator TAL1 is required for chromatin looping between the beta-globin LCR and human gamma-globin genes to activate transcription. Nucleic Acids Res. 2014;42:4283–4293. doi: 10.1093/nar/gku072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhodes J.M., McEwan M., Horsfield J.A. Gene regulation by cohesin in cancer: is the ring an unexpected party to proliferation. Mol. Cancer Res. 2011;9:1587–1607. doi: 10.1158/1541-7786.MCR-11-0382. [DOI] [PubMed] [Google Scholar]
- 24.Deb S., Xu H., Tuynman J., George J., Yan Y., Li J., Ward R.L., Mortensen N., Hawkins N.J., McKay M.J., et al. RAD21 cohesin overexpression is a prognostic and predictive marker exacerbating poor prognosis in KRAS mutant colorectal carcinomas. Br. J. Cancer. 2014;110:1606–1613. doi: 10.1038/bjc.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yadav S., Sehrawat A., Eroglu Z., Somlo G., Hickey R., Yadav S., Liu X., Awasthi Y.C., Awasthi S. Role of SMC1 in overcoming drug resistance in triple negative breast cancer. PLoS One. 2013;8:e64338. doi: 10.1371/journal.pone.0064338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morales C., Ribas M., Aiza G., Peinado M.A. Genetic determinants of methotrexate responsiveness and resistance in colon cancer cells. Oncogene. 2005;24:6842–6847. doi: 10.1038/sj.onc.1208834. [DOI] [PubMed] [Google Scholar]
- 27.Park J.G., Frucht H., LaRocca R.V., Bliss D.P., Jr, Kurita Y., Chen T.R., Henslee J.G., Trepel J.B., Jensen R.T., Johnson B.E., et al. Characteristics of cell lines established from human gastric carcinoma. Cancer Res. 1990;50:2773–2780. [PubMed] [Google Scholar]
- 28.Gravalos C., Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann. Oncol. 2008;19:1523–1529. doi: 10.1093/annonc/mdn169. [DOI] [PubMed] [Google Scholar]
- 29.Ku J.L., Park J.G. Biology of SNU cell lines. Cancer Res. Treat. 2005;37:1–19. doi: 10.4143/crt.2005.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song S.H., Hou C., Dean A. A positive role for NLI/Ldb1 in long-range beta-globin locus control region function. Mol. Cell. 2007;28:810–822. doi: 10.1016/j.molcel.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yun J., Song S.H., Park J., Kim H.P., Yoon Y.K., Lee K.H., Han S.W., Oh D.Y., Im S.A., Bang Y.J., et al. Gene silencing of EREG mediated by DNA methylation and histone modification in human gastric cancers. Lab. Invest. 2012;92:1033–1044. doi: 10.1038/labinvest.2012.61. [DOI] [PubMed] [Google Scholar]
- 32.Song S.H., Kim A., Ragoczy T., Bender M.A., Groudine M., Dean A. Multiple functions of Ldb1 required for beta-globin activation during erythroid differentiation. Blood. 2010;116:2356–2364. doi: 10.1182/blood-2010-03-272252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garnis C., Buys T.P., Lam W.L. Genetic alteration and gene expression modulation during cancer progression. Mol. Cancer. 2004;3:9. doi: 10.1186/1476-4598-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J.I., Ju Y.S., Park H., Kim S., Lee S., Yi J.H., Mudge J., Miller N.A., Hong D., Bell C.J., et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olshen A.B., Venkatraman E.S., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 36.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L., Feng Z., Wang X., Wang X., Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 38.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D., et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. New Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillou E., Ibarra A., Coulon V., Casado-Vela J., Rico D., Casal I., Schwob E., Losada A., Mendez J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010;24:2812–2822. doi: 10.1101/gad.608210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dekker J., Rippe K., Dekker M., Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 41.Tolhuis B., Palstra R.J., Splinter E., Grosveld F., de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 42.Spilianakis C.G., Lalioti M.D., Town T., Lee G.R., Flavell R.A. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 43.Hagege H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., Forne T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR) Nat. Protoc. 2007;2:1722–1733. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- 44.Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T., et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Abbas T., Keaton M.A., Dutta A. Genomic instability in cancer. Cold Spring Harb. Perspect. Biol. 2013;5:a012914. doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siddiqui K., On K.F., Diffley J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013;5:1–13. doi: 10.1101/cshperspect.a012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alabert C., Groth A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- 48.Okada N., Shimizu N. Dissection of the beta-globin replication-initiation region reveals specific requirements for replicator elements during gene amplification. PLoS One. 2013;8:e77350. doi: 10.1371/journal.pone.0077350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harada S., Sekiguchi N., Shimizu N. Amplification of a plasmid bearing a mammalian replication initiation region in chromosomal and extrachromosomal contexts. Nucleic Acids Res. 2011;39:958–969. doi: 10.1093/nar/gkq882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Neil N.J., van Pel D.M., Hieter P. Synthetic lethality and cancer: cohesin and PARP at the replication fork. Trends Genet. 