

ABSTRACT
Cortical interneurons are generated predominantly in the medial ganglionic eminence (MGE) and migrate through the ventral and dorsal telencephalon before taking their final positions within the developing cortical plate. Previously we demonstrated that interneurons from Robo1 knockout (Robo1−/−) mice contain reduced levels of neuropilin 1 (Nrp1) and PlexinA1 receptors, rendering them less responsive to the chemorepulsive actions of semaphorin ligands expressed in the striatum and affecting their course of migration (Hernandez‐Miranda et al. [2011] J. Neurosci. 31:6174–6187). Earlier studies have highlighted the importance of Nrp1 and Nrp2 in interneuron migration, and here we assess the role of PlexinA1 in this process. We observed significantly fewer cells expressing the interneuron markers Gad67 and Lhx6 in the cortex of PlexinA1 −/− mice compared with wild‐type littermates at E14.5 and E18.5. Although the level of apoptosis was similar in the mutant and control forebrain, proliferation was significantly reduced in the former. Furthermore, progenitor cells in the MGE of PlexinA1 −/− mice appeared to be poorly anchored to the ventricular surface and showed reduced adhesive properties, which may account for the observed reduction in proliferation. Together our data uncover a novel role for PlexinA1 in forebrain development. J. Comp. Neurol. 524:518–534, 2016. © 2015 The Authors The Journal of Comparative Neurology Published by Wiley Periodicals, Inc.
Keywords: neuronal migration, Plexin, interneurons, proliferation, forebrain
The medial ganglionic eminence (MGE) is the major source of interneurons to the mammalian cerebral cortex (Lavdas et al., 1999; Wichterle et al., 2001; Hansen et al., 2013). En route to the cortex, migrating MGE‐derived interneurons encounter the developing striatum and avoid it (Marin and Rubenstein, 2003; Métin et al., 2006). Work over the last decade has identified some of the cellular and molecular mechanisms that guide their migration through the ventral telencephalon (for review see Hernandez‐Miranda et al., 2010; Faux et al., 2012). These include class 3 semaphorin ligands and their receptors, the neuropilins (Nrps) and plexins.
Earlier studies have shown that migrating cortical interneurons express Nrp receptors (Marin et al., 2001; Tamamaki et al., 2003a). Blocking Nrp1 function with either anti‐Nrp1 antibody (Tamamaki et al., 2003a) or Nrp1 dominant negative focal electroporation in slice cultures disrupted their migration, with a significant number straying into the striatum (Marin et al., 2001). Similarly, loss of Nrp2 function in Nrp2−/− transgenic mice resulted in more cortical and striatal (neuropeptide Y [NPY]‐expressing) interneurons in the striatum (Marin et al., 2001). More recently, we have found that interneurons in mice lacking the roundabout receptor Robo1 have reduced levels of Nrp1 and PlexinA1 compared with control littermates and are less responsive to Sema3A and Sema3F, resulting in their aberrant migration through the striatum (Hernandez‐Miranda et al., 2011). This raises the question of whether the Nrp coreceptor PlexinA1 is important for cortical interneuron migration.
Here, we quantified the number and distribution of interneurons in the cortex of PlexinA1−/− mice and littermate controls in the middle and late stages of corticogenesis. We found significantly fewer cells in mice lacking the receptor, suggesting disrupted migration and/or reduced generation in the MGE. Further experiments showed a marked decrease in proliferation in ventral and dorsal forebrain, suggesting a reduction in the number of interneuron and pyramidal cell progenitors. Nestin staining in the proliferative zones of the MGE confirmed not only the reduction of progenitor cells in the knockout but also altered morphology, with cells often lacking attachments to the ventricular surface. Furthermore, adhesion assay experiments showed reduced attachment in PlexinA1−/− mice compared with controls. Together our data suggest that reduced adhesiveness of interneuron progenitors in PlexinA1−/− mice may underlie the observed reduction in proliferation, resulting in fewer interneurons (and pyramidal cells) in the cortex during development.
MATERIALS AND METHODS
Animals
All experimental procedures were performed in accordance with the U.K. Animals (Scientific Procedures) Act 1986 and institutional guidelines. Wild‐type animals were C57/bl6J mice obtained from Charles River, Ltd. PlexinA1−/− and GAD67‐GFPneo‐ mice were generated as described previously (Yoshida et al., 2006 [PMID: 17145500]; Tamamaki et al., 2003b [PMID: 14574680]). PlexinA1 mice were genotyped by polymerase chain reaction (PCR) with the following primers: WT‐forward (5′‐CCTGCAGATTGATGACGACTTCTGC‐3′), WT‐reverse (5′‐TCATGCAGACCCAGTCTCCCTGTCA‐3′), product size 200 bp; and mutant‐forward (5′‐GCATGCCTGTGACACTTGGCTCACT‐3′), mutant‐reverse (5′‐CCATTGCTCAGCGGTGCTGTCCATC‐3′), product size 600 bp. The day on which the vaginal plug was found was considered embryonic day (E) 0.5. Animals of both sexes were used in our experiments.
In situ hybridization
For in situ hybridization and immunohistochemistry, embryonic brains were dissected in phosphate‐buffered saline (PBS) and fixed in 4% paraformaldehyde (PFA), made by dissolving PFA in PBS for 4–8 hours at room temperature (RT). After fixation, embryonic brains were cryoprotected in 30% sucrose in diethyl pyrocarbonate (DEPC)‐treated PBS, embedded and frozen in a mixture of 15% sucrose/50% Tissue‐Tek OCT (Sakura Finetek), and sectioned in the coronal plane at 20 µm with a cryostat (Bright Instruments). Sections were dried at RT for 2 hours before overnight incubation at 65°C in hybridization buffer (a DEPC‐treated solution containing 200 mM NaCl, 5 mM EDTA, 10 mM Tris, pH 7.5, 5 mM NaH2PO4 · 2H2O, 5 mM Na2HPO4 [Sigma‐Aldrich, St. Louis, MO]; 50% deionized formamide [Ambion, Austin, TX]; 0.1 mg/ml RNase‐free yeast tRNA [Invitrogen, Carlsbad, CA]; 1× RNase/DNase‐free Denhardt's [Invitrogen]; 10% dextran‐sulfate [Sigma‐Aldrich]) containing 100–500 ng/ml DIG‐labeled RNA probes. Antisense probes were generated as described in Table 1. After hybridization, sections were washed three times in 50% formamide 1× SSC (Ambion) and 0.1% Tween‐20 (Sigma‐Aldrich) at 65°C and twice at RT in 1× MABT (20 mM maleic acid, 30 mM NaCl, 0.1% Tween‐20 [Sigma‐Aldrich]) before incubating in blocking solution [MABT containing 2% blocking reagent [Roche. Indianapolis, IN] and 10% normal goat serum [Vector, Burlingame, CA]), followed by overnight incubation in alkaline phosphatase‐conjugated anti‐DIG antibody (1:1,500; Roche). Nitroblue tetrazolium chloride/5‐bromo‐4‐chloro‐3‐indolyl phosphate (Roche) diluted 1:1,000 in MABT containing 5% polyvinyl alcohol (VWR International) was used for the colorimetric detection and Fast Red (Roche) dissolved in 100 mM Tris (pH 8.0) and 400 mM NaCl for fluorescent color detection by incubation at 37°C. Fluorescence in situ hybridization was followed by immunohistochemical detection of green fluorescent protein (GFP) as described below. Sections were mounted with Glycergel mounting medium (Dako, Carpinteria, CA).
Table 1.
In Situ Hybridization Probes
| Antisense probe | Source | Restriction enzyme | RNA polymerase |
|---|---|---|---|
| PlexinA1 | Dr. Nina Perälä, University of Helsinki (Finland) | XhoI | T3 |
| Lhx6 | Dr. Nicoletta Kessaris, UCL, UK | Not1 | T3 |
| ER81 | Dr. Thomas Jessell, Columbia University (U.S.) | SpeI | T7 |
| GAD67 | Dr. Brian Condie, University of Georgia (U.S.) | XhoI | T3 |
Immunohistochemistry
Embryonic brain sections were washed in PBS, blocked in a solution of 5% normal goat serum (v/v; Sigma‐Aldrich) containing 0.1% Triton X‐100 (v/v; Sigma‐Aldrich) in PBS at RT for 2 hours. They were subsequently incubated in primary antibodies at RT for 2 hours and, then, at 4°C overnight. After incubation in primary antibodies, sections were washed in PBS, incubated in biotinylated anti‐species (1:250; Vector) for 2 hours, and processed via immunohistochemistry protocols described previously (Andrews et al., 2008).
Antibody characterization
Details of the antibodies used in this study are summarized in Table 2.
Table 2.
