

Abstract
Structural variants (SVs) include all insertions, deletions, and rearrangements in the genome, with several common types of nucleotide repeats including single sequence repeats, short tandem repeats, and insertion‐deletion length variants. Polyallelic SVs provide highly informative markers for association studies with well‐phenotyped cohorts. SVs can influence gene regulation by affecting epigenetics, transcription, splicing, and/or translation.1 Accurate assays of polyallelic SV loci are required to define the range and allele frequency of variable length alleles.2
ACCURACY VS. PRECISION IN HUMAN GENETICS
Precision is hitting the same location on the target over and over again, while not necessarily hitting the bullseye. Accuracy is hitting the bullseye. This is a very relevant distinction in human genetics, since the publicly available human genome databases provide precise order and location of candidate genes to any investigator with a computer. Genome‐wide association studies (GWAS) provide low effect size association data for many gene regions, and do not distinguish many false‐positive signals. Without a secondary screen for biological confirmation, most reported GWAS‐associated disease genes have been implicated in disease more speculatively than proven. The paucity of specific disease‐based biology has contributed directly to the frequently stated conclusions that multiple small genetic hits across the genome may be relevant and additive for the pathogenesis of particular complex diseases.2 In contrast, the highly informative locus involving the linkage disequilibrium (LD) region containing both APOE and TOMM40 suggests a complex single locus for potential mechanisms of pathogenesis.
Polyallelic SVs in specific regions of association can also be used to associate phenotypic specificity of a disease. It is much easier to recruit well‐phenotyped cohorts of a sufficient size for association studies with a polyallelic marker, and without the necessary corrections based on a million single nucleotide polymorphism (SNP) tests. Most large GWAS studies collect cohorts of patients at multiple sites with little additional granular information available other than the name of the diagnosis. If well‐phenotyped cohorts are collected with standardized examinations and DNA banking, heterogeneity of specific phenotypes and effects of multiple participating ethnicities can be overcome.3, 4 The original association of both APOE and TOMM40 were based on accurate age of onset data. A distinct additional variable SV could also be associated with other specific phenotypic data, such as a variable pathology marker or physiology characteristic of the disease (see Lewy Bodies, below).
LATE‐ONSET ALZHEIMER'S DISEASE (LOAD)
In 1993 the APOE4 coding polymorphism was clearly associated with LOAD.5 The samples used for this original association were collected prospectively over more than a decade and all had standardized age of onset information. It was immediately possible to perform retrograde analyses with each APOE genotype (not simply the presence or absence of an APOE4 allele) and to demonstrate that each major genotype, including APOE3/3 and APOE2/3, had its own age of onset distribution (Figure 1). Within a very short time, many clinical centers confirmed the association of APOE4 with earlier onset of LOAD. APOE4 had a normal allele frequency of ∼0.15, representing only 29% of the population who carry one or two copies of APOE4. The larger proportion of the population, especially the 60% who carried the APOE3/3 genotype, became the “normal comparator group,” despite clear evidence that APOE3/3 had a clear age of onset distribution later than the APOE4/4 and APOE3/4 carriers, but earlier than APOE2/3 carriers.4
Figure 1.
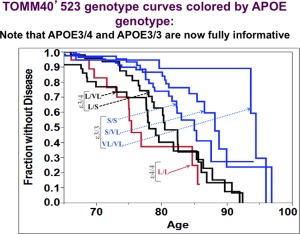
Age at onset of cognitive impairment as a function of TOMM40'523 genotype. The Bryan Alzheimer's Disease Research Center (ADRC) Memory, Health, and Aging cohort (n = 508, 106 conversion events) was followed prospectively at the Bryan ADRC at Duke University. Cognitive status was determined using standard neuropsychological tests. The age at which cognitive impairment occurred was retrospectively stratified by TOMM40'523 genotype, and Kaplan–Meier curves were constructed. TOMM40'523 genotype was determined using a sequencing‐based genotyping method (Polymorphic DNA Technologies). TOMM40'523 genotypes and the corresponding APOE genotypes are indicated in the figure. The red line corresponds to APOE ɛ4/4; the two green lines correspond to APOE ɛ3/4, and the three blue lines correspond to APOE ɛ3/3. Note that within this cohort there were 78 individuals who carried an APOE ɛ2 allele, but only five developed cognitive impairment during the study (VL/L (APOE ɛ2/4), n = 1; S/L (APOE ɛ2/4), n = 2; VL/VL (APOE ɛ3/3), n = 2). The numbers of subjects per TOMM40'523 genotype (number converted to case status) were: L/L, n = 23 (11); VL/L, n = 54 (24); S/L, n = 72 (23); S/S, n = 100 (20); S/ VL, n = 138 (22); VL/VL, n = 51 (6). Reprinted from ref. 4.
