

Abstract
Chronic hepatitis C virus (HCV) infection may progress to cirrhosis and hepatocellular carcinoma (HCC). Recently, two genetic variants, DEPDC5 rs1012068 and MICA rs2596542, were associated with the onset of HCC in Asian subjects with chronic HCV infection. The aim of the present study was to analyze whether DEPDC5 and MICA genetic variants were associated with liver disease progression in European subjects with chronic HCV infection. In a Northern Italian discovery cohort (n = 477), neither DEPDC5 rs1012068 nor MICA rs2596542 were associated with HCC (n = 150). However, DEPDC5 rs1012068 was independently associated with cirrhosis (n = 300; P = 0.049). The association of rs1012068 with moderate to severe fibrosis was confirmed in an independent cross‐sectional German cohort (n = 415; P = 0.006). Furthermore, DEPDC5 rs1012068 predicted faster fibrosis progression in a prospective cohort (n = 247; P = 0.027). Next, we examined the distribution of nonsynonymous DEPDC5 variants in the overall cross‐sectional cohort (n = 912). The presence of at least one variant increased the risk of moderate/severe fibrosis by 54% (P = 0.040). To understand the molecular mechanism underlying the genetic association of DEPDC5 variants with fibrosis progression, we performed in vitro studies on immortalized hepatic stellate cells (LX‐2). In these cells, down‐regulation of DEPDC5 resulted in increased expression of β‐catenin and production of its target matrix metallopeptidase 2 (MMP2), a secreted enzyme involved in fibrosis progression. Conclusion: DEPDC5 variants increase fibrosis progression in European subjects with chronic HCV infection. Our findings suggest that DEPDC5 down‐regulation may contribute to HCV‐related fibrosis by increasing MMP2 synthesis through the β‐catenin pathway. (Hepatology 2016;63:418–427)
Abbreviations
- CI
confidence interval
- DEPDC5
DEP domain‐containing 5
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- MICA
MHC class I polypeptide‐related sequence A
- MMP2
matrix metallopeptidase 2
- OR
odds ratio
- SMA
smooth muscle actin
- TIMP1
tissue inhibitor of metalloproteinase 1
- TIMP2
tissue inhibitor of metalloproteinase 2
Chronic hepatitis C virus (HCV) infection is an important health issue affecting approximately 3% of the population worldwide.1 Although many subjects show only mild or no symptoms in the early stages, chronic HCV infection frequently leads to cirrhosis and its complications, including hepatocellular carcinoma (HCC).2, 3, 4 The progression to cirrhosis and HCC is influenced by both environmental (e.g., age, coinfection, alcohol)5, 6, 7 and genetic factors.8, 9, 10, 11, 12, 13
To date, two studies have examined the susceptibility to HCC in subjects with chronic HCV infection at a genome‐wide level.14, 15 Those two genome‐wide association studies found that noncoding genetic variants in the DEP domain‐containing 5 (DEPDC5)14 and in the MHC class I polypeptide‐related sequence A (MICA)15 loci confer susceptibility to HCC in Asian subjects with chronic HCV infection. However, ethnicity influences the effect of genetic variants.16 Allele frequencies and haplotype patterns vary considerably, and the effect size of polymorphisms changes across different populations.17 Thus, it is important to study whether the effect of genetic variants may be applied to other populations.
The aim of the study was to examine the effect of DEPDC5 rs1012068 (T>G), and MICA rs2596542 (C>T) variants on HCC onset in Europeans with chronic HCV infection.
Subjects and Methods
Study Design
We tested our hypotheses in a discovery cohort (n = 477) and, subsequently, in independent validation cohorts, which comprised a cross‐sectional cohort (n = 415) and a prospective cohort (n = 247). Both the discovery and the validation cohorts consisted of therapy‐naïve subjects with chronic HCV infection and no HBV or HIV coinfection. Only the significant associations were carried on in the validation cohorts. Figure 1 illustrates the study design and the flow of the analyses. To analyze the association between genetic variants and HCC, we included only patients with cirrhosis, because cirrhosis is the main risk factor for HCC.18 To test the association with fibrosis and maximize the power of our analysis, we compared the genotype distribution of the DEPDC5 rs1012068 and MICA rs2596542 variants in the two extremes of fibrosis distribution in the discovery cohort: subjects with no/mild fibrosis (stage F0‐F1) versus those with cirrhosis (i.e., the most severe stage, F4). Next, we validated the association found with cirrhosis in the cross‐sectional validation cohort and extended it to the full spectrum of fibrosis. Finally, to confirm our findings in a prospective way, we analyzed the association between DEPDC5 rs1012068 and fibrosis progression rate (FPR) in the prospective validation cohort. The analysis of nonsynonymous variants was performed by pooling together the discovery and the cross‐sectional validation cohorts.
Figure 1.

Study design and flow of the analyses. This picture illustrates the study design and flow of analyses. Both the discovery and the validation cohorts consist of therapy‐naïve subjects with chronic HCV infection. The discovery cohort consisted of a cross‐sectional group of 477 subjects enrolled in Milan (Italy). In the analysis on HCC, only patients with cirrhosis were included. To test the association with fibrosis, we first analyzed the extremes of fibrosis distribution (stage F0‐F1 versus F4) to maximize the power by selecting two extremely different groups. Next, we examined the association with the full spectrum of fibrosis in the cross‐sectional validation cohort (the Leipzig and Bern cohorts pooled together [n = 415]) and with fibrosis progression in the prospective cohort (Milan prospective cohort [n = 247]). Only the significant associations were carried on in the validation cohorts. The analysis of nonsynonymous variants was performed by pooling together the discovery and the cross‐sectional validation cohorts.