Antibodies Useda
| Antibody ID | Antibody name | Antibody target | Vendor | Catalog No. | Clonality | Clone ID | Host organism | Proper citation | Reference |
|---|---|---|---|---|---|---|---|---|---|
| AB_306886 | Mouse anti‐BrdU monoclonal antibody, unconjugated, clone IIB5 | BrdU | Abcam | ab8955 | Monoclonal antibody | Clone IIB5 | Mouse | (Abcam catalog No. ab8955, RRID: AB_306886) | Morris and Solomon, 2004 |
| AB_10000340 | Rabbit anticalbindin D‐28k antibody | Rabbit anticalbindin D‐28k | Swant | CB 38 | Polyclonal antibody | Rabbit | (Swant catalog No. CB 38, RRID: AB_10000340) | Suárez et al., 2006 | |
| AB_331453 | Cleaved caspase‐3 (Asp175) (5A1E) rabbit mAb | Cleaved caspase‐3 (Asp175) (5A1E) rabbit mAb | Cell Signaling Technology | 9664S | Polyclonal antibody | Rabbit | (Cell Signaling Technology catalog No. 9664S, RRID: AB_331453) | Tokami et al., 2013 | |
| AB_2261231 | CDP (M‐222) antibody | CDP (M‐222) | Santa Cruz Biotechnology | sc‐13024 | Polyclonal antibody | Rabbit | (Santa Cruz Biotechnology catalog No. sc‐13024, RRID: AB_2261231) | Arellano et al., 2012 | |
| AB_2064130 | Rat anti‐Ctip2 monoclonal antibody, unconjugated, clone 25B6 | Ctip2 | Abcam | ab18465 | Monoclonal antibody | Clone 25B6 | Rat | (Abcam catalog No. ab18465, RRID: AB_2064130) | Stillman et al., 2009 |
| AB_2107107 | Rabbit anti‐FOXP2 polyclonal antibody, unconjugated | FOXP2 | Abcam | ab16046 | Polyclonal antibody | Rabbit | (Abcam catalog No. ab16046, RRID: AB_2107107) | Campbell et al., 2009 | |
| AB_10000240 | Chicken anti‐GFP antibodies | GFP | Aves Labs | GFP‐1020 | Polyclonal antibody | Chicken | (Aves Labs catalog No. GFP‐1020, RRID: AB_10000240) | Xu et al., 2006 | |
| AB_2235915 | Mouse anti‐rat nestin monoclonal antibody, unconjugated | rat nestin | Developmental Studies Hybridoma Bank | Rat‐401 | Monoclonal antibody | Mouse | (Developmental Studies Hybridoma Bank catalog No. Rat‐401, RRID: AB_2235915) | Wang et al., 2006 | |
| AB_1587367 | Rabbit anti‐PAX6, unconjugated antibody | PAX6 | Millipore | AB2237 | Polyclonal antibody | Rabbit | (Millipore catalog No. AB2237, RRID: AB_1587367) | Piper et al., 2011 | |
| AB_310177 | Rabbit antihistone H3 (phosphorylated at Ser10), mitosis marker polyclonal antibody, unconjugated | Histone H3 (phosphorylated at Ser10) | Millipore | 06–570 | Polyclonal antibody | Rabbit | (Millipore catalog No. 06–570, RRID: AB_310177) | Wang et al., 2006 | |
| AB_2166258 | PlexinA1 (H‐60) antibody | PlexinA1 (H‐60) | Santa Cruz Biotechnology | sc‐25639 | Polyclonal antibody | Rabbit | (Santa Cruz Biotechnology catalog No. sc‐25639, RRID: AB_2166258) | Cariboni et al., 2007 |
BrdU, bromodeoxyuridine; CB, calbindin; CDP, CCAAT displacement protein ovalbumin upstream promoter transcription factor‐interacting protein‐2; FOXP2, Forkhead box protein P2; Pax6, paired box; Ctip2, B‐cell leukemia/lymphoma 11B.
Bromodeoxyuridine antibody
A mouse monoclonal antibody raised against bromodeoxyuridine (BrdU) and conjugated to bovine serum albumin (BSA; Abcam, Cambridge, MA; catalog No. ab8955, RRID: AB_306886) was used to immunolabel proliferating progenitor cells in the developing forebrain following injection of BrdU into pregnant dams (Cavanagh et al., 1997). BrdU immunohistochemistry of wild‐type mouse forebrain sections showed no staining.
Calbindin antibody
The calbindin D‐28 (CB) antiserum (Swant, Belinzona, Switzerland; catalog No. CB 38, RRID: AB_10000340) recognized a single band of 28 kDa on Western blots of rat brain (manufacturer's data sheet) and stained a pattern of cellular morphology and distribution in the mouse developing cerebral cortex that is identical to that in previous reports (Andrews et al., 2008; Hernandez‐Miranda et al., 2011).
Cleaved caspase‐3 antibody
We used a rabbit monoclonal antibody (Cell Signaling Technology, Danvers, MA; catalog No. 9664S, RRID: AB_331453) raised against a synthetic peptide corresponding to amino‐terminal residues adjacent to Asp175 of human caspase‐3 to detect endogenous levels of the large fragment (17/19 kDa) of activated caspase‐3 (Yeh et al., 2014).
CDP antibody
CDP (M‐222) is a rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA; catalog No. sc‐13024, RRID: AB_2261231) raised against amino acids 1111–1332 of CDP/Cux1 of mouse origin. CDP immunohistochemistry of wild‐type embryonic mouse forebrain sections specifically labels upper layer pyramidal neurons (Arlotta et al., 2005; Yeh et al., 2014).
Ctip2 antibody
Rat monoclonal antibody (25B6; Abcam; catalog No. ab18465, RRID: AB_2064130) to Ctip2 recognizes an epitope between amino acids 1 and 150 of human CTIP2. Ctip2 immunohistochemistry carried out on wild‐type embryonic mouse forebrain sections specifically labels lower layer pyramidal neurons (Cubelos et al., 2008; Yeh et al., 2014).
FOXP2 antibody
Rabbit polyclonal FOXP2 antibody (Abcam; catalog No. ab16046, RRID: AB_210710) raised against a synthetic peptide in the C‐terminus of human FOXP2 was previously shown to immunolabel striatal projection neurons in the developing mouse forebrain (Takahashi et al., 2003; Hernandez‐Miranda et al., 2011).
GFP antibody
We used a GFP antiserum (Aves Labs; catalog No. GFP‐1020, RRID: AB_10000240) that recognizes the expected (27 kDa) band on Western blots of GFP‐positive transgenic mouse brain (manufacturer's data sheet). GFP immunohistochemistry, carried out on wild‐type mouse forebrain sections, showed no staining except for weak autofluorescence in the choroid plexus (data not shown).
Nestin antibody
Mouse nestin monoclonal antibody (Developmental Studies Hybridoma Bank; catalog No. Rat‐401, RRID: AB_2235915) raised against E15 Sprague‐Dawley rat spinal cord labeled radial glia progenitors, as previously described (Cavanagh et al., 1997).
Pax6 antibody
Pax6 rabbit polyclonal antibodies (Millipore, Bedford, MA; catalog No. AB2237, RRID: AB_1587367) raised against the C‐terminus of human PAX6 produced the characteristic labeling of cortical neuronal progenitors on embryonic mouse forebrain tissue, as previously described (Quinn et al., 2007).
Phospho‐histone H3 antibody
Phospho‐histone H3 (PH‐3) rabbit polyclonal antibodies (Millipore; catalog No. 06–570, RRID: AB_310177) raised against a linear peptide corresponding to human histone H3 phosphorylated at Ser10 was used to quantify cell proliferation in the developing cortex, as previously described (Andrews et al., 2008; Yeh et al., 2014).
PlexinA1 antibody
A PlexinA1 (H‐60) rabbit polyclonal antibody (Santa Cruz Biotechnology; catalog No. sc‐25639, RRID: AB_2166258) raised against amino acids 961–1020 of human PlexinA1, shown to recognize PlexinA1 in brain lysates by Western blotting, stained many cell types on wild‐type, but not knockout, forebrain sections (data not shown).
Interneuron counts in the cortex
Interneurons labeled with Lhx6, GAD67, or calbindin were counted in images collected with a Leica DM5000B light microscope. The images were of coronal strips (200 μm wide) spanning the thickness of the neocortex throughout its rostrocaudal extent at different ages (minimum of six sections from each of three animals for each genotype). Each coronal strip was divided into bins arranged parallel to the pial surface that corresponded to the different layers of the developing cortex.
Proliferation
The mothers of E14.5 embryos were injected intraperitoneally with 50 μg BrdU (Sigma‐Aldrich) per gram of body weight and culled 1 hour later. Brains were then fixed with PFA, and 20‐μm‐thick sections were cut with a cryostat. For BrdU immunolabeling, sections were first incubated with 2 N HCl at 37°C for 30 minutes to unmask the antigen, followed by three washes in PBS. PH‐3 staining was performed as described above. We counted all PH‐3+ cells in a 200‐µm‐wide strip perpendicular to the ventricular wall of the VZ and in a 4 × 104‐µm2 area within the SVZ of the MGE.
