


Abstract
Eukaryotic chromosomal DNA is assembled into regularly spaced nucleosomes, which play a central role in gene regulation by determining accessibility of control regions. The nucleosome contains ∼147 bp of DNA wrapped ∼1.7 times around a central core histone octamer. The linker histone, H1, binds both to the nucleosome, sealing the DNA coils, and to the linker DNA between nucleosomes, directing chromatin folding. Micrococcal nuclease (MNase) digests the linker to yield the chromatosome, containing H1 and ∼160 bp, and then converts it to a core particle, containing ∼147 bp and no H1. Sequencing of nucleosomal DNA obtained after MNase digestion (MNase-seq) generates genome-wide nucleosome maps that are important for understanding gene regulation. We present an improved MNase-seq method involving simultaneous digestion with exonuclease III, which removes linker DNA. Remarkably, we discovered two novel intermediate particles containing 154 or 161 bp, corresponding to 7 bp protruding from one or both sides of the nucleosome core. These particles are detected in yeast lacking H1 and in H1-depleted mouse chromatin. They can be reconstituted in vitro using purified core histones and DNA. We propose that these ‘proto-chromatosomes’ are fundamental chromatin subunits, which include the H1 binding site and influence nucleosome spacing independently of H1.
INTRODUCTION
Eukaryotic DNA is organized into chromatin, which is essentially an array of regularly spaced nucleosomes that is occasionally interrupted by non-histone protein complexes bound at regulatory regions, such as promoters, enhancers and replication origins. The nucleosome core is composed of a core histone octamer around which is wrapped ∼147 bp of DNA in ∼1.7 superhelical turns (1–4). The linker histone (H1) binds externally to the nucleosome core, where it interacts with the linker DNA between nucleosomes to direct the folding of the chromatin fibre (5,6) and also seals the DNA turns at the nucleosomal DNA entry/exit points (7), forming a stem-loop structure (8,9). The nucleosome is a compact and stable structure capable of inhibiting transcription, DNA replication and repair. Accordingly, the cell contains various chromatin remodelling activities that can modify histones or move nucleosomes. Accurate maps of nucleosome positions in cells are therefore essential to an understanding of gene regulation and other processes.
The DNA in the nucleosome core is strongly protected from digestion by micrococcal nuclease (MNase), which has a strong endonuclease activity and a relatively weak exonuclease activity (10). MNase cuts primarily in the linker DNA between nucleosome cores and then its exonuclease activity slowly trims the DNA ends from the initial cut sites. MNase digestion is temporarily impeded by H1, resulting in the transient formation of chromatosomes (∼160 bp), which contain the nucleosome core, H1 and some linker DNA (11). Eventually, MNase digests the linker DNA in the chromatosome, H1 dissociates, and the result is the nucleosome core particle (∼147 bp), which corresponds to the crystal structure (1–4). The protection of nucleosomal DNA from MNase digestion is made use of in MNase-seq, which involves massively parallel sequencing of mono-nucleosomal DNA (12). Paired-end sequencing represents a major advance in MNase-seq because it provides reads from both ends of each DNA molecule, such that the length of each molecule can be deduced after alignment to the genome sequence, resulting in more accurate position information (13–16).
Ideally, MNase digestion would involve cutting the linker on both sides of the nucleosome core, followed by digestion of the protruding linker DNA, resulting in DNA fragments of 145–147 bp. Such fragments would provide a very accurate and precise map of nucleosome positions throughout the genome. In practice, paired-end sequencing has shown that nucleosomal DNA is heterologous in length (13–16), often with a significant fraction of nucleosomes containing DNA longer than 147 bp, primarily due to incomplete trimming by MNase. To solve this problem, we have augmented the slow exonuclease activity of MNase with E. coli exonuclease III (ExoIII). We have shown previously that this approach is effective on a reconstituted nucleosome in vitro (17). Here, we apply it to budding yeast chromatin. We observe a major, sharp, core particle peak at 145–147 bp, which provides very accurate nucleosome position data. We also observe clearly defined intermediate nucleosomal particles containing 154 bp or 161 bp. Surprisingly, these particles are not chromatosomes because they are observed in yeast lacking H1 and in mouse liver chromatin from which H1 has been removed. Furthermore, they can be reconstituted in vitro using just purified core histones and DNA. We propose that these novel particles, which we term ‘proto-chromatosomes’, play a critical role in nucleosome spacing by fixing the minimum distance between nucleosomes at 15 bp, independently of H1.
MATERIALS AND METHODS
Plasmids and strains
p685 was constructed by inserting HHO1 (from −111 to +812 with respect to the start codon) as a KpnI-PmeI fragment obtained by PCR into pNEB193 (New England Biolabs) cut with the same enzymes. p687 was constructed by replacing the BamHI-StuI HHO1 fragment in p685 with the BamHI-PmeI URA3 fragment from pNEB-URA3 (18). pRS-ARG1-B (p688) was obtained by insertion of a PCR fragment containing ARG1 and flanking regions, from −1283 to +2936, relative to the start codon, at the NotI site in pRS414 (Stratagene), a CEN ARS TRP1 plasmid (ARG1 and TRP1 are transcribed in opposite directions). YVN381 (MATa can1–100 his3–11 leu2–3,112 lys2Δ trp1–1 ura3–1 hho1Δ::URA3) was constructed by transforming wild type strain JRY4012 (19) with a KpnI-PmeI digest of p687.
MNase-ExoIII digestion of yeast nuclei
Yeast cells (250 ml culture) were grown to log phase (A600 ∼0.8) and harvested. Nuclei were prepared (20) and resuspended in 5.2 ml Digestion Buffer (10 mM HEPES pH 7.5, 35 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 5 mM 2-mercaptoethanol, with protease inhibitors (Roche 04693159001)). Two series of six MNase digestions (15, 30, 60, 120, 240 and 480 Worthington units) were set up, with either 150 or 300 units of ExoIII (New England Biolabs). Nuclei (400 μl aliquots) were digested for 2 min at room temperature. The reaction was stopped by addition of Na-EDTA to 10 mM and SDS to 0.8%, mixing and incubating at room temperature for 5 min, before adding more SDS to a final concentration of 1.8%. The DNA was purified; 10% of each sample was analysed in an agarose gel. Appropriately digested samples were chosen for preparative gel electrophoresis.