Discovery Cohort
The discovery cohort consisted of 477 subjects with chronic HCV infection enrolled at Fondazione IRCCS Ca' Granda Milan, Italy. Subjects were selected according to the criteria defined above in the study design from a larger cohort that has been described previously.19 The degree of liver fibrosis was assessed histologically according to the METAVIR20 classification. We performed two different analyses by using two different subsets of this cohort. First, to test the association between DEPDC5 rs1012068, MICA rs2596542, and HCC, we analyzed only those subjects with cirrhosis (n = 150 with and n = 150 without HCC). Next, we examined subjects with no/mild fibrosis and those with cirrhosis. No/mild fibrosis was defined as stage F0‐F1 and cirrhosis as stage F4. All subjects gave informed consent to participate in the study, which was approved by the local ethics committee. The demographic and clinical characteristics of the discovery cohort are given in Table 1.
Table 1.
Demographic and Clinical Characteristics of the Cohorts
| Discovery Cohort | Validation Cohort | |||
|---|---|---|---|---|
| Milan | Leipzig | Bern | Milan Prospective | |
| N | 477 | 268 | 147 | 247 |
| Age, years | 63 ± 12 | 52 ± 12 | 48 ± 11 | 47 ± 11 |
| Sex | ||||
| Men | 317 (67) | 130 (49) | 93 (63) | 129 (52) |
| Women | 160 (33) | 138 (51) | 54 (37) | 118 (48) |
| Alcohol consumption | ||||
| None/milda | 433 (91) | 108 (77) | 110 (75) | 247 (100) |
| At‐riskb | 44 (9) | 32 (23)c | 37 (25) | 0 (0) |
| Liver fibrosis stage | ||||
| F0‐F1 | 177 (37) | 105 (39) | 68 (46) | 157 (64) |
| F2‐F4 | 300 (63)d | 163 (61) | 79 (54) | 90 (36) |
| Cirrhosis | 300 (63) | 72 (27) | 37 (25) | — |
| HCC | 150 (31) | 50 (19) | 8 (5) | 0 (0) |
Data are presented as the mean ± standard deviation or as n (%).
Defined as 0‐20 g/day for women and 30 g/day for men.
Defined as >20 g/day for women and >30 g/day for men.
Data on alcohol intake were not available in 128 subjects (48%).
All subjects in this cohort had stage F4 fibrosis.
Validation Cohorts
The validation cohorts consisted of subjects with chronic HCV infection and included a cross‐sectional cohort (n = 415) and a prospective cohort (n = 247). The cross‐sectional cohort was created by pooling together a cohort from Bern, Switzerland, and a cohort from Leipzig, Germany, the latter of which has been described previously.21 The current report includes 268 subjects with chronic HCV infection and the full spectrum of liver disease. The Bern cohort included 147 therapy‐naïve patients with chronic HCV from Switzerland. The stage of liver fibrosis was histologically assessed according to the METAVIR20 classification. The prospective cohort comprised 247 subjects that were enrolled at the University of Milan in Milan, Italy. The characteristics of this cohort have been described in detail previously.22, 23 The FPR was calculated as the ratio between the Ishak score24 and the disease duration (years) and was considered a continuous variable. The mean disease duration was 25 ± 10 years; log10 transformation of FPR was used to obtain linearity. Ethical approval to include patients for validation was obtained from all participating institutions, and all patients signed informed consent to be included in a genetic analysis. The demographic and clinical characteristics of each discovery cohort are given in Table 1.
Selection of DEPDC5 Nonsynonymous Variants
By using the Ensembl genome browser,25 we selected all the DEPDC5 nonsynonymous variants with a minor allele frequency ≥1% in Europeans according to 1000 Genomes Project data. We specifically focused on the nonsynonymous variants because they result in an amino acidic substitution and likely affect the protein function. Linkage disequilibrium estimations for the DEPDC5 variants were examined from the Ensembl genome browser data.25 Detailed information on these variants is reported in Supporting Table 1.
Genotyping of Genetic Variants
The fluorogenic 5′‐nucleotidase allelic discrimination assay (TaqMan Applied Biosystems, Foster City, CA) was used to genotype all the DEPDC5 variants and MICA rs2596542 in the discovery and the cross‐sectional cohorts according to the manufacturer's instructions, with a ≥95% success rate. In the prospective validation cohort, DEPDC5 rs1012068 genotyping was performed by Human660W‐Quad BeadChip (Illumina, San Diego, CA), followed by single‐nucleotide polymorphism calling using the default settings of Genome Studio software. All genotype distributions were in Hardy‐Weinberg equilibrium.
Gene Expression
DEPDC5 messenger RNA (mRNA) expression was assessed in seven immortalized human cell lines. To assess mRNA expression in cultured cells, total RNA was isolated from each cell type using an RNeasy Mini Kit (Qiagen, Valencia, CA). First‐strand complementary DNAs were synthesized from 1 μg RNA using a reverse‐transcription kit (Applied Biosystems). DEPDC5 and β‐catenin mRNA expression were assessed in LX‐2 control cells and in LX‐2 DEPDC5 knockout cells. TaqMan probe and master mix (Life Technologies, Carlsbad, CA, USA) were used in a total volume of 20 μL per reaction. Real‐time polymerase chain reaction (PCR) assay was performed on a CFX Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA). DEPDC5 expression levels were assessed using the delta‐delta Ct method and normalization to β‐actin.