Dissociated MGE cell cultures
Dissociated cell cultures were prepared from E12.5 mice as described previously (Cavanagh et al., 1997). Briefly, MGEs were dissected out in cold artificial cerebrospinal fluid (ACSF) under a stereomicroscope. They were incubated in neurobasal medium (Life Technologies, Grand Island, NY) containing 0.05% trypsin (Sigma‐Aldrich) and 100 µg/ml DNaseI (Roche) at 37°C for 15 minutes. Trypsinization was quenched with neurobasal medium containing 10% FBS (Life Technologies) at 37°C for 5 minutes. MGEs were then triturated by pipetting until no cellular aggregates were visible. The homogeneous cell suspensions were subsequently pelleted by centrifugation at 1,000g for 3 minutes. Cells were resuspended in DMEM/F12 culture media containing B27 supplement, 100 µg/ml penicillin/streptomycin, and 2 mM L‐glutamine (Life Technologies), and 100,000 cells were seeded onto 13‐mm coverslips coated previously with 10 µg/ml poly‐L‐lysine and 10 µg/ml laminin (Sigma‐Aldrich) and incubated in a the humidified incubator at 37°C. On the next day, the culture medium was changed.
Chemotaxis assays
Chemotaxis assays were performed with a 48‐well Boyden's chamber (NeuroProbe) as described previously (Hernandez‐Miranda et al., 2011). Briefly, 27 µl serum‐free dissociation media or 1% FBS dissociation media was placed into the lower compartment of the chamber. Dissociated MGE cells were resuspended in serum‐free medium (105 cells/50 µl) and placed in wells of the upper compartment of the chamber. These were separated from the lower chamber by a polycarbonate porous membrane (8‐µm pores), precoated with laminin (10 µg/ml). The chamber was kept in an incubator at 37°C overnight. After incubation, the migrated cells that adhered to the underside of the membrane were fixed and stained using the Diff‐Quick kit (Reagena). For quantitative analysis, the membranes were observed with an Olympus light microscope with a ×20 objective adapted with a 500 × 500‐µm grid. Four random fields of stained cells were counted for each well, and the mean number of migrating cells per square millimeter for each experimental condition was estimated.
Adhesion assay
MGEs were isolated from E12.5 PlexinA1+/+ and PlexinA1−/− littermates and dissociated cultures prepared as described above. Cell densities were adjusted to 2 × 106/ml in culture medium, and 50 µl cell suspension was seeded onto coated coverslips in triplicate. The coverslips were incubated at 37°C for 30 minutes, washed with PBS, and fixed with 2% PFA for 15 minutes. Attached cells were visualized with DAPI. Six pictures were taken per coverslip (×20 magnification), and attached cells were counted.
Angle measurements
E14.5 PlexinA1+/+ and PlexinA1−/− coronal brain sections (n = 3 both groups) were stained with the progenitor marker nestin. The degree of variation from the horizontal plane between the tip of apical and basal process within the ventricular zone of the MGE was measured in ImageJ 1.48 software (NIH; RRID: nif‐0000–30467).
In utero electroporation
In utero electroporation of the MGE was performed as described previously (Wu et al., 2011). Briefly, timed pregnant C57/BL6 mice at E12.5 were anesthetized, the abdomen opened, and the uterus was exposed. DNA vectors (1 µg/µl, 2 µl; siRNA) were injected into the third ventricle of embryos through a glass micropipette and introduced into the ventricular zone of the MGE by delivering electric pulses (40 V, 50 msec, 4 Hz) through the uterus. The uterus was repositioned in the abdominal cavity, and the abdominal wall and skin were sewn with surgical sutures. The embryos were fixed in PFA at E14.5. We quantified the number of GFP‐positive cells (in utero electroporated animals) in the MGE that were attached/unattached to the ventricular zone surface (n = 3/4 embryos for each siRNA construct).
Semaphorin binding assays
Alkaline phosphatase (AP)‐conjugated Sema3A (SEMA3A‐AP) was prepared as previously described (Vieira et al., 2007). Fresh frozen E14.5 brain sections were fixed in absolute methanol for 5 minutes, washed five times with PBS, incubated in PBS containing 0.1% Triton X‐100 (PBT) and 10% FBS for 30 minutes, and then reacted with SEMA3A‐AP at RT for 2 hours. Sections were then washed for 5 minutes with PBS, fixed in PFA at RT for 2 minutes, and washed again. Endogenous AP was heat inactivated by incubation at 65°C for 3 hours. Tissue‐bound, heat‐stable recombinant AP activity was detected as an insoluble reaction product after incubation with NBT and BCIP.
Digital image acquisition and processing
Brightfield and fluorescent images were collected with a light‐ or confocal microscope (Leica). Images were reconstructed and digitized in Photoshop CS2 (Adobe Systems, Mountain View, CA; RRID: SciRes_000161).
Statistical analysis
Statistical analyses were performed in GraphPad 3 (Graph‐Pad Software, San Diego, CA; RRID: nlx_156835). All data are reported as mean ± SEM. The statistical significance between group means was tested by one‐way ANOVA, followed by Bonferroni's post hoc test (for multiple‐comparisons tests). Significance was set at P = 0.05.
RESULTS
Expression of PlexinA1 in migrating interneurons
Previous studies have shown a function for Nrp receptors in cortical interneuron development but no clear role for their coreceptor, PlexinA1. To address this point, we first compared the expression of PlexinA1 relative to the interneuron marker Lhx6 (Alifragis et al., 2004) throughout early (E13.5), middle (E15.5), and late (E18.5) stages of corticogenesis.
At E13.5, the expression pattern of PlexinA1 overlapped partially with Lhx6 in the intermediate zone (IZ) of the cortex and more extensively in the mantle zone of the ventral telencephalon (Fig. 1A,D). PlexinA1 expression was also observed in the ventricular zone (VZ) and subventricular zone (SVZ) of the MGE. At E15.5, it showed a high degree of overlap with Lhx6 in the ventral telencephalon, although in the cortex it was strongly expressed in the cortical plate (CP) and overlapped with Lhx6 in the marginal zone (MZ) (Fig. 1B,E). At the end of corticogenesis (E18.5), PlexinA1 expression, similar to Lhx6, was evident in both dorsal and ventral forebrain (Fig. 1C,F), and in the former it was strongly present in the CP and subplate (SP; Fig. 1C).
Figure 1.
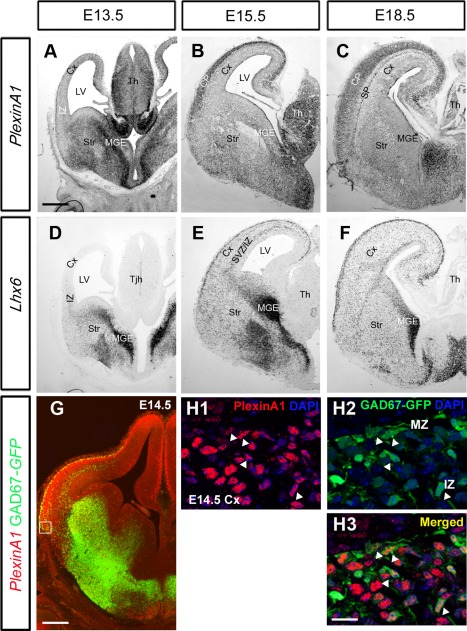
Expression patterns of PlexinA1 and Lhx6 in wild‐type mouse brain. A–F: In situ hybridization on coronal sections at E13.5 (A,D), E15.5 (B,E), and E18.5 (C,F) for PlexinA1 (A–C) and the interneuron marker Lhx6 (D–F). Overlapping patterns of expression were observed between PlexinA1 and Lhx6 at all ages. G–H3: Coronal section through the brain of a E14.5 GAD67‐GFP mouse, processed for immunohistochemistry with PlexinA1 antibody (red), showed the presence of the receptor within interneurons in the MGE, LGE, MZ, and IZ in the cortex (arrows H1–H3). CP, cortical plate; SP, subplate; Cx, cerebral cortex; IZ, intermediate zone; LV, lateral ventricle; MGE, medial ganglionic eminence; Str, striatum; SVZ, subventricular zone; Th, thalamus. Scale bars = 200 µm in A (applies to A–F); 200 µm in G; 30 µm in H.
To confirm that PlexinA1 is expressed in cortical interneurons, we used coronal sections from E14.5 GAD67‐GFP mice (Tamamaki et al., 2003b). Double labeling for PlexinA1 and GFP revealed colocalization in the mantle zones of the MGE and LGE as well as in the IZ and MZ of the cortex (Fig. 1G). In the cortex, approximately 50% (47.33% ± 4.11%) of GFP+ cells (presumptive interneurons) appeared to coexpress PlexinA1 (Fig. 1H1–H3). PlexinA1 expression in interneurons is in concordance with previous PCR (Hernandez‐Miranda et al., 2011) and Western blot analyses of MGE cells isolated by FACS (Andrews et al., 2013). Thus, it appears that cortical interneurons express PlexinA1, both at their sites of origin within the ventral forebrain and along their migratory paths to the cortex, suggesting that it may be crucial for their generation, migration, and development.