Mapping the LD area around APOE in 1998 clearly demonstrated that other genetic loci within the LD region also yielded increased association data.6 However, the discovery of a highly polymorphic single sequence repeat (SSR) located in intron 6 of TOMM40, the gene adjacent to APOE, increased the informed population for the age of distributions to >98%.3 Age of onset distributions for the TOMM40′523 alleles not only include all APOE4 carriers, but also demonstrate different distributions for two forms of APOE3/4 (TOMM40′523 S‐L and VL‐L). Distinct age of onset distributions are also now available for three previously uncharacterized APOE3/3 genotypes (S‐S, S‐VL, and VL‐VL) and contributed to mapping about 85% of the population. The APOE2/3 group added another 13%, with APOE2/4 and APOE2/2 too uncommon for onset of LOAD to construct a distribution curve.3
Like many genes in metabolic pathways, adjacent nearby genes are often interactive in the functional pathway. It was demonstrated that APOE(1‐272) metabolites of apoE3(1‐299) and apoE4(1‐299) bind to the outer surface of the mitochondria and alter mitochondrial dynamic mechanisms, including intracellular mobility, glucose‐utilization, oxygen consumption, and fission.7 Thus, while scientists were arguing whether APOE was a disease‐causing gene based on its precision in association studies, it is probably more accurate to examine the method of pathogenesis based on the effect of APOE‐TOMM40′523 cis‐haplotypes and their effect on energy metabolism and dynamic functions. An algorithm for estimating the 5‐year proximal risk for an individual in population‐based on APOE‐TOMM40 haplotype and age is now being validated in the TOMMORROW clinical trial and in several clinical centers. These studies can provide a clinical template for examination of the age of onset of other expressed biomarkers. In recent years, expressed biomarkers have been placed on theoretical age of onset templates for examining their expression preceding a diagnosis of AD.8 It is now possible to collect more accurate associations of expressed biomarkers based on TOMM40′523 and APOE cis‐haplotype templates.4, 5
LEWY BODIES (LB) AS A CONCOMITANT FEATURE OBSERVED IN LOAD NEUROPATHOLOGY
Over the course of the past 30 years, LBs have been detected commonly in autopsy brains from LOAD as well as Parkinson's disease patients; LBs contain several aggregated proteins but a large proportion of synuclein (SNCA gene on chromosome 4). There is a form of dementia called Lewy Body dementia (LBD), where there are dense depositions of LBs in the absence of AD plaque and tangle pathology. Investigators have questioned over the years whether the increased presence of LBs in patients with LOAD pathology is an additional causative factor, or provides a distinct disease phenotype. The presence of LBs in AD brain autopsies was observed in all major TOMM40′523 genotypes.