DEPDC5 Knockdown
LX‐2 cells were transfected with DEPDC5 small interfering RNA (siRNA) or negative control (scramble) siRNA (Ambion‐Life Technologies by Thermo Fisher Scientific, Rockford, IL) with TurboFect Transfection Reagent according to the manufacturer's instructions. The cells were transfected with 25 nM siRNA oligos for 48 hours in serum‐free medium. RNA was isolated from each cell type using an RNeasy Mini Kit (Qiagen). First‐strand complementary DNAs were synthesized from 1 μg RNA using a reverse‐transcription kit (Applied Biosystems) to confirm DEPDC5 knockdown.
Cell Proliferation
LX‐2 cells were seeded in six‐well plates. When 50% confluent, cells were transfected with DEPDC5 siRNA or scramble siRNA. Cell numbers were counted 24, 48, and 72 hours after transfection using the Nucleocounter NC‐100 (Chemometec A/S, Lillerød, Denmark).
Immunoblot Analysis
LX‐2 cells were seeded in T‐75 plates. When 50% confluent, cells were transfected with DEPDC5 siRNA or scramble siRNA. Cells were collected 48 hours after transfection and were lysed in M‐PER Mammalian Protein Extraction Reagent (Pierce, Thermo Fisher Scientific) containing complete protease inhibitor cocktail (Sigma‐Aldrich, St. Louis, MO). Immunoblot analysis was performed according to standard procedures. Bands were visualized using a Chemidoc XRS System and Image Lab Software (Bio‐Rad).
The following antibodies were used: rabbit anti‐collagen I (Sigma‐Aldrich), rabbit anti‐matrix metallopeptidase 2 (MMP2) (Sigma‐Aldrich), mouse anti‐α‐smooth muscle actin (SMA) (Sigma‐Aldrich), rabbit anti‐tissue inhibitor of metalloproteinase 1 (TIMP1) (Abcam, Cambridge, UK), mouse anti‐tissue inhibitor of metalloproteinase 2 (TIMP2) (Sigma‐Aldrich), and rabbit anti‐Calnexin (Abcam).
Statistical Analyses
Data are shown as the mean and standard deviation or number and proportion. The distribution of DEPDC5 rs1012068 and MICA rs2596542 genotypes between the different groups was compared using Fisher's exact test. In the analysis of nonsynonymous variants, we compared the proportion of subjects with at least one nonsynonymous allele between subjects with no/mild fibrosis and those with moderate/severe fibrosis by Fisher's exact test. Binary logistic regression was used to evaluate the effect of the DEPDC5 rs1012068 on severe fibrosis including other risk factors in the model. The association between DEPDC5 rs1012068 and FPR was performed by generalized linear model adjusted by age at liver biopsy and sex. Analyses were performed using IBM SPSS Statistics, Version 20.0 (IBM, Armonk, NY). DEPDC5 and β‐catenin expressions between LX‐2 control cells and LX‐2 DEPDC5 knockout cells were compared using an independent samples Mann‐Whitney U test.
Results
DEPDC5 rs1012068 and MICA rs2596542 Are Not Associated With HCC in Europeans With Chronic HCV Infection
To examine the association of DEPDC5 rs1012068 and MICA rs2596452 with HCC in Europeans, we tested whether the genotype distribution of these variants was different between patients with cirrhosis who did or did not also have HCC from the discovery cohort (n = 300). We specifically analyzed only patients with cirrhosis because cirrhosis is the main risk factor for HCC. We observed no difference in the DEPDC5 rs1012068 and MICA rs2596542 distribution between subjects with HCC and those with cirrhosis (Supporting Table 2). To increase the power of the analysis, we pooled together the discovery and the cross‐sectional validation cohorts (n = 403). No difference in DEPDC5 rs1012068 and MICA rs2596542 distribution was observed between subjects with HCC and those with cirrhosis in the pooled cohort (Supporting Table 2).
DEPDC5 rs1012068, But Not MICA rs2596542, Is Associated With Cirrhosis
Based on the lack of association between DEPDC5 rs1012068 and MICA rs2596542 with HCC, we hypothesized that these two genetic variants might be associated with fibrosis rather than HCC. To test this hypothesis and increase the power to detect the association, we analyzed the association between DEPDC5 rs1012068 and MICA rs2596542 and fibrosis by analyzing the extreme stages of fibrosis in the discovery cohort. Particularly, we compared the genotype distribution of these genetic variants between subjects with no/mild fibrosis and those with cirrhosis. Thus, based on the fibrosis grade defined histologically, we stratified the discovery cohort in subjects with no/mild fibrosis (n = 177; stage F0‐F1) and those with cirrhosis (n = 300; stage F4). The prevalence of DEPDC5 rs1012068 was higher in subjects with cirrhosis than in those with no/mild fibrosis (P = 0.049; Fig. 2A and Supporting Table 3). To confirm the association between DEPDC5 rs1012068 and cirrhosis, we analyzed the cross‐sectional validation cohort. We found that the prevalence of DEPDC5 rs1012068 was higher in subjects with cirrhosis (F4) than in those with no/mild (F0‐F1) fibrosis (P = 0.006; Fig. 2B and Supporting Table 3). The DEPDC5 rs1012068 G allele was associated with a 40% increased risk of cirrhosis (odds ratio [OR], 1.40; 95% confidence interval [CI], 1.08‐1.81; P = 0.011; Supplementary Table 4) after adjusting for age, sex, and recruitment center in the discovery and validation cohorts.