Deletion of PlexinA1 leads to a reduced number of interneurons in the developing cortex
Using in situ hybridization for the interneuron markers Lhx6 and Gad67 (Alifragis et al., 2004; Retaux et al., 1993), we next assessed the number of GABAergic cells within the developing cortex at middle (E14.5) and late (E18.5) developmental stages in PlexinA1+/+ and PlexinA1−/− littermates (n = 6 each). We counted labeled cells in 200‐µm‐wide coronal strips through the middle regions (along the rostral–caudal axis) of the cortex. At E14.5, we observed a significant decrease in the number of both Lhx6 (PlexinA1+/+ 106.12 ± 3.65, PlexinA1−/−83.47 ± 4.2; P < 0.0003) and Gad67 (PlexinA1+/+ 148 ± 4.9, PlexinA1−/−123.62 ± 5.9; P < 0.0002) cells in the cortex of mice lacking the receptor compared with control littermates (Fig. 2A,B,E,F). The reduction in the number of cells positive for both markers was evident across all cortical layers, with the exception of the SP (Fig. 2A,B,E,F).
Figure 2.
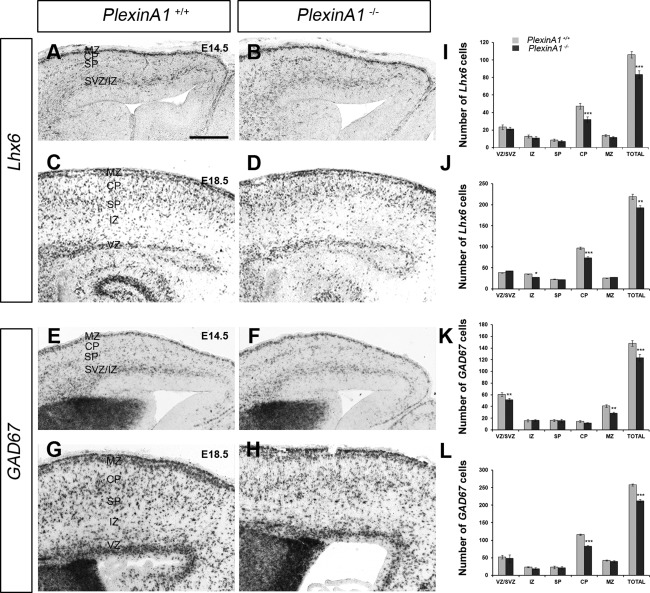
Deletion of PlexinA1 receptor leads to a decreased number of GABAergic interneurons in the cerebral cortex. A–H: Photomicrographs of in situ hybridization for Lhx6 (A–D) and Gad67 (E–H) on coronal sections through the cortex of PlexinA1+/+ (A,C,E,G) and PlexinA1−/− (B,D,F,H) mice at E14.5 (A,B,E,F) and E18.5 (C,D,G,H). I–L: Analysis of the number and distribution of Lhx6‐labeled cells at E14.5 (I) and E18.5 (J) and Gad67 cells at E14.5 (K) and E18.5 (L) in all layers of the cortex of PlexinA1+/+ and PlexinA1−/− mice. Counts were made in the middle region along the rostrocaudal extent of the cortex. Error bars indicate SEM (*P < 0.01, **P < 0.001, ***P < 0.0001). CP, cortical plate; IZ, intermediate zone; MZ, marginal zone; SP, subplate; SVZ, subventricular zone; VZ, ventricular zone. Scale bar = 150 µm.
Analysis at a later stage of corticogenesis (E18.5) similarly revealed significant reductions in the numbers of cells positive for Lhx6 (PlexinA1+/+ 219.34 ± 5.72, PlexinA1−/− 193.1 ± 4.91; P < 0.004) or Gad67 (PlexinA1+/+ 257.8 ± 2.7, PlexinA1−/− 211.95 ± 4.32; P < 0.0006) in the cortex of PlexinA1−/− mice compared with control littermates across all layers, with the exception of the SP (Fig. 2C,D,G,H). This finding is surprising because PlexinA1 is distinctly expressed in the SP at this age (Fig. 1C). These observations were confirmed by immunohistochemistry for another interneuron marker, calbindin (data not shown). Taken together, these findings suggest alterations in the generation and/or migration of cortical interneurons in PlexinA1−/− mice.
No change in migratory potential of PlexinA1−/− MGE cells
We have recently shown, by using electroporation of siRNAs in brain slices and in utero, that downregulation of either Limk2 or PlexinA1 results in aberrant migration of interneurons through the striatum (Andrews et al., 2013), presumably as a result of perturbed semaphorin signaling. To assess directly whether loss of plexinA1 function affects interneuron migration independently of the responsiveness to semaphorin, we carried out a migratory assay with the Boyden chemotaxis chamber and dissociated MGE cells prepared from E13.5 PlexinA1+/+ and PlexinA1−/− littermates (n = 3 each). In the absence of serum, we observed similar levels of migration between PlexinA1+/+ and PlexinA1−/− mice (PlexinA1+/+ 1,264 ± 167 MGE cells/mm2, PlexinA1−/− 1,315 ± 143 cells/mm2; P < 0.3). Although addition of serum increased the basal level of migration, in line with previous observations (Rakić et al., 2009), we did not observe any significant differences between the two groups (PlexinA1+/+ 3,121 ± 447 cells/mm2, PlexinA1−/− 3,251 ± 343 cells/mm2; P < 0.4), suggesting that loss of PlexinA1 function does not alter the migratory potential of MGE‐derived neurons.
Reduced number of striatal projection neurons in PlexinA1−/− mice
To assess whether projection neuron numbers were also affected within the striatum, we immunostained coronal sections from PlexinA1−/− mice and PlexinA1+/+ littermates at E14.5 and E18.5 (n = 3 per age for each genotype) for the transcription factor Forkhead box protein P2 (FOXP2), a marker of developing striatal projection neurons (Takahashi et al., 2003). Counts of labeled cells throughout the rostrocaudal extent of the striatum showed a significant reduction in mutants compared with control littermates at E14.5 (medial levels PlexinA1+/+ 711.21 ± 4.53 cells/105 µm2, PlexinA1−/− 548.96 ± 4.57 cells/105 µm2; P < 0.00049) and at E18.5 (PlexinA1+/+ 840.45 ± 5.44 cells/105 µm2, PlexinA1−/− 658.75 ± 5.48 cells/105 µm2; P < 0.00054; Fig. 3A,B,E,F). A similar reduction in the number of striatal projection neurons was observed with another marker, ER81 (Stenman et al., 2003; Fig. 3C,D).
Figure 3.
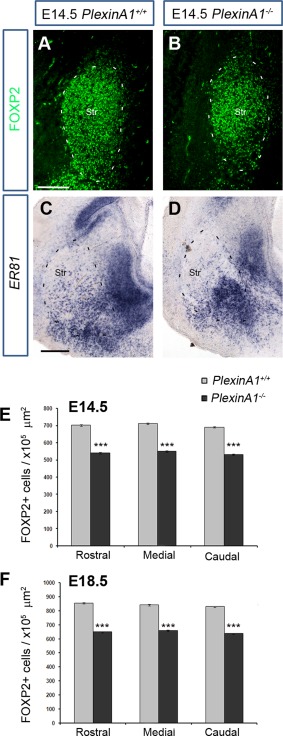
Reduced number of FOXP2+ cells in the developing striatum of PlexinA1−/− mice. Coronal brain sections from PlexinA1+/+ (A,C) and PlexinA1−/− (B,D) mice at E14.5 were immunostained for FOXP2 (A,B) or processed for in situ hybridization for ER81 (C,D). Quantification of FOXP2+ cells in the striatum of PlexinA1−/− animals showed reduced numbers of labeled cells compared with PlexinA1+/+ littermates at E14.5 (E) and E18.5 (F). Error bars indicate SEM (***P < 0.0005). Str, striatum. Scale bars = 100 µm in A (applies to A,B); 150 µm in C (applies to C,D).
Reduced proliferation in the developing forebrain of PlexinA1−/− mice
The reduced number of cortical interneurons and striatal projection cells could be due to either increased apoptosis or reduced proliferation. To assess these possibilities, we immunostained coronal sections from E14.5 PlexinA1−/− and control littermate mice (n = 4 for each genotype) for the apoptotic marker cleaved caspase 3 (CC3) and the mitotic marker PH‐3. At this age, we observed very little CC3 staining in the forebrains of control or PlexinA1−/− mutant mice, in accordance with previous studies (Thomaidou et al., 1997; Yeh et al., 2014), suggesting that the observed reduction in neuronal numbers is unlikely to be due to increased apoptosis. In contrast, analysis of the number of PH‐3‐positive cells in the proliferative zones throughout the rostrocaudal extent of the developing cortex showed a significant decrease in mutants compared with controls (VZ medial level: PlexinA1+/+ 17.54 ± 0.20, PlexinA1−/− 13.19 ± 0.11 per 100 µm [P < 0.0002[; SVZ medial level: PlexinA1+/+ 5.28 ± 0.26, PlexinA1−/− 2.28 ± 0.15 per 104 µm2 [P < 0.0023]; Fig. 4A–D). We also observed a similar decrease in the number of PH‐3 cells along the VZ of the CGE, LGE, and MGE in mutants compared with controls (MGE PlexinA1+/+ 12.16 ± 0.24, PlexinA1−/− 9.59 ± 0.11 100 µm; P < 0.0073; Fig. 4E–G).