MNase-ExoIII digestion of mouse chromatin
Livers were dissected from female NIH/S mice (E13.5) and stored at −80°C. All buffers contained 15 mM 2-mercaptoethanol and protease inhibitors as above. One liver (0.8 g) was disrupted in 6 ml Buffer A+ (0.34 M sucrose, 60 mM KCl, 15 mM NaCl, 15 mM TrisHCl pH 8.0, 0.5 mM spermidine–HCl, 0.15 mM spermine–HCl, 1 mM Na–EDTA) in a glass homogeniser on ice by 10 strokes with a teflon pestle and filtered through four layers of cheese cloth. The homogenate was adjusted to 12.5 ml Buffer A+ with 0.92 M sucrose in a 30-ml polycarbonate high-speed centrifuge tube and spun for 15 min at 2°C in a Sorvall SA600 rotor at 12 500 rpm. The pellet was resuspended in 5 ml Buffer A+ and spun for 5 min as above. The washed nuclei were resuspended in Buffer A+ to A260 = 50 measured in 1 M NaOH (∼4 ml). A 1.4 ml-aliquot of nuclei in a microfuge tube was adjusted to 2 mM CaCl2 ( = 1 mM excess over EDTA), warmed briefly to 37°C, and digested with MNase (20 Worthington units) for 3 min. The digestion was terminated by adding 0.1 M Na-EDTA pH 7.5 to a final concentration of 10 mM. The digested nuclei were recovered by a 5 min spin in a microfuge at 4°C and lysed by thorough resuspension in 1 ml 0.2 mM NaEDTA pH 7.5. The debris was removed by a 5 min spin. The chromatin concentration in the supernatant was measured by A260 in 1% SDS. Two 400-μl aliquots of chromatin at ∼0.2 mg/ml were diluted with an equal vol. of 160 mM NaCl, 2 mM Na-phosphate pH 7.2, 0.4 mM Na-EDTA. To remove H1 (21), 200 μl AG50W-X2 resin (BioRad 142–1241; washed and equilibrated in 80 mM NaCl, 1 mM Na-phosphate pH 7.2, 0.2 mM Na-EDTA as described (22)) was added to one aliquot. Both aliquots were subjected to gentle rotation for 1 h at 4°C. The resin was allowed to settle and the H1-depleted chromatin was removed to a fresh tube on ice. The NaCl concentration was reduced to 50 mM by a 1.6-fold dilution in a pre-mixed buffer resulting in final concentrations of 10 mM HEPES pH 7.5, 2 mM MgCl2, 2 mM CaCl2. Histones in 100 μl aliquots were TCA-precipitated and analysed in a protein gel stained with Coomassie Blue. The chromatin was depleted of ∼85% of its H1 (estimated by protein gel analysis of H1 extracted by 5% perchloric acid). Six 100 μl-aliquots (∼5.5 μg) of native and H1-depleted chromatin were warmed briefly to 30°C and digested with 0, 0.1, 0.2, 0.4, 0.8 or 1.6 units MNase and 16.5 units ExoIII (all aliquots) for 3 min at 30°C. The digestion was stopped by adding 20 μl 50 mM Na-EDTA pH 7.5, 5% SDS. DNA was extracted as described above (nucleosomal DNA was not gel-purified).
Reconstituted chromatin
Reconstituted nucleosomes were made using recombinant yeast H3, H4, H2A and H2B purified from E. coli (23). Histone octamers were formed and purified by gel filtration (23). The octamer concentration was determined initially by amino acid composition analysis and then correlated with Bradford protein assay measurements using IgG as standard (BioRad): 1 mg/ml in the Bradford assay corresponds to an actual concentration of 0.36 mg/ml. pRS-ARG1-B was purified from E. coli lacking DNA methylases (dam- dcm-; New England Biolabs C2925). Briefly, 10 μg DNA and 4 μg octamer were mixed in 100 μl 2 M NaCl, 10 mM HEPES-K pH 7.5, 0.1 mM Na-EDTA and transferred to a Slide-A-Lyzer mini-dialysis unit with a molecular weight cut-off of 10 000 (ThermoScientific 69574). The solution was dialysed overnight at 4°C against 500 ml of 2 M NaCl in R-buffer (10 mM HEPES-K pH 7.5, 0.1 mM Na-EDTA, 0.05% NP40, 1 mM 2-mercaptoethanol) and then against R-buffer containing 1.5 M NaCl (2 h), 1 M NaCl (2 h), 0.75 M NaCl (3 h), and finally against 50 mM NaCl in R-buffer overnight. Alternatively, nucleosomes were reconstituted on pRS-ARG1-B at the same ratio using native chicken erythrocyte histones, as described (17). Reconstituted chromatin (1 μg) in 30 μl R-buffer containing 50 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, was digested with 3.125 units ExoIII plus either 0.1, 0.125 or 0.2 units MNase for 3 min at 30°C. Reactions were stopped by addition of Na-EDTA pH 7.5–6.7 mM and cooling on ice. Libraries of gel-purified nucleosomal DNA were subjected to Illumina paired-end sequencing (20). The length of each sequenced DNA fragment was deduced after alignment of each pair of 50-nt reads to the yeast genome (SacCer2) or the mouse genome (mm10) using Bowtie 2 (24). A summary of the data is provided in Supplementary Table S1.
RESULTS
MNase–ExoIII digestion yields precisely trimmed core particles
We added ExoIII to facilitate removal of linker DNA protruding from nucleosomes liberated from yeast nuclei by MNase. ExoIII is a partly processive 3′-to-5′ exonuclease which hydrolyses only one DNA strand, leaving a single-stranded 5′-overhang (25). Its activity is mildly sequence-dependent, in the order C > A, T > G (26). To remove the 5′ overhang, we initially included mung bean nuclease in the digestions, but it was found to be unnecessary, presumably because MNase digests single-stranded DNA much more rapidly than double-stranded DNA (10). Thus, the endonuclease activity of MNase cuts the linker DNA, ExoIII rapidly removes one strand of the linker until it is blocked by the histones, and then MNase destroys the single-stranded overhang (Figure 1A). Nuclei were digested at different MNase concentrations in the presence of a fixed concentration of ExoIII (Figure 1B) and paired-end sequencing libraries were prepared from gel-purified mono-nucleosomal DNA.
Figure 1.
