

Abstract
Objective
Melanopsin retinal ganglion cells (mRGCs) are photoreceptors driving circadian photoentrainment, and circadian dysfunction characterizes Alzheimer disease (AD). We investigated mRGCs in AD, hypothesizing that they contribute to circadian dysfunction.
Methods
We assessed retinal nerve fiber layer (RNFL) thickness by optical coherence tomography (OCT) in 21 mild‐moderate AD patients, and in a subgroup of 16 we evaluated rest–activity circadian rhythm by actigraphy. We studied postmortem mRGCs by immunohistochemistry in retinas, and axons in optic nerve cross‐sections of 14 neuropathologically confirmed AD patients. We coimmunostained for retinal amyloid β (Aβ) deposition and melanopsin to locate mRGCs. All AD cohorts were compared with age‐matched controls.
Results
We demonstrated an age‐related optic neuropathy in AD by OCT, with a significant reduction of RNFL thickness (p = 0.038), more evident in the superior quadrant (p = 0.006). Axonal loss was confirmed in postmortem AD optic nerves. Abnormal circadian function characterized only a subgroup of AD patients. Sleep efficiency was significantly reduced in AD patients (p = 0.001). We also found a significant loss of mRGCs in postmortem AD retinal specimens (p = 0.003) across all ages and abnormal mRGC dendritic morphology and size (p = 0.003). In flat‐mounted AD retinas, Aβ accumulation was remarkably evident inside and around mRGCs.
Interpretation
We show variable degrees of rest–activity circadian dysfunction in AD patients. We also demonstrate age‐related loss of optic nerve axons and specifically mRGC loss and pathology in postmortem AD retinal specimens, associated with Aβ deposition. These results all support the concept that mRGC degeneration is a contributor to circadian rhythm dysfunction in AD. ANN NEUROL 2016;79:90–109
Alzheimer disease (AD) is the most common age‐related dementia, characterized by accumulation in the brain of amyloid‐β protein (Aβ) plaques and aggregates of hyperphosphorylated tau as neurofibrillary tangles.1 Previous studies have shown circadian dysfunction of rest–activity, melatonin, and temperature rhythms in AD patients.2 In particular, actigraphic studies demonstrated an increased intradaily variability, and reduced interdaily stability and relative amplitude of the rest–activity rhythm in AD.3, 4 Moreover, in AD patients there has been observed neuronal loss in the suprachiasmatic nucleus (SCN), the circadian pacemaker.5, 6, 7 These abnormalities, which do not correlate tightly with severity of dementia, may be ameliorated by light therapy.8
Postmortem studies in AD, besides the well‐known occurrence of neurodegeneration in the brain, also highlighted retinal and optic nerve tissue pathology.9, 10, 11 In vivo studies with optical coherence tomography (OCT) corroborated these findings, showing a reduction of the retinal nerve fiber layer (RNFL) thickness in AD patients.12, 13, 14, 15, 16, 17, 18, 19, 20 Accumulation of Aβ in the retina of AD patients has also been reported in a few studies. The original study by Koronyo‐Hamaoui et al21 identified Aβ pathology in postmortem retinas of definite and early stage AD patients. They demonstrated various types of deposits, including diffuse, immature, and mature plaques, those including Aβ1–40 and Aβ1–42 species, and those associated with lipid deposits.21 Elevated Aβ1–42 levels and Aβ deposits were also found by 2 other independent groups in AD patients' retina22, 23 and in various transgenic rodent models of AD.21, 22, 23, 24, 25, 26, 27 Thus, there is multiple evidence that the neuroretina is affected by the pathological process of AD.
In the past decade a second light‐detecting system has been identified in the mammalian eye mediating non–image‐forming functions, such as photoentrainment of circadian rhythms, regulation of pupil size, sleep, alertness, and pineal melatonin synthesis.28, 29, 30 These cells represent a subpopulation (about 1–2%) of retinal ganglion cells (RGCs) that are intrinsically photosensitive due to the expression of the photopigment melanopsin (mRGCs).31 The mRGCs send information on ambient light to the brain via a monosynaptic neuronal pathway, known as the retinohypothalamic tract,32 projecting to the SCN of the hypothalamus.33, 34 Loss of mRGCs has been described with aging35, 36 and in some ocular diseases, such as glaucoma,37 and has been suggested to contribute to circadian dysfunction in these conditions.38
These observations prompted us to hypothesize that mRGC loss may contribute to circadian dysfunction in AD. Thus, we studied AD patients assessing the integrity of the optic nerve by OCT and rest–activity circadian rhythms by actigraphic recordings. We also objectively evaluated the RGCs and mRGCs by postmortem histological analyses in tissue samples from AD patients and age‐matched controls, including retinal immunohistochemistry (IHC) for Aβ and Aβ–melanopsin costaining for mRGCs.
Subjects and Methods
OCT Evaluation of Retinal Nerve Fiber Layer Thickness in AD Patients and Controls
All participants gave their informed consent to this study, which was approved by the internal review board at the University of Bologna (Independent Ethics Committee, Local Health Authority of Bologna, Protocol #656/CE).
We included patients with a diagnosis of AD according to National Institute of Neurological and Communication Disorders–Alzheimer's Disease and Related Disorders Association criteria,39 at mild–moderate stage (Mini‐Mental State Examination [MMSE] > 11).40 Absence of cognitive dysfunction was ascertained in controls.