Figure 4.
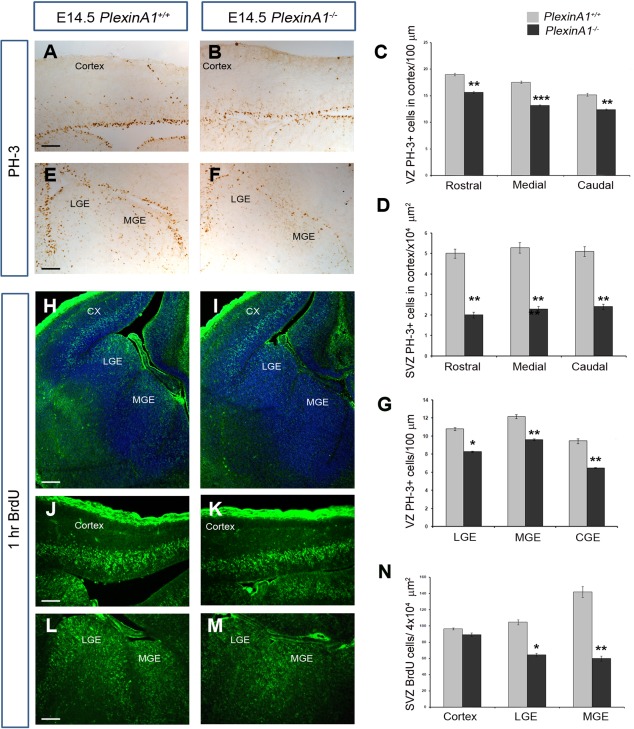
Reduced proliferation in PlexinA1−/− mice. Coronal brain sections from PlexinA1+/+ (A,E,H,J,L) and PlexinA1−/− (B,F,I,K,M) mice at E14.5 were immunostained for PH‐3 (A,B,E,F) and BrdU (H–M). Quantification of PH‐3+ (C,D,G) and BrdU+ (N) cells in the proliferative zones of PlexinA1−/− animals showed reduced numbers compared with PlexinA1+/+ littermates. Error bars indicate SEM (*P < 0.01, **P < 0.001, ***P < 0.0001). Cx, cortex; CGE, caudal ganglionic eminence; MGE, medial ganglionic eminence; LGE, lateral ganglionic eminence. Scale bars = 50 µm in A (applies to A,B); 150 µm in E (applies to E,F); 150 µm in H (applies to H,I); 50 µm in J (applies to J,K); 150 µm in L (applies to L,M).
To confirm these findings, PlexinA1−/− mice and their wild‐type littermates at E14.5 (n = 4 each) were pulse labeled for 1 hour with BrdU, a thymidine analogue that becomes incorporated into DNA during S phase of the cell cycle. We observed a reduced number of BrdU‐positive cells in PlexinA1−/− mutant mice compared with control littermates in the LGE (PlexinA1+/+ 104.44 ± 3.07, PlexinA1−/− 64.51 ± 1.87 per 4 × 104 µm2; P < 0.01) and MGE (PlexinA1+/+ 141.76 ± 6.83, PlexinA1−/− 59.84±.3.12 per 4 × 104 µm2; P < 0.001; Fig. 4H–N). These observations suggest that the decrease in the numbers of striatal projection neurons and cortical interneurons in the forebrains of PlexinA1‐deficient mice likely is due to reduced proliferation of their progenitors in the LGE and MGE.
To assess whether the decreased number of mitotically active progenitor cells in the cortex of PlexinA1−/− mice results in a decrease in pyramidal neurons, we carried out immunostaining for the cortical progenitor cell marker Pax6 and in situ hybridization for its downstream target, ER81, a layer 5 neuronal marker (Tuoc and Stoykova, 2008) at E14.5. We observed reduced expression of both markers in the cortex of PlexinA1−/− mice compared with control littermates (n = 3, both groups; Fig. 5A–D). We also observed a significantly reduced number of Ctip2 (early‐born)‐ and Cux1 (late‐born)‐positive neurons (Arlotta et al., 2005; Cubelos et al., 2008) in the developing cortex at E18.5 (Ctip2: PlexinA1+/+ 41.93 ± 15.7, PlexinA1−/− 28.41 ± 6.7 [P < 0.3]; Cux1: PlexinA1+/+ 82.07 ± 6.4, PlexinA1−/− 58.24 ± 4.3 [P < 0.005]; Fig. 5E–I). This reduction correlated with significantly decreased cortical thickness (PlexinA1+/+ 711.6 ± 6.04, PlexinA1−/− 666 ± 11.72 µm; P < 0.07). Thus our findings suggest that loss of PlexinA1 function impairs the generation of neurons contributing to the cortex.
Figure 5.
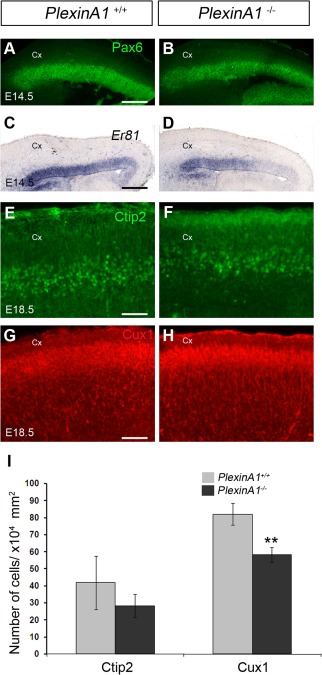
Reduced number of pyramidal neurons in the cortex of PlexinA1−/− mice. Coronal brain sections from PlexinA1+/+ (A,C,E,G) and PlexinA1−/− (B,D,F,H) mice at E14.5 (A–D) and E18.5 (E–H) were immunostained for Pax6 (A,B), Ctip2 (E,F), and Cux1 (G,H) or processed for in situ hybridization for Er81 (C,D). I: Quantification of Ctip2 and Cux1+ cells in the cortex of PlexinA1−/− animals showed reduced numbers compared with PlexinA1+/+ littermates. Error bars indicate SEM (**P < 0.001). Cx, cortex. Scale bars = 200 µm in A (applies to A,B); 200 µm in C (applies to C,D); 100 µm in E (applies to E,F); 100 µm in G (applies to G,H).
To explore further the apparent decline in proliferation in the MGE, we examined the morphology of progenitors cells along the VZ by immunostaining E14.5 ventral forebrain sections of PlexinA1+/+ and PlexinA1−/− mice (n = 3 both groups) with an antibody to the neuronal progenitor marker nestin. As expected, nestin‐positive cells from PlexinA1+/+ mice appeared to be anchored and positioned perpendicular to the ventricular surface (52/57 cells, corresponding to 91.23%, were located at 85–95°); in contrast, in PlexinA1−/− mice, fewer nestin‐positive cells were attached to the ventricular wall, and fewer were positioned perpendicular to the ventricular surface (17/79 cells, corresponding to 21.52%, were located at 85–95°; n = 3 each; Fig. 6A–C). There were also fewer nestin‐positive cells in PlexinA1−/− mice, in agreement with our PH‐3 data. The lack of receptor appeared to have a similar effect on the number and orientation of progenitor cells in the VZ of the dorsal forebrain (data not shown).
Figure 6.