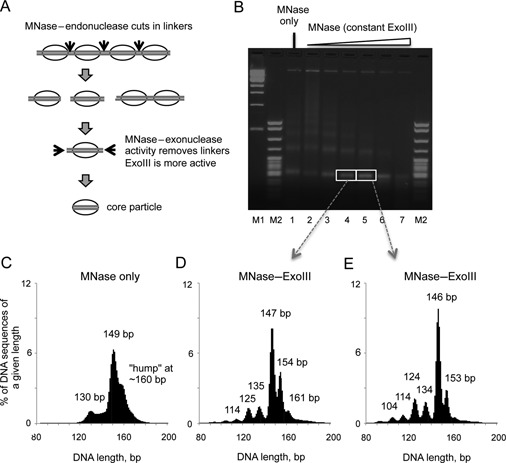
Simultaneous digestion of yeast chromatin with MNase and ExoIII results in a set of DNA fragments of discrete lengths, corresponding to the nucleosome core particle and its intermediates. (A) Digestion of chromatin with MNase and ExoIII. Ovals: nucleosome cores; black arrows: cut sites. (B) Agarose gel electrophoresis of chromatin digested with MNase and ExoIII. Markers: 1-kb ladder (M1); pBR322 MspI digest (M2). (C) Length distribution histograms for nucleosome sequences obtained by paired-end sequencing of the mono-nucleosome band from wild type strain JRY4012: MNase-only. (D, E) Length histograms for MNase-ExoIII at two different MNase concentrations, corresponding to the mono-nucleosome bands in the gel.
The length distributions for an MNase-only sample and two levels of MNase-ExoIII digestion are plotted as histograms (Figure 1C, D, E). The MNase-only sample has a typical length distribution with a fairly broad peak at 149 bp, corresponding to core particles, and a weak hump at ∼160 bp corresponding to particles with protruding linker DNA, as well as a set of poorly resolved peaks around 130 bp, corresponding to internally digested nucleosomes, sub-nucleosomes (i.e. nucleosomes lacking one or both H2A–H2B dimers; 27) and perhaps non-histone complexes (16). In the presence of ExoIII, much sharper peaks were observed in the length distribution, which shifted to slightly lower values with increased MNase digestion (Figure 1D, E). The major peak was at 147 bp or 146 bp, corresponding to precisely trimmed core particles. There were also minor peaks at sub-nucleosomal lengths of 134, 124, 114 and 104 bp, which show a 10 bp periodicity. This observation is consistent with internal digestion of nucleosomes by MNase and/or invasion of the nucleosome by ExoIII (28). Unexpectedly, there were also sharp peaks at 153/154 and 161 bp, apparently corresponding to defined nucleosomal particles containing more DNA than is present in the core particle. The periodicity in this case was 7 or 8 bp. The relative amounts of these particles decreased with MNase digestion, consistent with the hypothesis that they were eventually converted into core particles. Thus, the effect of ExoIII is to resolve and sharpen the peaks observed in the MNase-only digest; the resolution of the weak hump at ∼160 bp in the MNase-only digest into sharp peaks at 154 and 161 bp is particularly striking.
Proto-chromatosomes
The structural explanation for the pauses in digestion external to the nucleosome core is of interest. The most obvious possibility is that they represent chromatosomes: nucleosomes with additional linker DNA protected by the globular domain of histone H1, typically ∼160 bp (11,29). However, unlike higher organisms, yeast has much less H1 than nucleosomes (30), implying that it could not protect a large fraction of the total nucleosomes. Nevertheless, we tested the possibility that the 154-bp and 161-bp particles are chromatosomes by repeating the experiment using a strain in which the gene for H1 had been deleted (hho1Δ). The length distribution was very similar to that of wild type nucleosomes, yielding the same peaks, including those at 153/154 and 161 bp (Figure 2A). Since the 154-bp and 161-bp particles were observed in the absence of H1, they cannot be chromatosomes. Accordingly, we will refer to the particles that contain 154 bp and 161 bp as ‘proto-chromatosomes’, because they contain a precise amount of linker DNA but lack H1.
Figure 2.
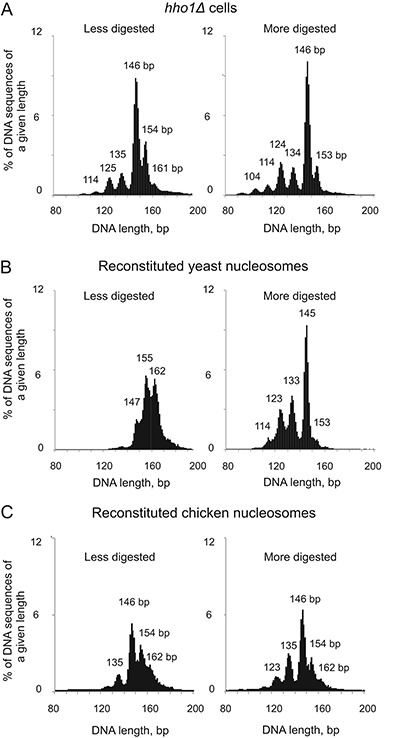
The 154-bp and 161-bp nucleosomal particles are present in yeast cells lacking linker histone and can be reconstituted in vitro using only core histones and DNA. (A) The 154-bp and 161-bp particles are not chromatosomes. Length distribution histograms for nucleosome sequences obtained by sequencing of mono-nucleosomes from hho1Δ cells. (B, C) The 154-bp and 161-bp particles can be formed using recombinant yeast core histones or native chicken erythrocyte core histones and plasmid DNA in vitro. Length distribution histograms for nucleosome sequences obtained at two different levels of MNase-ExoIII digestion.
Proto-chromatosomes might result from protection of linker DNA by non-histone proteins, but this explanation is difficult to eliminate in vivo. Instead, we determined whether the 154-bp and 161-bp particles are observed after digestion of chromatin reconstituted in a purified system in vitro. We assembled nucleosomes on a large yeast plasmid using recombinant yeast histones and subjected them to MNase-ExoIII digestion (Figure 2B). Less digested mono-nucleosomes included a weak peak at 147 bp, corresponding to core particles, and stronger peaks at 155 bp and 162 bp, corresponding to proto-chromatosomes. More digested nucleosomes gave the expected sharp core particle peak at 145 bp and only a very minor peak at 153 bp. Some invasion of the nucleosomes was also observed (peaks at 133, 123 and 114, again with a ∼10 bp period). Clearly, the 162-bp and 155-bp particles were converted to core particles (145 bp) after extended digestion. We obtained the same result when native chicken erythrocyte core histones were used to reconstitute nucleosomes on the same plasmid, indicating that recombinant yeast histones are not atypical (Figure 2C). Thus, protection of the extra DNA in proto-chromatosomes requires only core histone-DNA interactions.