2013;29:290–297. doi: 10.1016/j.tig.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terret M.E., Sherwood R., Rahman S., Qin J., Jallepalli P.V. Cohesin acetylation speeds the replication fork. Nature. 2009;462:231–234. doi: 10.1038/nature08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacAlpine H.K., Gordan R., Powell S.K., Hartemink A.J., MacAlpine D.M. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res. 2010;20:201–211. doi: 10.1101/gr.097873.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Courbet S., Gay S., Arnoult N., Wronka G., Anglana M., Brison O., Debatisse M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature. 2008;455:557–560. doi: 10.1038/nature07233. [DOI] [PubMed] [Google Scholar]
- 54.Hashimoto Y., Puddu F., Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2012;19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa A., Ilves I., Tamberg N., Petojevic T., Nogales E., Botchan M.R., Berger J.M. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat. Struct. Mol. Biol. 2011;18:471–477. doi: 10.1038/nsmb.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X., Liu G., Leffak M. Activation of a human chromosomal replication origin by protein tethering. Nucleic Acids Res. 2013;41:6460–6474. doi: 10.1093/nar/gkt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waga S., Stillman B. The DNA replication fork in eukaryotic cells. Ann. Rev. Biochem. 1998;67:721–751. doi: 10.1146/annurev.biochem.67.1.721. [DOI] [PubMed] [Google Scholar]
- 58.Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010;11:728–738. doi: 10.1038/nrm2976. [DOI] [PubMed] [Google Scholar]
- 59.Moyer S.E., Lewis P.W., Botchan M.R. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mannini L., Menga S., Musio A. The expanding universe of cohesin functions: a new genome stability caretaker involved in human disease and cancer. Hum. Mutat. 2010;31:623–630. doi: 10.1002/humu.21252. [DOI] [PubMed] [Google Scholar]
- 61.Kon A., Shih L.Y., Minamino M., Sanada M., Shiraishi Y., Nagata Y., Yoshida K., Okuno Y., Bando M., Nakato R., et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013;45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
- 62.Deardorff M.A., Wilde J.J., Albrecht M., Dickinson E., Tennstedt S., Braunholz D., Monnich M., Yan Y., Xu W., Gil-Rodriguez M.C., et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 2012;90:1014–1027. doi: 10.1016/j.ajhg.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ottini L., Falchetti M., Lupi R., Rizzolo P., Agnese V., Colucci G., Bazan V., Russo A. Patterns of genomic instability in gastric cancer: clinical implications and perspectives. Ann. Oncol. 2006;17(Suppl. 7):97–102. doi: 10.1093/annonc/mdl960. [DOI] [PubMed] [Google Scholar]
- 64.Schaaf C.A., Kwak H., Koenig A., Misulovin Z., Gohara D.W., Watson A., Zhou Y., Lis J.T., Dorsett D. Genome-wide control of RNA polymerase II activity by cohesin. PLoS Genet. 2013;9:e1003382. doi: 10.1371/journal.pgen.1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nitzsche A., Paszkowski-Rogacz M., Matarese F., Janssen-Megens E.M., Hubner N.C., Schulz H., de Vries I., Ding L., Huebner N., Mann M., et al. RAD21 cooperates with pluripotency transcription factors in the maintenance of embryonic stem cell identity. PLoS One. 2011;6:e19470. doi: 10.1371/journal.pone.0019470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coquelle A., Rozier L., Dutrillaux B., Debatisse M. Induction of multiple double-strand breaks within an hsr by meganucleaseI-SceI expression or fragile site activation leads to formation of double minutes and other chromosomal rearrangements. Oncogene. 2002;21:7671–7679. doi: 10.1038/sj.onc.1205880. [DOI] [PubMed] [Google Scholar]
- 67.Negi L.M., Talegaonkar S., Jaggi M., Ahmad F.J., Iqbal Z., Khar R.K. Role of CD44 in tumour progression and strategies for targeting. J. Drug Target. 2012;20:561–573. doi: 10.3109/1061186X.2012.702767. [DOI] [PubMed] [Google Scholar]
- 68.Meyer N., Penn L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 69.Katoh M., Katoh M. Recombination cluster around FGFR2-WDR11-HTPAPL locus on human chromosome 10q26. Int. J. Mol. Med. 2003;11:579–583. [PubMed] [Google Scholar]
- 70.Martin M.M., Ryan M., Kim R., Zakas A.L., Fu H.Q., Lin C.M., Reinhold W.C., Davis S.R., Bilke S., Liu H.F., et al. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011;21:1822–1832. doi: 10.1101/gr.124644.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.