Figure 2.
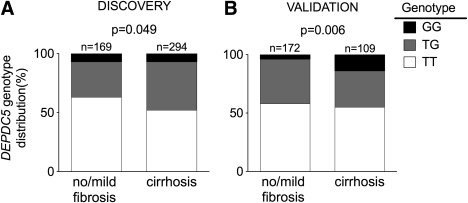
DEPDC5 rs1012068 is associated with cirrhosis in European subjects with chronic HCV infection. (A) Genotype distribution of DEPDC5 rs1012068 in the discovery cohort. (B) Genotype distribution of DEPDC5 rs1012068 in the cross‐sectional validation cohort. The validation cohort consisted of subjects from the Leipzig and Bern cohorts pooled together. Carriers of the G allele also had a higher risk of cirrhosis (OR, 1.46; 95% CI, 1.04‐2.04; P = 0.027 after pooling together the discovery and the validation cohorts and adjusting for age, sex, and recruitment center). The exact frequencies are shown in Supporting Table 3.
On the contrary, the distribution of MICA rs2596542 genotypes was not significantly different according to the presence or absence of cirrhosis (P = 0.075; Supporting Table 5).
The DEPDC5 rs1012068 Genetic Variant Is Associated With Fibrosis Severity in the Validation Cohorts
To confirm our hypothesis that DEPDC5 rs1012068 variant is associated not only with cirrhosis but, in general, with a more severe fibrosis, we examined the cross‐sectional validation cohort including subjects with the entire spectrum of fibrosis. To confirm the association with fibrosis progression, we next analyzed the prospective validation cohort where the fibrosis progression rate has been assessed.
First, we examined the cross‐sectional cohort and stratified it in subjects with no/mild fibrosis (n = 173; stage F0‐F1) and those with moderate/severe fibrosis (n = 242; stage F2‐F4). Consistent with the findings on cirrhosis, the prevalence of DEPDC5 rs1012068 was higher in subjects with moderate/severe fibrosis than in those with no/mild fibrosis (P = 0.006; Fig. 3A and Supporting Table 3). DEPDC5 rs1012068 variant was an independent risk factor for moderate/severe fibrosis (OR, 1.52; 95% CI, 1.09‐2.12; P = 0.015 per each G allele; Supporting Table 6) after adjusting for age, sex, and recruitment center. Results were virtually identical after adjusting for diabetes and at‐risk alcohol intake (Supporting Table 6).
Figure 3.
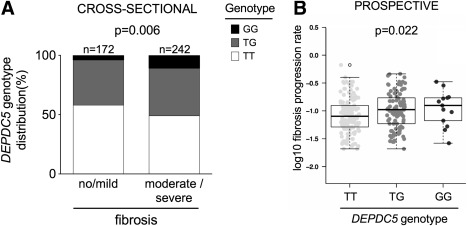
DEPDC5 rs1012068 is associated with fibrosis progression in Europeans with chronic HCV infection. The genotype distribution of DEPDC5 rs1012068 is shown in the cross‐sectional (A) and in the prospective (B) validation cohorts. The cross‐sectional validation cohort consisted of subjects from Leipzig and Bern cohorts pooled together. Carriers of the G allele also had a higher risk of moderate/severe fibrosis (OR, 1.46; 95% CI, 1.04‐2.04; P = 0.027 after adjusting for age, sex, and recruitment center). The association between DEPDC5 rs1012068 and FPR was calculated by multivariate generalized linear models under an additive genetic model (see Table 2 for further details).
Next, we tested whether DEPDC5 rs1012068 was associated with higher FPR in the prospective cohort. Carriers of this variant had a greater FPR (adjusted P = 0.027; Fig. 3B and Table 2), independently of age at liver biopsy and sex.
Table 2.
DEPDC5 rs1012068 Is Associated With Higher Fibrosis Progression Rate in European Subjects With Chronic HCV Infection
| Estimatea | Standard Error | P | Estimateb | Standard Error | P | |
|---|---|---|---|---|---|---|
| DEPDC5, G alleles | 0.078 | 0.034 | 0.022 | 0.074 | 0.033 | 0.027 |
| Age at biopsy, years | 0.001 | 0.001 | 0.54 | 0.002 | 0.002 | 0.31 |
| Sex, men | 0.045 | 0.020 | 0.026 | 0.047 | 0.020 | 0.020 |
Unadjusted.
Adjusted for age at biopsy and sex.