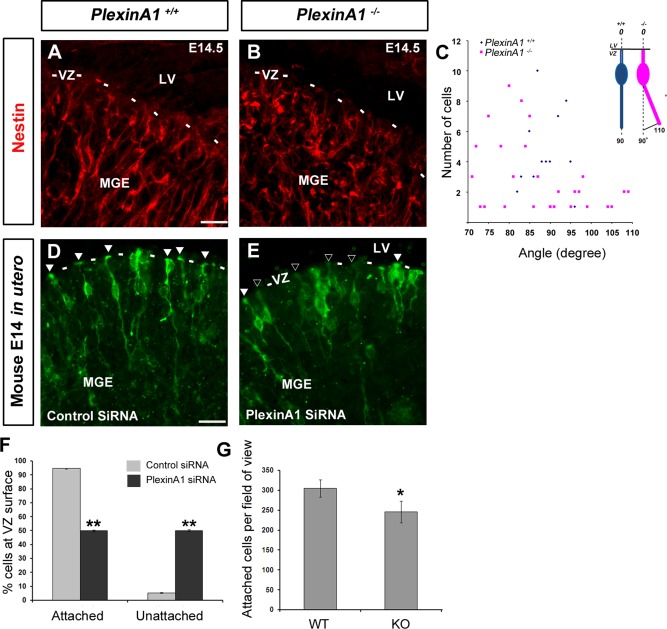
Reduced attachment and altered morphology of progenitor cells in the MGE of PlexinA1−/− mice. Coronal brain sections from PlexinA1+/+ (A) and PlexinA1−/− (B) mice at E14.5 were immunostained for the progenitor cell marker Nestin (A‐B). (A,B) Fewer Nestin‐positive cells appear to be attached to the ventricular wall and positioned perpendicular to the ventricular surface in PlexinA1−/− mice. (C) Quantification of the orientation of the basal process of labelled cells in the VZ/SVZ in PlexinA1−/− mice. (D,E) Mouse embryos electroporated in utero at E12.5 with control‐(GFP)siRNA (D) or PlexinA1‐(GFP)siRNA (E) into the MGE and harvested at E14.5. Fewer cells appear to be attached to the ventricular surface following PlexinA1 knockdown (black arrowheads) compared to control (white arrowheads), quantified in F. (G) Quantification of adhesion assay, showing fewer E12.5 dissociated MGE cells from PlexinA1−/− mice attached to coated coverslips compared to control littermates. Scale bars in A‐B, 20 μm; and D‐E, 50 μm. (*P < 0.01). Error bars indicate SEM. Abbreviation LV, lateral ventricle; MGE, medial ganglionic eminence; VZ, ventricular zone.
To determine whether the observed effect is cell autonomous, we carried out in utero electroporation of PlexinA1 and control GFP‐siRNA constructs into the MGE (n = 4 both groups). We quantified the percentage of GFP‐positive cells (186 control siRNA and 260 PlexinA1 siRNA cells) that were anchored to the ventricular surface. Most control GFP‐labeled cells (94.5% ± 0.27%) appeared to have pronounced end feet, anchored to the ventricular wall (white arrowheads, Fig. 6D), and long radial processes; in contrast, PlexinA1‐knockdown cells were infrequently attached to the ventricular surface (49.87% ± 0.68%, P < 0.003) and had shorter and thinner processes (black arrowheads, Fig. 6E). This finding is consistent with a previous study that demonstrated reduced proliferation and alteration of progenitor cell morphology in response to inflammation (Stolp et al., 2011).
Plexins were initially identified as cell adhesion molecules (Ohta et al., 1995), so we wondered whether loss of PlexinA1 function reduced attachment of neural progenitors to the extracellular matrix at the ventricular surface. We observed that fewer nestin‐positive cells from PlexinA1−/− mice attached to laminin/poly‐L‐lysine‐coated coverslips 30 minutes after seeding, compared with cells from control littermates (n = 3 both groups; Fig. 6G; PlexinA1+/+ 305.17 ± 21.78, PlexinA1−/− 245.78 ± 27.07; P < 0.007). These observations suggest that PlexinA1 promotes cell adhesion to maintain the number and correct arrangement of progenitor cells in the developing forebrain.
DISCUSSION
Cortical interneurons, generated predominantly in the MGE, migrate through the ventral and dorsal telencephalon before reaching their final positions within the CP (Métin et al., 2006; Faux et al., 2012). Previously we demonstrated that interneurons in Robo1 knockout (Robo1−/−) mice express reduced levels of Nrp1 and PlexinA1, rendering them less responsive to the chemorepulsive effects of Sema3A and Sema3F and causing their aberrant migration through the developing striatum en route to the cortex (Hernandez‐Miranda et al., 2011). Even though earlier studies had demonstrated the importance of Nrps in this process (Marin et al., 2001; Tamamaki et al., 2003a), a potential role for PlexinA1 in interneuron development had not previously been shown.
To complement and extend a previous study (Perälä et al., 2005) demonstrating PlexinA1 mRNA expression in the developing mouse forebrain, we used in situ hybridization to define its precise expression pattern. These experiments revealed the presence of PlexinA1 in progenitor cells lining the ventricular surface of the ganglionic eminences as early as E13.5 and in migrating interneurons expressing GAD67. Consistent with an important role for PlexinA1 in interneuron development, a significant decrease in the number of these cells in the cortex of PlexinA1−/− mice at E14.5 and E18.5 was observed. We first considered the possibility that this might be due to altered migration, but our migration assay failed to detect any significant differences in their migratory potential. We also considered whether changes in apoptosis or proliferation contribute to the decreased number of cortical interneurons in PlexinA1−/− mice. Previous studies have highlighted the importance of programmed cell death in shaping the cortex throughout development (Thomaidou et al., 1997; Haydar et al., 1999). However, we observed little cell death in the cortex of PlexinA1−/− mice, suggesting that increased apoptosis is unlikely the cause for the reduced interneuron number. We then considered the possibility of alterations in proliferation of forebrain progenitors of mice lacking the receptor. Plexins have been shown to play key roles in regulating cytoskeletal architecture and cell proliferation. PlexinA1 or PlexinA4 knockdown studies in endothelial cells resulted in prominent rearrangements of the actin cytoskeleton that were accompanied by inhibition of cell proliferation (Kigel et al., 2011). More recently, PlexinB2 has been shown to regulate the proliferation and migration of neuroblasts in the postnatal and adult SVZ (Saha et al., 2012). Consistent with such role for PlexinA1 in forebrain progenitor cells, reduced proliferation and progenitor cell numbers in the cortex, LGE, and MGE of PlexinA1−/− mice was observed, which would account for the reduction in the number of cortical pyramidal neurons and interneurons and of striatal projection cells in these mice.
Plexins and their coreceptors, Nrps, play important roles in vascular as well as neuronal development (Vieira et al., 2007; Gu and Giraudo, 2013). Thus, we wondered whether altered blood vessel formation within the developing forebrain of PlexinA1 mutant mice might contribute to the observed decrease in neurogenesis. However, staining for vascular markers did not show any obvious changes in the number or patterning of blood vessels in the developing forebrain in mutants compared with control littermates (data not shown). This observation indicated that vascular patterning is relatively normal in these mice and is unlikely to contribute to altered neurogenesis. However, our findings do not exclude the possibility of a more intimate interaction between vasculature and progenitors. This can be studied only with detailed analysis of the vasculature together with the progenitors, similar to that described in a study by Stubbs et al. (2009), who charted the movements and neurite extensions of live, individually labeled neuronal progenitors and/or newly born neurons in cortical slice cultures in relation to labeled vasculature.
PlexinA1 has been shown to have a function in different semaphorin signaling pathways. Thus, it acts as receptor for Sema6D and forms a complex with the vascular endothelial growth factor receptor 2 (VEGFR2) to convey signals for this semaphorin during cardiac morphogenesis (Toyofuku et al., 2004). Furthermore, a complex formed by Sema3A, Nrp1, and PlexinA1 signaling promotes lymphatic valve formation (Bouvrée et al., 2012). In the nervous system, perturbations in this signaling pathway result in defects in the projection of statoacoustic ganglion neurons (Katayama et al., 2013). We, therefore, examined the expression of semaphorins and VEGF receptors within the developing forebrain using data available in the GenePaint database. Sema6D and VEGFR2 both appeared to be expressed in the vasculature, and not in the proliferative zones (Fig. 7A,B), suggesting that they are unlikely to play a role in the present PlexinA1‐mediated events. Also, Sema3A was expressed very strongly throughout the cortex, in the VZ of the LGE, and along the routes of migrating interneurons (Fig. 7C,D). In contrast, it was only weakly expressed in the VZ of the MGE. To examine whether a defective Sema3A signaling pathway might be involved in proliferation, we carried out a Sema3A‐AP binding assay, which failed to show any differences between wild‐type and PlexinA1−/− mice (Fig. 7E,F). Thus, although we cannot definitively rule out involvement of these two semaphorin signaling pathways, this finding indicates that the effect on proliferation from loss of PlexinA1 function is likely to occur via another mechanism(s).
Figure 7.
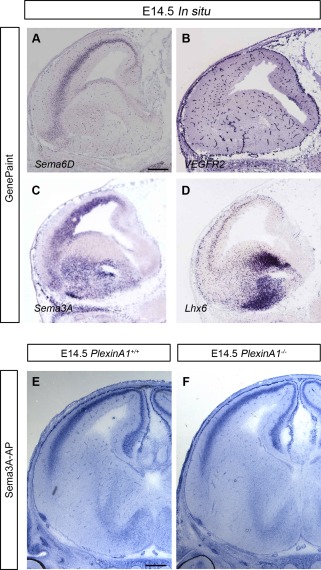
Expression patterns of semaphorins and VEGFR2 in wild‐type mouse brain. A–D: In situ hybridization of parasagittal sections at E14.5 for Sema6D (A) VEGFR2 (B), Sema3A (C), and the interneuron marker Lhx6 (D). Images were downloaded from the GenePaint server. E,F: Sema3A‐AP was added to coronal brain sections from PlexinA1+/+ (E) and PlexinA1−/− (F) mice at E14.5. Similar levels of Sema3A‐AP binding were observed in both sets of animals. Scale bars = 300 µm in A (applies to A–D); 200 µm in E (applies to E,F).