We explored the relationship between native yeast proto-chromatosomes (152–155 bp) and core particles (144–147 bp) by comparing their sequences using cross-correlation analysis (31). The ends of the core particle and proto-chromatosome sequences were compared with one another: for every core particle sequence, the distance ‘d'’ of its right-hand end (with respect to the chromosome) to the first nucleotide of all proto-chromatosome sequences was summed. If the core particle and the proto-chromatosome have the same right-hand end, d = 0 (Figure 3A). A comparison of core particles (145–147 bp) and 152–155 bp proto-chromatosomes from wild-type cells (Figure 1E) revealed strong peaks at 0 and +8 bp, indicating that proto-chromatosomes tend to have one end that is identical to that of the corresponding core particle and a 7 or 8-bp extension at the other end (Figure 3B). That is, the extra 8 bp in the 154-bp particles are asymmetrically located on one side of the nucleosome. The additional correlation peaks at 19 and 30 bp etc. (Figure 3B), are shifted by 10 bp and represent the ends of rotationally related translational positions (31). A similar analysis of 161-bp proto-chromatosomes supports the proposal that, in most cases, the extra 15 bp in these particles correspond to 7 or 8 bp protruding from both sides of the nucleosome core (Figure 3C). There might be a fraction of asymmetric 161-bp particles, with 15 bp protruding from one side of the core, because the peak at 16–17 bp is quite wide, but such particles cannot be reliably distinguished from particles shifted translationally by 10 bp, which give rise to a peak at 19 bp, as observed for 154-bp particles (Figure 3B). The same results were obtained by cross-correlation analysis of core particles and proto-chromatosomes from less digested nucleosomes (Supplementary Figure S1).
Figure 3.
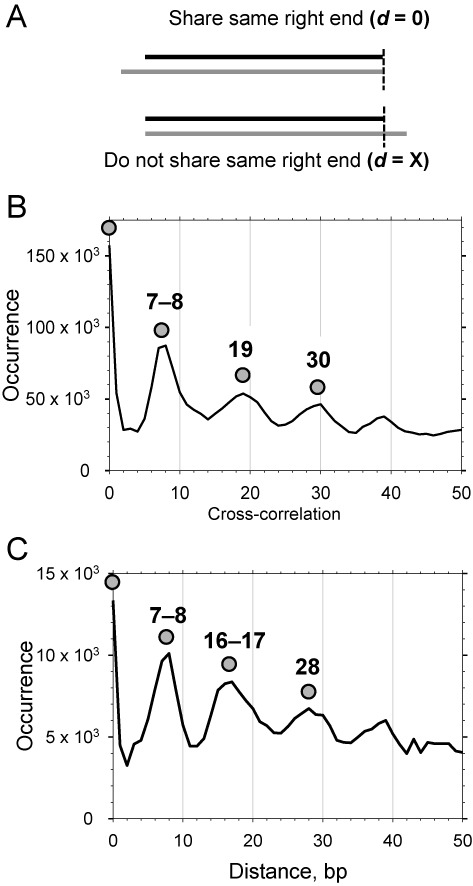
Proto-chromatosomes have a 7 or 8-bp extension on one side (154-bp particles) or both sides (161-bp particles) of the nucleosome core in wild type cells. (A) Calculation of cross-correlation between the right-hand end of a core particle sequence (black line) and the right-hand end of a proto-chromatosome sequence (grey line). The right-hand end refers to the location of the sequence with respect to the chromosome. (B) Cross-correlation between nucleosome core particles (145–147 bp) and proto-chromatosomes (152–155 bp) (data in Figure 1E). (C) Cross-correlation between core particles (145–147 bp) and proto-chromatosomes (160–162 bp) (data in Figure 1E).
In conclusion, MNase and ExoIII pause during digestion of the linker at a point ∼7 bp from the boundary of the nucleosome core, resulting in metastable proto-chromatosomes, which are eventually digested to core particles. This scenario implies that proto-chromatosomes have the same genomic distribution as core particles, and do not derive from specific regions of the genome. We confirmed that this was the case by comparing the nucleosome phasing patterns of core particles and proto-chromatosomes relative to the transcription start site (TSS) (12,32). Both core particles and proto-chromatosomes showed very similar phasing profiles (Supplementary Figure S2). Since genes are very close together in the budding yeast genome, most nucleosomes are represented by these phasing patterns.
Proto-chromatosomes are present in mouse liver
We determined whether proto-chromatosomes are present in the chromatin of higher organisms by MNase–ExoIII digestion of native and H1-depleted chromatin prepared from mouse liver. Nuclei were initially digested with MNase only to obtain long chromatin fragments. Some of this chromatin was depleted of H1 using a cation exchange resin; another aliquot was mock-treated (‘native’ chromatin). H1-depleted chromatin was digested more rapidly by MNase and ExoIII than native chromatin, consistent with protection of linker DNA by H1 (Figure 4A). Nucleosomal DNA was sequenced and aligned to the mouse genome to obtain the length distributions, which revealed the presence of the 154-bp and ∼161-bp peaks in both native and H1-depleted chromatin (Figure 4C). In native chromatin, the broad chromatosome peak at 163–166 bp is converted into 154 bp-particles and core particles (146 bp). Particles of the same size were observed in H1-depleted chromatin. The 154-bp and 161-bp particles were converted into a strong, sharp, core particle peak at 146 bp, consistent with conversion of proto-chromatosomes into core particles as digestion proceeds. Thus, the chomatosome is simply the proto-chromatosome with bound H1, which confers additional protection to the 7-bp extensions on either side of the classical core particle.
Figure 4.
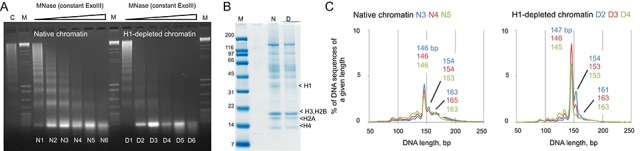
Proto-chromatosomes are present in mouse chromatin. (A) Agarose gel electrophoresis of DNA from native (N) and H1-depleted (D) mouse liver chromatin digested with MNase and ExoIII (lanes 1–6). ‘C’, long chromatin obtained by initial digestion of nuclei with MNase only. ‘M’, λ-BstEII digest mixed with pBR322 MspI digest (NEB). (B) Depletion of H1 shown by SDS-PAGE analysis of proteins in native (‘N’) and H1-depleted (‘D’) chromatin (M: marker). Stained with Coomassie blue. (C) Length histograms for the MNase-ExoIII digests of native (lanes N4, N5 and N6 in A) and H1-depleted chromatin (D3, D4 and D5).