DEPDC5 Nonsynonymous Variants Confer Susceptibility to Liver Fibrosis
We next investigated whether the DEPDC5 gene locus is directly associated with the development of moderate/severe liver fibrosis. We examined the distribution of and risk conferred by DEPDC5 nonsynonymous variants at the onset of moderate/severe fibrosis. The genotype distribution of the DEPDC5 nonsynonymous variants is shown in Supporting Table 7. To increase the power of our analysis, we analyzed the discovery and the cross‐sectional validation cohorts pooled together. Specifically, we compared the presence of DEPDC5 nonsynonymous variants between subjects with no/mild fibrosis (stage F0‐F1) and those with moderate/severe fibrosis (stage F2‐F4). Subjects with severe fibrosis had a higher, albeit nonsignificant (P = 0.097) prevalence of at least one DEPDC5 nonsynonymous variants than those with no/mild fibrosis (67% versus 60%, respectively).
Our analysis of the risk of developing moderate/severe fibrosis revealed that carriage of at least one nonsynonymous variant was associated with a 54% increase of the risk of moderate/severe fibrosis (OR, 1.54; 95% CI, 1.02‐2.34; P = 0.040; Table 3).
Table 3.
Carriers of DEPDC5 Nonsynonymous Variants Have Higher Risk of Moderate/Severe Fibrosis
| OR | 95% CI | P | |
|---|---|---|---|
| Age, years | 1.05 | 1.04‐1.07 | <0.001 |
| Sex, men | 1.33 | 0.98‐1.81 | 0.066 |
| Recruitment center | 0.88 | 0.76‐1.02 | 0.095 |
| Nonsynonymous variants | 1.54 | 1.02‐2.34 | 0.040 |
DEPDC5 Is Highly Expressed in Immortalized Hepatic Stellate Cells and Its Knockout Increases β‐catenin Expression and MMP2 Production
DEPDC5 has a DEP domain involved in several cellular functions, including the Wnt/β‐catenin pathway.26, 27 The Wnt/β‐catenin pathway is involved in liver fibrogenesis by affecting hepatic stellate cells activation.28, 29 Therefore, to test the hypothesis that DEPDC5 increases fibrosis through this pathway, we first assessed DEPDC5 expression in seven different human immortalized cell types. We found that DEPDC5 is highly expressed in LX‐2 cells, a human hepatic stellate cell line spontaneously immortalized through culture in low serum30 (Fig. 4A). Next, to test whether DEPDC5 is involved in hepatic stellate cell activation through the Wnt/β‐catenin pathway, we incubated LX‐2 cells with and without DEPDC5 siRNA (Ambion) for 48 hours. DEPDC5 knockdown induces an increase in β‐catenin expression in LX‐2 cells (P = 0.002; Fig. 4B).
Figure 4.
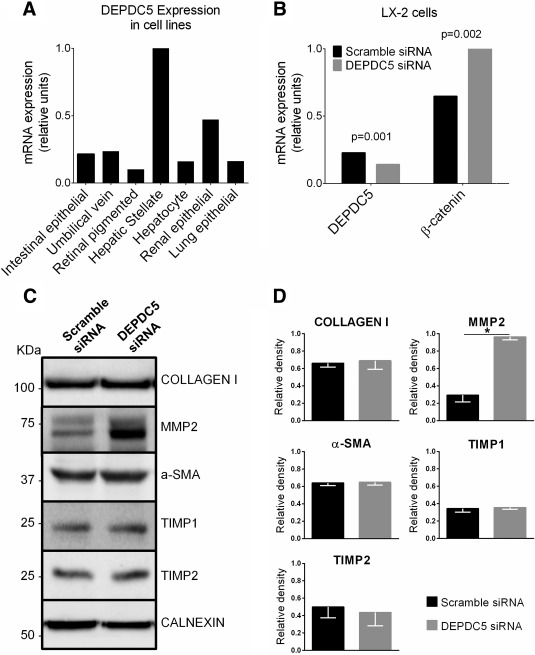
DEPDC5 is highly expressed in LX‐2 cells and DEPDC5 silencing induces β‐catenin levels and MMP2 production in LX‐2 cells. (A) DEPDC5 mRNA expression in human tissues assessed by quantitative PCR. The tissue with the highest Ct value was assigned a value of 1. (B) β‐catenin and DEPDC5 mRNA expression in LX‐2 cells incubated with negative control (scramble) siRNA or DEPDC5 siRNA for 48 hours. The experiments were repeated four times. DEPDC5 reduction (P = 0.001) and β‐catenin increase (P = 0.002) were significant. (C) Lysate fractions of LX‐2 cells transfected with scramble siRNA or DEPDC5 siRNA for 48 hours analyzed by western blotting. Calnexin was used as loading control. (D) Quantified relative protein levels are shown as column charts. The data are the means ± standard deviation of four independent experiments (P < 0.05).
Next, we tested the effect of DEPDC5 knockdown on cell proliferation. Cell numbers were counted 24, 48, and 72 hours after transfection, and we did not observe any increase on LX‐2 cell proliferation after DEPDC5 knockdown (Supporting Fig. 1).
Furthermore, we tested whether DEPDC5 knockdown affects the synthesis of molecules involved in liver fibrogenesis, namely collagen, MMP2, α‐SMA, TIMP1, and TIMP2.30, 31 DEPDC5 knockdown resulted in an increase in MMP2 production (P < 0.05; Fig. 4C,D) and in no differences in collagen, α‐SMA, TIMP1, and TIMP2 production (Fig. 4C,D).
Discussion
In this study, we examined the association between severe liver disease and DEPDC5 rs1012068 in European subjects with chronic HCV infection. We showed that DEPDC5 variants confer susceptibility to severe fibrosis.