Slit–Robo and semaphorin–plexin signaling pathways participate in various developmental and pathogenic processes. We have previously described a cross‐talk between the two signaling pathways, with Robo1 playing a role in semaphorin signaling within the ventral forebrain, in part by regulating the expression of Nrp and plexin receptors in interneurons in Robo1−/− mice (Hernandez‐Miranda et al., 2011). PlexinA1 has also recently been shown to modulate Slit signaling (Delloye‐Bourgeois et al., 2015). We and others have shown that Slit–Robo signaling plays a role in neurogenic events within the developing forebrain (Borrell et al., 2012; Yeh et al., 2014). These findings raise the possibility that PlexinA1 might also modulate Slit signaling during corticogenesis. However, we think this is unlikely because Slit and PlexinA1 mutants affect neurogenesis differently, with an increased number of proliferating progenitors in the forebrain of mice lacking both Slit1 and Slit2 or Robo1 (Yeh et al., 2014) and fewer in PlexinA1−/− mice.
Complex interactions between neural stem/progenitor cells and extracellular matrix (ECM) proteins regulate their proliferation and differentiation, and many of these interactions involve transmembrane integrin receptors (Wojcik‐Stanaszek et al., 2011). The PlexinA1 coreceptor Nrp1 was also originally identified as a surface protein mediating cellular adhesion (Shimizu et al., 2000). Nrp1 was found to interact with β1 integrin in pancreatic carcinoma cells and to be important for integrin‐mediated anchorage‐independent growth and adhesion (Fukasawa et al., 2007). Knockdown of Nrp1 has also been shown to inhibit endothelial cell adhesion to fibronectin by altering the functional activity of α5β1 integrin (Valdembri et al., 2009). Plexins were also initially identified as cell surface molecules, and their exogenous expression on the surface of L cells promotes cell adhesion via a homophilic binding mechanism (Ohta et al., 1995; Fujisawa et al., 1997). Subsequent studies have shown that Sema4D activation of PlexinB1 and PlexinA1 hinders cell attachment to adhesive substrates, resulting in cell collapse and inhibition of cell migration (Barberis et al., 2004).
Dissociated neuronal progenitor cells attach to tissue culture dishes or glass coverslips only when coated with a permissive substrate such as laminin. Thus, reduced adhesion of PlexinA1 knockdown cells would suggest that loss of receptor function perturbs attachment to laminin. Plexins associate with integrins (Basile et al., 2007; Choi et al., 2014), which are known laminin receptors (Tomaselli et al., 1988). Thus, it is possible that PlexinA1 could be acting as an integrin coreceptor required for laminin binding and attachment to the ECM. Interestingly, ephrin B1 signaling has been shown to enhance ECM adhesion of neuroepithelial cells by promoting apical localization of integrin β1 (Arvanitis et al., 2013). By analogy, loss of PlexinA1 function might impair the apical distribution of integrins in progenitor cells and, thereby, affect their ability to attach properly to the ECM and consequently impair their capacity to divide. Further studies will be required to assess the possibility that PlexinA1 regulates integrin function in cortical neural progenitors.
In summary, we report here that progenitor cells lining the telencephalic ventricles express PlexinA1 and that absence of this receptor impairs neurogenesis in the developing forebrain, likely because of impaired ECM attachment of neural progenitors to the ventricular surface. Ultimately, this defect hinders the formation of striatal projection neurons and cortical pyramidal cells and interneurons during corticogenesis.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ROLE OF AUTHORS
Designed the research: WDA, JGP. Performed the research: WDA, KD, NT. Analyzed data: WDA. Wrote the manuscript: WDA, CR, JGP
ACKNOWLEDGMENTS
We thank the following for supplying transgenic mouse lines: Drs Yuchio Yanagawa and Kunihiko Obata (GAD67‐GFP), and Dr Yutaka Yoshida (PlexinA1).
REFERENCES
- Alifragis P, Liapi A, Parnavelas JG. 2004. Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their GABA phenotype. J Neurosci 24:5643–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews W, Barber M, Hernadez‐Miranda LR, Xian J, Rakic S, Sundaresan V, Rabbitts TH, Pannell R, Rabbitts P, Thompson H, Erskine L, Murakami F, Parnavelas JG. 2008. The role of Slit–Robo signaling in the generation, migration and morphological differentiation of cortical interneurons. Dev Biol 313:648–658. [DOI] [PubMed] [Google Scholar]
- Andrews WD, Zito A, Memi F, Jones G, Tamamaki N, Parnavelas JG. 2013. Limk2 mediates semaphorin signaling in cortical interneurons migrating through the subpallium. Biology Open 2:277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arellano JI, Guadiana SM, Breunig JJ, Rakic P, Sarkisian MR. 2012. Development and distribution of neuronal cilia in mouse neocortex. J Comp Neurol 520:848–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. 2005. Neuronal subtype‐specific genes that control corticospinal motor neuron development in vivo. Neuron 45:207–221. [DOI] [PubMed] [Google Scholar]
- Arvanitis DN, Béhar A, Tryoen‐Tóth P, Bush JO, Jungas T, Vitale N, Davy A. 2013. Ephrin B1 maintains apical adhesion of neural progenitors. Development 140:2082–2092. [DOI] [PubMed] [Google Scholar]
- Barberis D, Artigiani S, Casazza A, Corso S, Giordano S, Love CA, Jones EY, Comoglio PM, Tamagnone L. 2004. Plexin signaling hampers integrin‐based adhesion, leading to Rho‐kinase independent cell rounding, and inhibiting lamellipodia extension and cell motility. FASEB J 18:592–594. [DOI] [PubMed] [Google Scholar]
- Basile JR, Gavard J, Gutkind JS. 2007. Plexin‐B1 utilizes RhoA and Rho kinase to promote the integrin‐dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem 282:34888–34895. [DOI] [PubMed] [Google Scholar]
- Borrell V, Cárdenas A, Ciceri G, Galcerán J, Flames N, Pla R, Nóbrega‐Pereira S, García‐Frigola C, Peregrín S, Zhao Z, Ma L, Tessier‐Lavigne M, Marín O. 2012. Slit/Robo signaling modulates the proliferation of central nervous system progenitors. Neuron 76:338–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvrée K, Brunet I, Del Toro R, Gordon E, Prahst C, Cristofaro B, Mathivet T, Xu Y, Soueid J, Fortuna V, Miura N, Aigrot MS, Maden CH, Ruhrberg C, Thomas JL, Eichmann A. 2012. Semaphorin3A, Neuropilin‐1, and PlexinA1 are required for lymphatic valve formation. Circ Res 111:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P, Reep RL, Stoll ML, Ophir AG, Phelps SM. 2009. Conservation and diversity of Foxp2 expression in muroid rodents: functional implications. J Comp Neurol 512:84–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariboni A, Hickok J, Rakic S, Andrews W, Maggi R, Tischkau S, Parnavelas JG. 2007. Neuropilins and their ligands are important in the migration of gonadotropin‐releasing hormone neurons. J Neurosci 27:2387–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh JF, Mione MC, Pappas IS, Parnavelas JG. 1997. Basic fibroblast growth factor prolongs the proliferation of rat cortical progenitor cells in vitro without altering their cell cycle parameters. Cereb Cortex 7:293–302. [DOI] [PubMed] [Google Scholar]
- Choi YI, Duke‐Cohan JS, Chen W, Liu B, Rossy J, Tabarin T, Ju L, Gui J, Gaus K, Zhu C, Reinherz EL. 2014. Dynamic control of β1 integrin adhesion by the plexinD1–sema3E axis. Proc Natl Acad Sci U S A 111:379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubelos B, Sebastian‐Serrano A, Kim S, Moreno‐Ortiz C, Redondo JM, Walsh CA, Nieto M. 2008. Cux‐2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb Cortex 18:1758–1770. [DOI] [PubMed] [Google Scholar]
- Delloye‐Bourgeois C, Jacquier A, Charoy C, Reynaud F, Nawabi H, Thoinet K, Kindbeiter K, Yoshida Y, Zagar Y, Kong Y, Jones YE, Falk J, Chédotal A, Castellani V. 2015. PlexinA1 is a new Slit receptor and mediates axon guidance function of Slit C‐terminal fragments. Nat Neurosci 18:36–45. [DOI] [PubMed] [Google Scholar]
- Faux C, Rakic S, Andrews W, Britto JM. 2012. Neurons on the move: migration and lamination of cortical interneurons. Neuro‐Signals 20:168–189. [DOI] [PubMed] [Google Scholar]
- Fujisawa H, Ohta K, Kameyama T, Murakami Y. 1997. Function of a cell adhesion molecule, plexin, in neuron network formation. Dev Neurosci 19:101–105. [DOI] [PubMed] [Google Scholar]
- Fukasawa M, Matsushita A, Korc M. 2007. Neuropilin‐1 interacts with integrin beta1 and modulates pancreatic cancer cell growth, survival and invasion. Cancer Biol Ther 6:1173–1180. [DOI] [PubMed] [Google Scholar]