Enhanced WW and SS patterns in MNase–ExoIII yeast nucleosome sequences
The initial aim of this study was to increase the accuracy of nucleosome position data by eliminating linker DNA protruding from mono-nucleosomes. This should improve alignment of nucleosomal DNA sequences and enhance the periodicities in the distributions of various dinucleotide and trinucleotide motifs relative to the nucleosome dyad (33). The strongest patterns are the 10 bp periodicities displayed by the distributions of A or T-containing dinucleotides (AT, TA, TT and AA = WW) and G or C-containing dinucleotides (GC, CG, GG and CC = SS), which are exactly out of phase with one another (33). The patterns have been attributed to the relative ease with which A-T base pairs can bend into the minor groove and the tendency for G-C base pairs to bend into the major groove, as the DNA is strongly bent around the core histone octamer (1,33).
We compared the WW and SS patterns for MNase-only and MNase–ExoIII core particle DNA by aligning the sequences by their midpoints and calculating the frequencies of WW and SS dinucleotides within the nucleosome (Figure 5A) (34). The MNase-only data were restricted to the sequences in the major peak (147–152 bp), close to core particle length; likewise, the MNase–ExoIII data were restricted to the major peak (144–147 bp). In the case of the WW motif, both MNase-only and MNase–ExoIII core particles showed a clear ∼10 bp modulation in frequency across most of the nucleosome, with peaks at 7, 17, 27, 38, 48 and 59 bp from the boundary (i.e. 14, 25, 35, 46, 56 and 66 bp from the dyad). The amplitudes of all of these peaks were enhanced by ExoIII. In addition, two weak peaks at 64 and 70 bp in the MNase-only data, located near the dyad at 73 bp, were clearly enhanced by ExoIII. Although the peak at 70 bp is in phase with the other 10 bp-modulated peaks, the peak at 64 bp is exactly out of phase with them and therefore represents an anomaly, or ‘rogue’ peak, observed previously in chemical maps (35,36).
Figure 5.
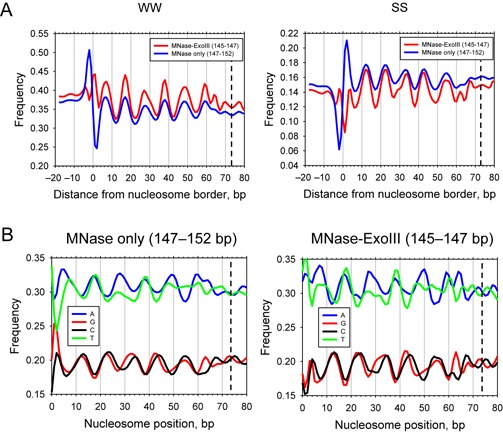
Enhanced dinucleotide periodicities and strand-specific preferences in MNase–ExoIII nucleosomes. (A) WW versus SS sequence patterns in MNase–ExoIII nucleosomes aligned at their dyads. WW versus SS patterns for MNase–ExoIII and MNase-only nucleosomes in wild type cells. (B) Single-nucleotide distributions in MNase–ExoIII and MNase-only nucleosomes (derived from the peaks in Figure 1C, E). The data were smoothed using a 3-bp running average.
The amplitude of the SS signal was weaker than the WW signal, and is centered on lower dinucleotide frequencies because the yeast genome is AT-rich. SS dinucleotides showed a clear 10-bp modulation, with peaks at 12, 22, 34, 43 and 54 bp in both data sets (i.e. 19, 30, 39, 51 and 61 bp from the dyad), which are ∼5 bp out of phase with the WW peaks and enhanced by ExoIII. Additional peaks at 62 and 67 bp emerged in the MNase-ExoIII data, which flank the additional peaks observed in the WW profile. Thus there is a weak alternation between WW and SS with a ∼5 bp period close to the dyad (WW at 59, SS at 62, WW at 64, SS at 67, WW at 70 and SS at the dyad).
At the nucleosome border (set at 0 in Figure 5), the MNase-only data show a large WW peak at −2 and similarly large trough at +2, which are resolved by ExoIII into two relatively small flanking peaks at −4 and +1 (Figure 5A). Neither of these peaks is in phase with the 10-bp WW period. Similarly, the large SS peak at +2 in the MNase-only data are absent from MNase–ExoIII nucleosomes, and the trough at −2 decreased in amplitude and shifted to −2, resulting in two new weak SS peaks at −1 and 4 bp in MNase–ExoIII nucleosomes, neither of which are in phase with the 10-bp signal. The MNase-only nucleosome boundary peak coincides with the MNase cut site and has therefore been attributed to its preference for AT-rich DNA (31,35), although this seems unlikely given its absence from MNase–ExoIII nucleosomes. In summary, the increased accuracy of nucleosome positions deduced from the MNase–ExoIII data results in a more accurate alignment of nucleosome sequences, enhancing the dinucleotide signals, such that strong peaks are sharpened and weak peaks are resolved.
Strand-specific preferences for A versus T
We also determined the distributions of all four mono-nucleotides within nucleosomes (Figure 5B). Intriguingly, all four nucleotides showed quite strong 10-bp periodicities, in line with those observed for the relevant dinucleotides, with A and T out of phase relative to G and C, although the amplitudes of the signals were not as strong as those observed for WW and SS. Surprisingly, the distributions of A and T are not identical, and neither are those of G and C. In the case of G and C, the differences are small, except near the dyad, where the weak G and C peaks are out of phase with one another; there is a G-peak at 63 that is not matched in the C profile, and a C peak that is stronger than the G peak at 67. These differences occur in the region next to the dyad, where the SS distribution shows a 5-bp period. The A and T distributions are more different from one another than the G and C distributions. The A profile is more periodic than that of T, such that A shows stronger peaks than T at 7, 27, 50, 59 and 64, essentially because the T peaks at these locations are split in two, suggesting that T tends to flank an A on both sides at these locations. Furthermore, the region near the dyad shows weak A and T peaks that are out of phase with one another, analogous to those observed for G and C in this region. The A peak at 64 is relatively strong and coincides with the rogue WW peak that is out of phase with the other WW peaks, indicating that WW at this location tends to be AA rather than TT (33).
DISCUSSION
We present a significant improvement on the MNase method for mapping nucleosomes, involving the combination of MNase and ExoIII for efficient removal of residual linker DNA from mono-nucleosomes. This results in a sharply defined peak of core particles (144–147 bp). More accurate nucleosome position data will facilitate nucleosome sequence analysis and may be critical in deciding whether a specific sequence, such as a transcription factor binding site, is located inside a nucleosome or in the more accessible linker DNA. The quality of MNase–ExoIII data is comparable with that of data obtained from the chemical mapping method, which involves sequencing DNA fragments generated by chemical nicking of DNA near the nucleosome dyad in cells carrying the S47C mutation in H4 (35,37). We find that dinucleotide periodicities are enhanced and that the WW signal at 64 is now apparent. Furthermore, the artifactual MNase-associated WW and SS peaks at the boundary of the nucleosome core are largely eliminated by ExoIII. The only significant discrepancy concerns the A peak located 3 bp from the dyad in chemically mapped nucleosomes (35). Given that this A-peak is far from the MNase cleavage sites, it may reflect a problem with the chemical cleavage method, perhaps involving the assignment of minor cleavage sites, or perhaps from an unexpected sequence preference of the DNA cleavage reagent and/or a structural alteration due to the H4-S47C mutation (38).