Genome‐wide association studies identified DEPDC5 rs1012068 and MICA rs2596542 as loci of susceptibility to HCC in Asian subjects with chronic HCV infection.14, 15 In the current study, we examined whether genotype distribution was different in European subjects with cirrhosis who did and did not also have HCC. We examined those two specific groups because HCC occurs primarily in patients with cirrhosis.18 We found that neither DEPDC5 rs1012068 nor MICA rs2596542 are associated with HCC in Europeans with chronic HCV infection. To understand the discrepancy between our results and those reported in Asian subjects,14, 15 we analyzed the association between those two variants and liver fibrosis. To maximize the power of our analysis, we compared the genotype distribution of the DEPDC5 rs1012068 and MICA rs2596542 variants in the two extremes of fibrosis distribution: no/mild fibrosis (F0‐F1) versus the most severe stage of fibrosis (F4 [i.e., cirrhosis]). We found that MICA rs2596542 was not associated with cirrhosis, but we observed an enrichment of DEPDC5 rs1012068 in subjects with cirrhosis. This enrichment was confirmed in an independent cross‐sectional validation cohort.
To confirm our hypothesis that the DEPDC5 rs1012068 variant is associated with, in general, a more severe fibrosis, we examined two validation cohorts: the cross‐sectional and prospective cohorts, which included subjects with the entire spectrum of fibrosis. In the cross‐sectional cohort, DEPDC5 rs1012068 conferred an approximately 50% increased risk of fibrosis independently of age and sex. This association was confirmed in the prospective cohort by observing that carriers of DEPDC5 rs1012068 had a higher fibrosis progression rate. Taken together, our data suggest that DEPDC5 rs1012068 confers susceptibility to the development of severe fibrosis, rather than HCC, in European subjects with chronic HCV infection.
The apparent discrepancy between our results and those reported by the two genome‐wide association studies in Asian subjects14, 15 might be due to a key difference in the study design, as also pointed out by Hoshida et al.32 Cirrhosis, which is the most severe grade of fibrosis, is the main risk factor for HCC,18 and it is highly likely that most of the subjects with HCC in the study by Miki et al.14 had also cirrhosis. However, the investigators did not use subjects with HCV‐related cirrhosis without HCC as control group, so it is likely that the association they found actually refers to severe fibrosis rather than to HCC.
Our results are different from those reported by a previous study in Asian subjects in which DEPDC5 rs1012068 was found to be associated with lower fibrosis stage.33 However, that study was conducted only on a small and selected cohort of subjects who underwent hepatectomy for HCC. Furthermore, there was no replication cohort, and the association analysis on fibrosis was performed differently. On the other hand, our data are in line with a recent study on HCV‐positive subjects from Saudi Arabia in which homozygotes for DEPDC5 rs1012068 were found to have a 1.7‐fold higher risk of HCV‐related cirrhosis.34
The rs1012068 is an intronic variant of DEPDC5,14 and the role of DEPDC5 in hepatic fibrogenesis is currently unknown. Therefore, to test association of the DEPDC5 locus with the severity of liver fibrosis by an independent approach, we examined DEPDC5 nonsynonymous variants. We specifically selected nonsynonymous variants because, leading to an amino acidic change, they are biologically more likely to result in a functional change of the protein. To increase the power of our study, we examined whether carriage of at least one nonsynonymous variant increased the risk of fibrosis in the pooled cross‐sectional discovery and validation cohort. We found that the presence of nonsynonymous variants increased the risk of moderate/severe fibrosis, suggesting that DEPDC5 might be involved in liver fibrogenesis. A limitation of this analysis is that two of the nonsynonymous variants are in linkage disequilibrium (D′ = 1).
DEPDC5 rs1012068 does not affect gene expression.14 By using the data on linkage disequilibrium available from Ensembl genome browser, we found that DEDPC5 rs1012068 is in linkage disequilibrium with two nonsynonymous variants (rs61731662 and rs146449468). We therefore speculate that the association found for the rs1012068 intronic variant might mirror the effect of one of these two amino acidic changes (and possibly others) on the protein structure and/or function or that this variant is associated with DEPDC5 expression.
Other DEPDC5 variants have been associated with familial focal epilepsy,35, 36, 37 but the function of the DEPDC5 protein is still not known. DEPDC5 has a DEP domain that is involved in different cellular functions, including Wnt signaling.26, 27 Wnt/β‐catenin signaling is involved in liver fibrogenesis, inducing hepatic stellate cell activation.28, 29 We showed that DEPDC5 is highly expressed in immortalized LX‐2 human hepatic stellate cells. We also showed that DEPDC5 down‐regulation increases β‐catenin expression and induces MMP2 production in these cells. MMP2 is a downstream target of β‐catenin.38 In addition, several lines of evidence suggest that MMP2 is a profibrotic molecule: (1) it is highly expressed in liver fibrosis,39 (2) it colocalizes with activated HSCs,40 (3) it induces a remodeling of the extracellular matrix in favor of type 1 collagen,41 and (4) it may have an autocrine action inducing HSC activation.42
We speculate that this noncoding variant, or the causal variant in linkage with it, determines a reduction in DEPDC5 expression or activity. This reduction determines an increase in the Wnt/β‐catenin pathway followed by an increase in the MMP2 synthesis, resulting in a higher degree of liver fibrosis.