A search for causal variants for variable LB pathology started by looking for SVs in the large SNCA gene. Using a database of SVs (dbSV), a CT‐rich region of ∼600 bases within intron 4 of the SNCA gene was rich in SVs.9 This region also had histone modification signals, suggesting a regulatory role. The cloning and sequencing of this region found nine different polymorphisms, with potentially 512 haplotype associations. However, in studying 188 chromosomes, only four haplotype combinations were observed and designated as haplotypes 1–4 (Figure 2). The LB brain pathology was dense when haplotype #3 was inherited on both chromosomes, and less dense or absent for other haplotype combinations—independent of APOE‐TOMM40′523 haplotypes. Further studies to characterize the haplotype combinations in LBD, without AD plaques and tangles, are currently in progress. The cluster of four adjacent SVs is associated with haplotype combinations of SNCA and differs from the result of TOMM40′523 haplotypes in LOAD. Variants of both gene haplotypes are carried in the population but specificity to LOAD and LB pathology can be assigned accurately to distinct SVs.9
Figure 2.

Observed haplotypes of the CT‐rich region. The portion of the sequences shown corresponds to chr4:90,742,421‐90,742,492 (GRCh37/hg19) on the minus strand. Identical sequences shared among the haplotypes are color coded. Reprinted from ref. 10.
CONCLUSION
The devil is in the details when comparing disease association studies of highly polyallelic SV markers with the results of GWAS methods. There are more than 7 million SVs in the human genome that can also be used to map much of the understudied noncoding regions, including introns, regulatory sites, and untranslated regions (UTRs). The cis‐linkage of variants within a region of linkage disequilibrium can provide the opportunity to return to the discovery mode of historic linkage studies of the 1990s, except that the size of the identified LD regions are markedly smaller than the large regions of chromosome mapping data during the early 1990s. Polyallelic SVs can provide more highly variant, focused allelic markers for disease association studies, and can make use of much smaller‐sized cohorts than GWAS. In fact, with highly phenotyped cohorts, secondary association testing can be based on age, rate of change, and symptoms and signs that define the clinical characteristics and possible heterogeneity of the disease classification. Polyallelic SV length frequencies can be extremely variable between different ethnic groups.10 Current next‐generation sequencing can detect the presence and location of SVs but the variations of allele size of most polyallelic SVs are not yet well characterized at each locus. Therefore, while the exact localization of SVs can be recognized and mapped, the accurate variability at polyallelic SV loci is currently understudied in human populations, and requires focused experimentation to accurately define variable length alleles and their relative allele frequencies for each SV. Polyallelic SVs can serve as highly informative and accurate tags related to specific characteristics of disease expression and will become increasingly important in dissecting the genetics and the mechanisms of action in common complex diseases, as well as uncommon and rare diseases.
CONFLICT OF INTEREST
The author declares no conflicts of interest.
References
- 1. Frazer, K.A. , Murray, S.S. , Schork, N.J. & Topol, E.J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251 (2009). [DOI] [PubMed] [Google Scholar]
- 2. Naj, A.C. et al Age‐at‐onset in late onset Alzheimer disease is modified by multiple genetic loci. JAMA Neurol. 71, 1394–1404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crenshaw, D.G. et al Using genetics to enable studies on the prevention of Alzheimer's disease. Clin. Pharmacol. Ther. 93, 177–185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lutz M.W. et al., editors. TOMM40/APOE variation and age of onset of mild cognitive impairment and dementia in a prospective longitudinal study. Alzheimer's Association International Conference (AAIC); 2015; July 18–23, 2015, Washington, DC.
- 5. Corder, E. et al Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 6. Martin, E.R. et al SNPing away at complex diseases: analysis of single‐nucleotide polymorphisms around APOE in Alzheimer disease. Am. J. Hum. Genet. 67, 383–394 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang, S. et al Lipid‐ and receptor‐binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 102, 18694–18699 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jack, C.R., Jr. & Holtzman, D.M. Biomarker modeling of Alzheimer's disease. Neuron. 80, 1347–1358 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lutz, M.W. et al A CT‐rich haplotype in intron 4 of SNCA confers risk for Lewy body pathology in Alzheimer's disease and affects SNCA expression. Alzheimers Dement. 11, 1133–1143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roses, A.D. et al African‐American TOMM40'523‐APOE haplotypes are admixture of West African and Caucasian alleles. Alzheimers Dement. 10, 592–601 e2 (2014). [DOI] [PubMed] [Google Scholar]