We also observe a striking preference for A over T in the top strand at 7, 27, 50, 59 and 64 bp relative to the nucleosome boundary (Figure 5B), where A is located on the inside of the bend, just after the small kinks directed into the minor groove (Supplementary Figure S3). A differential distribution of A and T mono-nucleotides was reported previously, although the period was irregular, perhaps because the data were reduced to relatively few, idealized, nucleosome positions prior to analysis (39). The preference may reflect wedge formation by AA:TT dimeric steps, which occurs because the TT strand is slightly more extended than the AA strand (40), perhaps facilitating anisotropic bending (41). Alternatively, the fact that most arginine residues contacting DNA are closer to the bottom strand than the top strand may be important (42,43). However, neither of these explanations account for the opposite preference, for T over A, at 38 bp from the border. Thus, an additional factor is required to account completely for the A versus T strand asymmetry.
We have identified clearly defined particles larger than the core particle, which have 7 or 8 bp projecting from one side (154-bp) or both sides (161-bp) of the nucleosome core. These proto-chromatosomes contain a similar amount of DNA to the chromatosome, but do not contain H1 and can be reconstituted in vitro using just DNA and core histones. Our observation is consistent with early papers showing that MNase digestion of H1-depleted chromatin yields a weak, transient band similar in size to the chromatosome (17,44–46). We propose that a steric block to MNase and ExoIII is located in the linker ∼7 bp from the nucleosome core (Figure 6A). In the case of the typical yeast linker of ∼15 bp, the putative steric blocks between two nucleosomes would coincide in the middle of the linker, as shown (Figure 6A). The ‘10n +5’ rule for the linker length (35,45) predicts linker lengths of 5, 15, 25 bp, etc. If the linker is 25 bp then the steric blocks would be separated by 10 bp. We envisage that MNase cuts within one of the linkers, between the block and a core, resulting in one nucleosome with some protruding DNA that is trimmed away by MNase and ExoIII until it is stopped by the core particle boundary, and a second nucleosome which is trimmed until the exonucleases reach the block, resulting in a proto-chromatosome. Eventually, MNase cuts the proto-chromatosome between the block and the core, and the residual linker is digested, resulting in a core particle (Figure 6B).
Figure 6.
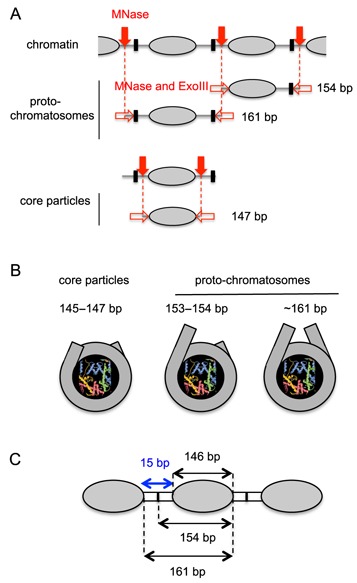
Nucleosome core particles, proto-chromatosomes and linker length. (A) Provenance of proto-chromatosomes. The endonuclease activity of MNase (solid red arrows) cuts the linker DNA between nucleosome cores (grey ovals). ExoIII and the exonuclease activity of MNase (open red arrows) trim the linker, stopping either at the border of the nucleosome core, or at a putative block to digestion (black box), located in the linker ∼7 bp from the border of the nucleosome core. Another cut by MNase in the linker between the block and the core allows the exonucleases to remove the remaining linker and convert proto-chromatosomes to core particles. The typical linker length in yeast is 15 bp ( = 10n +5; n = 1); in this case, the block due to each flanking nucleosome would be at the same location (in the middle of the linker, as shown). (B) Views of core particle and proto-chromatosomes from above, drawn approximately to scale, based on the nucleosome structure (1). The final 10 bp on each side of the nucleosome core are almost straight, projecting a short distance out of the particle. Proto-chromatosomes are shown with an extra 7 bp on one side (154 bp) or both sides (161 bp) with a continuing straight trajectory. (C) Potential role of the proto-chromatosome in determining linker length. The average nucleosome spacing is 161 bp (Supplementary Figure S4B). We propose that the proto-chromatosome fixes the minimum distance between nucleosomes at 15 bp, independently of H1.
The 161-bp proto-chromatosome is significant for two reasons. Firstly, 161 bp is sufficient to complete two turns of DNA in the nucleosome. This observation might be coincidental, particularly in view of the tetra-nucleosome structure, which does not show additional DNA coiled around the nucleosome (47), but it is intriguing. Secondly, the average distance between MNase–ExoIII core particles determined by auto-correlation analysis of core particle sequences is also 161 bp (Supplementary Figure S4), suggesting that the proto-chromatosome might determine the distance of closest approach between two nucleosome cores, which corresponds to a minimum linker of ∼15 bp (Figure 6C) and may account for the ‘10n +5’ rule, setting n > 0.
What is the structural nature of the steric block? One possibility is a histone–DNA contact located ∼7 bp external to the nucleosome core, possibly involving the histone tail domains, since they interact with linker DNA and affect chromatin folding in vitro (48,49). In particular, it has been shown recently using ChIP-exo, a technique for mapping protein–DNA contacts genome-wide, that H3 makes a major contact with the linker in vivo, which is partly dependent on its N-tail domain (50). We are currently investigating whether the H3 tail domain is involved in proto-chromatosome formation. It should be noted that ChIP-exo works on a different principle to MNase–ExoIII: the locations of histone-specific cross-links on immuno-precipitated DNA are mapped using λ-exonuclease, which digests one strand until it is blocked by a histone–DNA cross-link. The method provides valuable information on individual histone occupancy at relatively high resolution. However, it cannot be used to obtain an accurate nucleosome dyad or boundary because the histone residue involved in each cross-link is unknown. Instead, the method provides an average dyad position, corresponding to the weighted average of the various rotationally related overlapping nucleosome positions within each nucleosome occupancy peak.