In conclusion, DEPDC5 variants increase fibrosis progression in European subjects with chronic HCV infection. Our findings suggest that DEPDC5 down‐regulation may contribute to HCV‐related fibrosis by increasing MMP2 synthesis trough the β‐catenin pathway.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28322/suppinfo.
Supporting Information
Potential conflict of interest: Felix Stickel has received grants from Gilead and AbbVie and is a consultant for BioTech.
S.R. was supported by the Swedish Research Council (VR, 254439006), the Swedish Heart Lung Foundation (244439007), the Swedish federal government funding under the LUA/ALF agreement (76290), and the Novo Nordisk Foundation Grant for Excellence in Endocrinology (244439012). L.V. was supported by Ricerca Corrente Fondazione Ca'Granda IRCCS Policlinico of Milan, Associazione Malattie Metaboliche del Fegato ONLUS, and Bando Medicina Molecolare Fondazione IRCCS Ca'Granda‐Istituto Nazionale Genetica Molecolare 2015‐2016. R.M.M. was supported by Nilsson‐Ehle funds from the Fysiografiska Sällsk‐apet in Lund, Sweden. S.M.L. and V.L. were supported by a cofinanced grant from the European Commission, the European Social Fund, and the Calabria Region. J.F. and T.B. were supported by the German Competence Network for Viral Hepatitis (Hep‐Net), funded by the German Ministry of Education and Research (BMBF) (grant no. 01 KI 0437, project no. 10.1.3, and core project no. 10.1 [Genetic Host Factors in Viral Hepatitis and Genetic Epidemiology Group in Viral Hepatitis]); the EU‐Vigilance Network of Excellence Combating Viral Resistance (VIRGIL) (project no. LSHM‐CT‐2004‐503359); and the BMBF Project: Host and Viral Determinants for Susceptibility and Resistance to Hepatitis C Virus Infection (grant no. 01KI0787). F.S. was supported by unrestricted research support from AbbVie AG (Switzerland) and Gilead (Switzerland) and additional funding from the Swiss National Funds (grant no. 310030_138747).
References
- 1. Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect 2011;17:107‐115. [DOI] [PubMed] [Google Scholar]
- 2. Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow‐up study of 384 patients. Gastroenterology 1997;112:463‐472. [DOI] [PubMed] [Google Scholar]
- 3. Tremolada F, Casarin C, Alberti A, Drago C, Tagger A, Ribero ML, et al. Long‐term follow‐up of non‐A, non‐B (type C) post‐transfusion hepatitis. J Hepatol 1992;16:273‐281. [DOI] [PubMed] [Google Scholar]
- 4. Yano M, Kumada H, Kage M, Ikeda K, Shimamatsu K, Inoue O, et al. The long‐term pathological evolution of chronic hepatitis C. Hepatology 1996;23:1334‐1340. [DOI] [PubMed] [Google Scholar]
- 5. Poynard T, Mathurin P, Lai CL, Guyader D, Poupon R, Tainturier MH, et al. A comparison of fibrosis progression in chronic liver diseases. J Hepatol 2003;38:257‐265. [DOI] [PubMed] [Google Scholar]
- 6. Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups . Lancet 1997;349:825‐832. [DOI] [PubMed] [Google Scholar]
- 7. Burza MA, Molinaro A, Attilia ML, Rotondo C, Attilia F, Ceccanti M, et al. PNPLA3 I148M (rs738409) genetic variant and age at onset of at‐risk alcohol consumption are independent risk factors for alcoholic cirrhosis. Liver Int 2014;34:514‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Estrabaud E, Vidaud M, Marcellin P, Asselah T. Genomics and HCV infection: progression of fibrosis and treatment response. J Hepatol 2012;57:1110‐1125. [DOI] [PubMed] [Google Scholar]
- 9. Hu S, Zhao L, Yang J, Hu M. The association between polymorphism of P53 codon 72 Arg/Pro and hepatocellular carcinoma susceptibility: evidence from a meta‐analysis of 15 studies with 3704 cases. Meta Gene 2013;1:126‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He J, Yu G, Li Z, Liang H. Influence of interleukin‐28B polymorphism on progression to hepatitis virus‐induced hepatocellular carcinoma. Tumour Biol 2014;35:8757‐8763. [DOI] [PubMed] [Google Scholar]
- 11. Guo PF, Jin J, Sun X. Influence of IL10 gene polymorphisms on the severity of liver fibrosis and susceptibility to liver cirrhosis in HBV/HCV‐infected patients. Infect Genet Evol 2015;30:89‐95. [DOI] [PubMed] [Google Scholar]
- 12. Sato M, Kondo M, Tateishi R, Fujiwara N, Kato N, Yoshida H, et al. Impact of IL28B genetic variation on HCV‐induced liver fibrosis, inflammation, and steatosis: a meta‐analysis. PLoS One 2014;9:e91822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trépo E, Nahon P, Bontempi G, Valenti L, Falleti E, Nischalke HD, et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: evidence from a meta‐analysis of individual participant data. Hepatology 2014;59:2170‐2177 . [DOI] [PubMed] [Google Scholar]
- 14. Miki D, Ochi H, Hayes CN, Abe H, Yoshima T, Aikata H, et al. Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers. Nat Genet 2011;43:797‐800. [DOI] [PubMed] [Google Scholar]
- 15. Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, et al. Genome‐wide association study identifies a susceptibility locus for HCV‐induced hepatocellular carcinoma. Nat Genet 2011;43:455‐458. [DOI] [PubMed] [Google Scholar]