- Gu C, Giraudo E. 2013. The role of semaphorins and their receptors in vascular development and cancer. Exp Cell Res 319:1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Flandin P, Yoshikawa K, Rubenstein JL, Alvarez‐Buylla A, Kriegstein AR. 2013. Non‐epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nat Neurosci 16:1576–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar TF, Kuan CY, Flavell RA, Rakic P. 1999. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex 9:621–626. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Miranda LR, Parnavelas JG, Chiara F. 2010. Molecules and mechanisms involved in the generation and migration of cortical interneurons. ASN Neuro 2:e00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Miranda LR, Cariboni A, Faux C, Ruhrberg C, Cho JH, Cloutier JF, Eickholt BJ, Parnavelas JG, Andrews WD. 2011. Robo1 regulates semaphorin signaling to guide the migration of cortical interneurons through the ventral forebrain. J Neurosci 31:6174–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama K, Imai F, Suto F, Yoshida Y. 2013. Deletion of Sema3a or plexinA1/plexinA3 causes defects in sensory afferent projections of statoacoustic ganglion neurons. PLoS One 8:e72512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigel B, Rabinowicz N, Varshavsky A, Kessler O, Neufeld G. 2011. Plexin‐A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 118:4285–4296. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. 1999. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci 19:7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. 2003. Cell migration in the forebrain. Annu Rev Neurosci 26:441–483. [DOI] [PubMed] [Google Scholar]
- Marin O, Yaron A, Bagri A, Tessier‐Lavigne M, Rubenstein JL. 2001. Sorting of striatal and cortical interneurons regulated by semaphorin–neuropilin interactions. Science 293:872–875. [DOI] [PubMed] [Google Scholar]
- Métin C, Baudoin JP, Rakic S, Parnavelas JG. 2006. Cell and molecular mechanisms involved in the migration of cortical interneurons. Euro J Neurosci 23:894–900. [DOI] [PubMed] [Google Scholar]
- Morris JR, Solomon E. 2004. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet 13:807–817. [DOI] [PubMed] [Google Scholar]
- Ohta K, Mizutani A, Kawakami A, Murakami Y, Kasuya Y, Takagi S, Tanaka H, Fujisawa H. 1995. Plexin: a novel neuronal cell surface molecule that mediates cell adhesion via a homophilic binding mechanism in the presence of calcium ions. Neuron 14:1189–1199. [DOI] [PubMed] [Google Scholar]
- Perälä NM, Immonen T, Sariola H. 2005. The expression of plexins during mouse embryogenesis. Gene Expr Patterns 5:355–362. [DOI] [PubMed] [Google Scholar]
- Piper M, Harris L, Barry G, Heng YH, Plachez C, Gronostajski RM, Richards LJ. 2011. Nuclear factor one X regulates the development of multiple cellular populations in the postnatal cerebellum. J Comp Neurol 519:3532–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD, Price DJ. 2007. Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol 302:50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakić S, Yanagawa Y, Obata K, Faux C, Parnavelas JG, Nikolić M. 2009. Cortical interneurons require p35/Cdk5 for their migration and laminar organization. Cereb Cortex 19:1857–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retaux S, Caboche J, Rogard M, Julien JF, Penit‐Soria J, Besson MJ. 1993. GABA interneurons in the rat medial frontal cortex: characterization by quantitative in situ hybridization of the glutamic acid decarboxylase (GAD67) mRNA. Brain Res 611:187–196. [DOI] [PubMed] [Google Scholar]
- Saha B, Ypsilanti AR, Boutin C, Cremer H, Chédotal A. 2012. Plexin‐B2 regulates the proliferation and migration of neuroblasts in the postnatal and adult subventricular zone. J Neurosci 32:16892–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Murakami Y, Suto F, Fujisawa H. 2000. Determination of cell adhesion sites of neuropilin‐1. J Cell Biol 148:1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J, Toresson H, Campbell K. 2003. Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J Neurosci 23:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman AA, Krsnik Z, Sun J, Rasin MR, State MW, Sestan N, Louvi A. 2009. Developmentally regulated and evolutionarily conserved expression of SLITRK1 in brain circuits implicated in Tourette syndrome. J Comp Neurol 513:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolp HB, Turnquist C, Dziegielewska KM, Saunders NR, Anthony DC, Molnár Z. 2011. Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain 134:3236–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs D, DeProto J, Nie K, Englund C, Mahmud I, Hevner R, Molnár Z. 2009. Neurovascular congruence during cerebral cortical development. Cereb Cortex 19 Suppl 1:i32–i41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez J, Dávila JC, Real MA, Guirado S, Medina L. 2006. Calcium‐binding proteins, neuronal nitric oxide synthase, and GABA help to distinguish different pallial areas in the developing and adult chicken. I. Hippocampal formation and hyperpallium. J Comp Neurol 497:751–771. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Liu FC, Hirokawa K, Takahashi H. 2003. Expression of Foxp2, a gene involved in speech and language, in the developing and adult striatum. J Neurosci Res 73:61–72. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Fujimori K, Nojyo Y, Kaneko T, Takauji R. 2003a. Evidence that Sema3A and Sema3F regulate the migration of GABAergic neurons in the developing neocortex. J Comp Neurol 455:238–248. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. 2003b. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67‐GFP knock‐in mouse. J Comp Neurol 467:60–79. [DOI] [PubMed] [Google Scholar]
- Thomaidou D, Mione MC, Cavanagh JF, Parnavelas JG. 1997. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci 17:1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokami H, Ago T, Sugimori H, Kuroda J, Awano H, Suzuki K, Kiyohara Y, Kamouchi M, Kitazono T. 2013. RANTES has a potential to play a neuroprotective role in an autocrine/paracrine manner after ischemic stroke. Brain Res 1517:122–132. [DOI] [PubMed] [Google Scholar]
- Tomaselli KJ, Damsky CH, Reichardt LF. 1988. Purification and characterization of mammalian integrins expressed by a rat neuronal cell line (PC12): evidence that they function as alpha/beta heterodimeric receptors for laminin and type IV collagen. J Cell Biol 107:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Zhang H, Kumanogoh A, Takegahara N, Suto F, Kamei J, Aoki K, Yabuki M, Hori M, Fujisawa H, Kikutani H. 2004. Dual roles of Sema6D in cardiac morphogenesis through region‐specific association of its receptor, Plexin‐A1, with off‐track and vascular endothelial growth factor receptor type 2. Genes Dev 18:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuoc TC, Stoykova A. 2008. Er81 is a downstream target of Pax6 in cortical progenitors. BMC Dev Biol 8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdembri D, Caswell PT, Anderson KI, Schwarz JP, König I, Astanina E, Caccavari F, Norman JC, Humphries MJ, Bussolino F, Serini G. 2009. Neuropilin‐1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol 7:e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira JM, Schwarz Q, Ruhrberg C. 2007. Selective requirements for NRP1 ligands during neurovascular patterning. Development 134:1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TW, Stromberg GP, Whitney JT, Brower NW, Klymkowsky MW, Parent JM. 2006. Sox3 expression identifies neural progenitors in persistent neonatal and adult mouse forebrain germinative zones. J Comp Neurol 497:88–100. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez‐Buylla A. 2001. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development 128:3759–3771. [DOI] [PubMed] [Google Scholar]
- Wojcik‐Stanaszek L, Gregor A, Zalewska T. 2011. Regulation of neurogenesis by extracellular matrix and integrins. Acta Neurobiol Exp 71:103–112. [DOI] [PubMed] [Google Scholar]
- Wu S, Esumi S, Watanabe K, Chen J, Nakamura KC, Nakamura K, Kometani K, Minato N, Yanagawa Y, Akashi K, Sakimura K, Kaneko T, Tamamaki N. 2011. Tangential migration and proliferation of intermediate progenitors of GABAergic neurons in the mouse telencephalon. Development 138:2499–2509. [DOI] [PubMed] [Google Scholar]
- Xu X, Roby KD, Callaway EM. 2006. Mouse cortical inhibitory neuron type that coexpresses somatostatin and calretinin. J Comp Neurol 499:144–160. [DOI] [PubMed] [Google Scholar]
- Yeh ML, Gonda Y, Mommersteeg MT, Barber M, Ypsilanti AR, Hanashima C, Parnavelas JG, Andrews WD. 2014. Robo1 modulates proliferation and neurogenesis in the developing neocortex. J Neurosci 34:5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Han B, Mendelsohn M, Jessell TM. 2006. PlexinA1 signaling directs the segregation of proprioceptive sensory axons in the developing spinal cord. Neuron 52:775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]