Alternatively, the block might be due to a steric clash between the nucleosome core and the approaching MNase or ExoIII as it rotates around the DNA helix. The outer 10 bp on each side of the nucleosome core are almost straight and therefore deviate from the superhelix (Figure 6B) (1). If the linker maintains this straight trajectory in proto-chromatosomes, then a clash between ExoIII and the DNA at the dyad may occur (Supplementary Figure S5). On the other hand, MNase is smaller than ExoIII, reducing the probability of a clash, but it still detects the steric block, albeit less precisely. Irrespective of the structural origin of the steric block to exonuclease digestion, the binding of transcription factors to cognate sites located within 7 bp of the core (i.e. within the proto-chromatosome) may be inhibited, particularly if the rotational setting of the site is such that the factor must bind to the inner surface of the linker DNA.
We propose that a critical function of the proto-chromatosome is to fix the linker length and geometry of the yeast chromatin fibre, resulting in compaction (51), even though it is H1-deficient (30). The proto-chromatosome would provide the correct DNA topography for H1 binding. In higher eukaryotes, longer linkers tend to de-stabilize the chromatin zigzag (52); this effect is offset by the binding of H1 (6), which forms a stem-loop structure in the linker (8,9). Nevertheless, linker histone is not essential for chromosome condensation or cell viability in vertebrates; complete loss of H1 results in chromatin with closely-spaced nucleosomes (53), as observed in yeast and in vitro (54). Thus, the proto-chromatosome discovered here may represent an essential chromatin subunit, influencing both H1 binding and nucleosome spacing.
ACCESSION NUMBERS
GEO database GSE65889.
Supplementary Material
Acknowledgments
We thank the NHLBI Sequencing Core Facility (Yan Luo, Poching Lu, Yoshi Wakabayashi and Jun Zhu) and the Tufts University Core Facility for paired-end sequencing. We thank Rohinton Kamakaka for JRY4012, Will Huffman and Doug Fields for mouse livers, and Razvan Chereji for helpful comments on the manuscript.
Author contributions: H.A.C. developed the method and performed experiments. F.C. analysed the data. T.L.B., J.O., T.N., V.N. and N.K. performed experiments. V.B.Z. and D.J.C. directed research and wrote the manuscript.
Footnotes
Present addresses:
V. Nagarajavel, Genomic Medicine Institute, Lerner Research Institute NE-50, 9500 Euclid Avenue, Cleveland, OH 44195, USA.
Naoe Kotomura, Department of Biochemistry, Fujita Health University School of Medicine, Toyoake, Aichi, Japan.
Victor B. Zhurkin, Building 37, Room 3035A, National Cancer Institute, NIH, Bethesda, MD 20892, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institutes of Health (NICHD and NCI). Funding for open access charge: Intramural Research Program of the National Institutes of Health (NICHD).
Conflict of interest statement. None declared.
REFERENCES
- 1.Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.White C.L., Suto R.K., Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasudevan D., Chua E.Y., Davey C.A. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 4.Davey C.A., Sargent D.F., Luger K., Maeder A.W., Richmond T.J. Solvent-mediated interactions in the structure of the nucleosome core particle at 1.9Å resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 5.Thoma F., Koller T., Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark D.J., Kimura T. Electrostatic mechanism of chromatin folding. J. Mol. Biol. 1990;211:883–896. doi: 10.1016/0022-2836(90)90081-V. [DOI] [PubMed] [Google Scholar]
- 7.Allan J., Hartman P.G., Crane-Robinson C., Aviles F.X. The structure of histone H1 and its location in chromatin. Nature. 1980;288:675–679. doi: 10.1038/288675a0. [DOI] [PubMed] [Google Scholar]
- 8.Bednar J., Horowitz R.J., Grigoryev S.A., Carruthers L.M., Hansen J.C., Koster A.J., Woodcock C.L. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14173–14178. doi: 10.1073/pnas.95.24.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syed S.H., Goutte-Gattat D., Becker N., Meyer S., Shukla M.S., Hayes J.J., Everaers R., Angelov D., Bednar J., Dimitrov S. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9620–9625. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anfinsen C.B., Cuatrecasas P., Taniuchi H. Staphylococcal nuclease, chemical properties and catalysis. In: Boyer PD, editor. The Enzymes. 3rd edn. Vol. 4. 1971. pp. 177–204. [Google Scholar]
- 11.Simpson R.T. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochem. 1978;17:5524–5531. doi: 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- 12.Mavrich T.N., Ioshikhes I.P., Venters B.J., Jiang C., Tomsho L.P., Qi J., Schuster S.C., Albert I., Pugh B.F. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kent N.A., Adams S., Moorhouse A., Paszkiewicz K. Chromatin particle analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res. 2011;39:e26. doi: 10.1093/nar/gkq1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole H.A., Howard B.H., Clark D.J. The centromeric nucleosome of budding yeast is perfectly positioned and covers the entire centromere. Proc. Natl. Acad. Sci. U.S.A. 2011;108:12687–12692. doi: 10.1073/pnas.1104978108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole H.A., Howard B.H., Clark D.J. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res. 2011;39:9521–9535. doi: 10.1093/nar/gkr643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henikoff J.G., Belsky J.A., Krassovsky K., MacAlpine D.M., Henikoff S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. U.S.A. 2011;108:18318–18323. doi: 10.1073/pnas.1110731108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikitina T., Wang D., Gomberg M., Grigoryev S.A., Zhurkin V.B. Combined micrococcal nuclease and exonuclease III digestion reveals precise positions of the nucleosome core/linker junctions: implications for high-resolution nucleosome mapping. J. Mol. Biol. 2013;425:1946–1960. doi: 10.1016/j.jmb.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen C.H., Leblanc B.P., Alfieri J.A., Clark D.J. Remodeling of yeast CUP1 chromatin involves activator-dependent repositioning of nucleosomes over the entire gene and flanking sequences. Mol. Cell. Biol. 2001;21:534–547. doi: 10.1128/MCB.21.2.534-547.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox C.A., Loo S., Dillin A., Rine J. The origin recognition complex has essential functions in transcriptional silencing and chromosomal replication. Genes Dev. 1995;9:911–924. doi: 10.1101/gad.9.8.911. [DOI] [PubMed] [Google Scholar]
- 20.Cole H.A., Howard B.H., Clark D.J. Genome-wide mapping of nucleosomes in yeast using paired-end sequencing. Methods Enzymol. 2012;513:145–168. doi: 10.1016/B978-0-12-391938-0.00006-9. [DOI] [PubMed] [Google Scholar]