- 16. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet 2009;10:241‐251. [DOI] [PubMed] [Google Scholar]
- 18. Sangiovanni A, Prati GM, Fasani P, Ronchi G, Romeo R, Manini M, et al. The natural history of compensated cirrhosis due to hepatitis C virus: a 17‐year cohort study of 214 patients. Hepatology 2006;43:1303‐1310. [DOI] [PubMed] [Google Scholar]
- 19. Valenti L, Rumi M, Galmozzi E, Aghemo A, Del Menico B, De Nicola S, et al. Patatin‐like phospholipase domain‐containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology 2011;53:791‐799. [DOI] [PubMed] [Google Scholar]
- 20. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group . Hepatology 1996;24:289‐293. [DOI] [PubMed] [Google Scholar]
- 21. Fischer J, Böhm S, Scholz M, Müller T, Witt H, George J, et al. Combined effects of different interleukin‐28B gene variants on the outcome of dual combination therapy in chronic hepatitis C virus type 1 infection. Hepatology 2012;55:1700‐1710. [DOI] [PubMed] [Google Scholar]
- 22. De Nicola S, Dongiovanni P, Aghemo A, Cheroni C, D'Ambrosio R, Pedrazzini M, et al. Interaction between PNPLA3 I148M variant and age at infection in determining fibrosis progression in chronic hepatitis C. PLoS One 2014;9:e106022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marabita F, Aghemo A, De Nicola S, Rumi MG, Cheroni C, Scavelli R, et al. Genetic variation in the interleukin‐28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011;54:1127‐1134. [DOI] [PubMed] [Google Scholar]
- 24. Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696‐699. [DOI] [PubMed] [Google Scholar]
- 25. Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2014. Nucleic Acids Res 2014;42:D749‐D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei W, Li M, Wang J, Nie F, Li L. The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol Cell Biol 2012;32:3903‐3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tauriello DV, Jordens I, Kirchner K, Slootstra JW, Kruitwagen T, Bouwman BA, et al. Wnt/β‐catenin signaling requires interaction of the Dishevelled DEP domain and C terminus with a discontinuous motif in Frizzled. Proc Natl Acad Sci U S A 2012;109:E812‐E820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li W, Zhu C, Li Y, Wu Q, Gao R. Mest attenuates CCl4‐induced liver fibrosis in rats by inhibiting the Wnt/β‐catenin signaling pathway. Gut Liver 2014;8:282‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol 2008;294:G39‐G49. [DOI] [PubMed] [Google Scholar]
- 30. Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX‐1 and LX‐2: new tools for analysis of hepatic fibrosis. Gut 2005;54:142‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoshida Y, Fuchs BC, Tanabe KK. Genomic risk of hepatitis C‐related hepatocellular carcinoma. J Hepatol 2012;56:729‐730. [DOI] [PubMed] [Google Scholar]
- 33. Motomura T, Ono Y, Shirabe K, Fukuhara T, Konishi H, Mano Y, et al. Neither MICA nor DEPDC5 genetic polymorphisms correlate with hepatocellular carcinoma recurrence following hepatectomy. HPB Surg 2012;2012:185496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Al‐Anazi MR, Matou‐Nasri S, Abdo AA, Sanai FM, Khan MQ, Albenmousa A, et al. Variations in DEPDC5 gene and its association with chronic hepatitis C virus infection in Saudi Arabia. BMC Infect Dis 2014;14:3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berkovic SF, Serratosa JM, Phillips HA, Xiong L, Andermann E, Díaz‐Otero F, et al. Familial partial epilepsy with variable foci: clinical features and linkage to chromosome 22q12. Epilepsia 2004;45:1054‐1060. [DOI] [PubMed] [Google Scholar]
- 36. Callenbach PM, van den Maagdenberg AM, Hottenga JJ, van den Boogerd EH, de Coo RF, Lindhout D, et al. Familial partial epilepsy with variable foci in a Dutch family: clinical characteristics and confirmation of linkage to chromosome 22q. Epilepsia 2003;44:1298‐1305. [DOI] [PubMed] [Google Scholar]
- 37. Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 2013;45:546‐551. [DOI] [PubMed] [Google Scholar]
- 38. Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity 2007;26:227‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Preaux AM, Mallat A, Nhieu JT, D'Ortho MP, Hembry RM, Mavier P. Matrix metalloproteinase‐2 activation in human hepatic fibrosis regulation by cell‐matrix interactions. Hepatology 1999;30:944‐950. [DOI] [PubMed] [Google Scholar]
- 40. Milani S, Herbst H, Schuppan D, Grappone C, Pellegrini G, Pinzani M, et al. Differential expression of matrix‐metalloproteinase‐1 and ‐2 genes in normal and fibrotic human liver. Am J Pathol 1994;144:528‐537. [PMC free article] [PubMed] [Google Scholar]
- 41. Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis 2001;21:373‐384. [DOI] [PubMed] [Google Scholar]
- 42. Benyon RC, Hovell CJ, Da Gaca M, Jones EH, Iredale JP, Arthur MJ. Progelatinase A is produced and activated by rat hepatic stellate cells and promotes their proliferation. Hepatology 1999;30:977‐986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28322/suppinfo.
Supporting Information