- 21.Muyldermans S., Lasters I., Wyns L. Histone H1 can be removed selectively from chicken erythrocyte chromatin at near physiological conditions. Nucleic Acids Res. 1980;8:731–739. [PMC free article] [PubMed] [Google Scholar]
- 22.Bolund L.A., Johns E.W. The selective extraction of histone fractions from deoxyribonucleoprotein. Eur. J. Biochem. 1973;35:546–553. doi: 10.1111/j.1432-1033.1973.tb02871.x. [DOI] [PubMed] [Google Scholar]
- 23.Luger K., Rechsteiner T.J., Richmond T.J. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 24.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richardson C.C., Lehman I.R., Kornberg A. A deoxyribonucleic acid phosphatase-exonuclease from Escherichia coli. J. Biol. Chem. 1964;239:251–258. [PubMed] [Google Scholar]
- 26.Linxweiler W., Hörz W. Sequence specificity of exonuclease III from E. coli. Nucleic Acids Res. 1982;10:4845–4859. doi: 10.1093/nar/10.16.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole H.A., Ocampo J., Iben J.R., Chereji R.V., Clark D.J. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res. 2014;42:12512–12522. doi: 10.1093/nar/gku1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riley D., Weintraub H. Nucleosomal DNA is digested to repeats of 10 bases by exonuclease III. Cell. 1978;13:281–293. doi: 10.1016/0092-8674(78)90197-6. [DOI] [PubMed] [Google Scholar]
- 29.Patterton H.G., Landel C.C., Landsman D., Peterson C.L. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of Saccharomyces cerevisiae. J. Biol. Chem. 1998;273:7268–7276. doi: 10.1074/jbc.273.13.7268. [DOI] [PubMed] [Google Scholar]
- 30.Friedkin I., Katcoff D. Specific distribution of the Saccharomyces cerevisiae linker histone homologue Hho1p in the chromatin. Nucleic Acids Res. 2001;29:4043–4051. doi: 10.1093/nar/29.19.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui F., Cole H.A., Clark D.J., Zhurkin V.B. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res. 2012;40:10753–10764. doi: 10.1093/nar/gks870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganguli D., Chereji R.V., Iben J.R., Cole H.A., Clark D.J. RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res. 2014;24:1637–1649. doi: 10.1101/gr.177014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satchwell S.C., Drew H.R., Travers A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- 34.Cui F., Zhurkin V.B. Distinctive sequence patterns in metazoan and yeast nucleosomes: Implications for linker histone binding to AT-rich and methylated DNA. Nucleic Acids Res. 2009;37:2818–2829. doi: 10.1093/nar/gkp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brogaard K., Xi L., Wang J.P., Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012;486:496–501. doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davey C.A. Does the nucleosome break its own rules. Curr. Opin. Struc. Biol. 2013;23:311–313. doi: 10.1016/j.sbi.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Flaus A., Luger K., Tan S., Richmond T.J. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1370–1375. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleming A.B., Pennings S. Antagonistic remodelling by Swi-Snf and Tup1-Ssn6 of an extensive chromatin region forms the background for FLO1 gene regulation. EMBO J. 2001;20:5219–5231. doi: 10.1093/emboj/20.18.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reynolds S.M., Bilmes J.A., Noble W.S. Learning a weighted sequence model of the nucleosome core and linker yields more accurate predictions in Saccharomyces cerevisiae and Homo sapiens. PLoS Comp. Biol. 2010;6:e1000834. doi: 10.1371/journal.pcbi.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ulanovsky L.E., Trifonov E.N. Estimation of wedge components in curved DNA. Nature. 1987;326:720–722. doi: 10.1038/326720a0. [DOI] [PubMed] [Google Scholar]
- 41.Zhurkin V.B., Lysov Y.P., Ivanov V.I. Anisotropic flexibility of DNA and the nucleosomal structure. Nucleic Acids Res. 1979;6:1081–1096. doi: 10.1093/nar/6.3.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D., Ulyanov N.B., Zhurkin V.B. Sequence-dependent kink and slide deformations of nucleosomal DNA facilitated by histone arginines bound in the minor groove. J. Biomol. Struc. Dyn. 2010;27:843–859. doi: 10.1080/07391102.2010.10508586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tolstorukov M.Y., Jernigan R.L., Zhurkin V.B. Protein-DNA hydrophobic recognition in the minor groove is facilitated by sugar switching. J. Mol. Biol. 2004;337:65–76. doi: 10.1016/j.jmb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 44.Weischet W.O., Allen J.R., Riedel G., van Holde K.E. The effects of salt concentration and H1-depletion on the digestion of calf thymus chromatin by micrococcal nuclease. Nucleic Acids Res. 1979;6:1843–1862. doi: 10.1093/nar/6.5.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Holde K.E. Chromatin. NY: Springer-Verlag; 1988. [Google Scholar]
- 46.Lindsey G.G., Thompson P. Isolation and characterisation of a 167 bp core particle from stripped chicken erythrocyte chromatin. Biochim. Biophys. Acta. 1989;1009:257–263. doi: 10.1016/0167-4781(89)90111-5. [DOI] [PubMed] [Google Scholar]
- 47.Schalch T., Duda S., Sargent D.F., Richmond T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 48.Angelov D., Vitolo J.M., Mutskov V., Dimitrov S., Hayes J.J. Preferential interaction of the core histone tail domains with linker DNA. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6599–6604. doi: 10.1073/pnas.121171498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon F., Luger K., Hansen J.C. The core histone N-terminal tail domains function independently and additively during salt-dependent oligomerization of nucleosomal arrays. J. Biol. Chem. 2005;280:33701–33706. doi: 10.1074/jbc.M507048200. [DOI] [PubMed] [Google Scholar]
- 50.Rhee H.S., Bataille A.R., Zhang L., Pugh B.F. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell. 2014;159:1377–1388. doi: 10.1016/j.cell.2014.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowary P.T., Widom J. Higher-order structure of Saccharomyces cerevisiae chromatin. Proc. Natl. Acad. Sci. U.S.A. 1989;86:8266–8270. doi: 10.1073/pnas.86.21.8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Correll S.J., Schubert M.H., Grigoryev S.A. Short nucleosome repeats impose rotational modulations on chromatin fibre folding. EMBO J. 2012;31:2416–2426. doi: 10.1038/emboj.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashimoto H., Takami Y., Sonoda E., Iwasaki T., Iwano H., Tachibana M., Takeda S., Nakayama T., Shinkai Y. Histone H1 null vertebrate cells exhibit altered nucleosome architecture. Nucleic Acids Res. 2010;38:3533–3545. doi: 10.1093/nar/gkq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blank T.A., Becker P.B. Electrostatic mechanism of nucleosome spacing. J. Mol. Biol. 1995;252:305–313. doi: 10.1006/jmbi.1995.0498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.