Exclusion criteria were: (1) spherical or cylindrical refractive errors higher than 3 or 2 diopters, respectively; and (2) posterior pole ocular pathology such as age‐related macular degeneration and optic neuropathies, including open‐angle high intraocular pressure glaucoma.
Overall, in vivo ocular studies were performed in 21 AD patients (age = 71.2 ± 10.5 years, range = 51–84; 11 males) and 74 controls (age = 69.1 ± 8.1 years, range = 52–85 years; 31 males). All subjects underwent RNFL thickness measurements by OCT (StratusOCT, software version 4.0.1; Carl Zeiss Meditec, Dublin, CA) with the 3.4 acquisition protocol.41 We retrieved the average RNFL thickness (360 ° measure), as well as temporal (316–45 ° unit circle), superior (46–135 °), nasal (136–225 °), and inferior quadrant thickness (226–315 °). At the beginning of the examination, lenses were adjusted for the patients' refractive errors. Polarization was optimized to maximize the reflective signal, and the best centration of the scan with respect to the optic disk was used. For the statistical analysis, 1 eye was randomly chosen, after verification that there was no statistical difference between the 2 eyes, as previously reported.41
Average and single sector RNFL thickness values were used for comparisons between AD patients and controls, and for correlation with clinical data (see Statistical Analysis).
Actigraphic Evaluation of Rest–Activity Circadian Rhythm in AD Patients and Controls
In a subgroup of AD patients and controls, belonging to the same cohorts investigated by OCT, we measured spontaneous motor activity by a wrist actigraph (Actigraph Mini Motionlogger; Ambulatory Monitoring, Ardsley, NY) worn on the nondominant wrist for 7 days. We also evaluated the occurrence of sleep disturbances with the following self‐administered questionnaires: Epworth Sleepiness Scale (ESS), Pittsburgh Sleep Quality Index (PSQI), and Berlin Questionnaire.
Overall, 16 AD patients (age = 70.25 ± 10.2 years, range = 52–84 years; 8 males) and 10 controls (age = 65.8 ± 7.5 years, range = 54–80 years; 5 males) were studied. AW2 software (Ambulatory Monitoring) was used to analyze the data collected. This software allowed retrieving the total sleep time (TST; minutes of rest activity during the night period) and sleep efficiency (SE; sleep time divided by the time in bed multiplied by 100). Each participant also completed a sleep diary over the 7 days of actigraphic monitoring indicating bed/rise time, time to sleep onset, and eventual daytime naps.
Actigraphic data were inspected to detect the correspondence with the sleep diary and possible artifacts (low activity due to the actigraph removal). Each single artifactual epoch was marked as a “bad” epoch. Furthermore, to allow the comparison between subjects, all the recordings were aligned with the starting time at 12:00 am. Given that 5 subjects wore the actigraph for <7 days, to adequately compare subjects, 120 consecutive hours (5 days) of recordings were considered for subsequent analysis. Data were then exported to MATLAB (MathWorks, Natick, MA), and the mean of each hour was computed as the mean of the “good” epochs. If consecutive bad epochs occupied an entire hour, these periods were treated as missing data and the hourly mean was calculated as the weighted mean of the pre‐ and postactivity counts.
Nonparametric methods were applied for the analysis of circadian measurements.42, 43 We thus calculated the interdaily stability (IS; an index ranging from 0 to 1 reflecting the strength of the rhythm to supposedly stable environmental zeitgebers), the intradaily variability (IV; an index ranging from 0 to 2 measuring the frequency and extent of transitions between rest and activity), and the relative amplitude (RA; the normalized difference between the most active 10‐hour period [M10] of the rest–activity rhythm and the least active 5‐hour period [L5] in the average 24‐hour pattern).
Circadian (IS, IV, RA, M10, and L5) and sleep (TST and SE) parameters were used for comparisons between AD patients and controls, and correlation with clinical data (see Statistical Analysis).
Postmortem Studies of Retinas and Optic Nerves in AD Patients and Controls
Postmortem control eyes were collected both from the eye tissue bank Lions VisionGift (Portland, OR) and the Alzheimer's Disease Research Center (ADRC) Neuropathology Core at the University of Southern California (USC; Los Angeles, CA). Postmortem AD eyes were obtained from the ADRC at USC. Clinical charts of controls and AD patients were reviewed.
Overall, we collected and studied 17 neuropathologically confirmed AD cases (age = 80.6 ± 13.8 years, range = 51–98 years; 10 males) and 13 age‐matched controls (age = 77.8 ± 16.1, range = 54–105; 9 males). The inclusion criteria for AD cases were a neuropathological diagnosis of high‐likelihood AD according to National Institute on Aging–Reagan Institute criteria,44 frequent neuritic plaques according to modified Consortium to Establish a Registry for Alzheimer's Disease criteria,45 and Braak stage V or VI,46 based on the neuropathological reports (Dr C. Miller, ADRC at USC). Exclusion criteria were the presence of significant posterior pole ocular pathology such as age‐related macular degeneration and optic neuropathies including glaucoma, ischemic optic neuropathy, and other significant ocular pathology such as diabetic retinopathy. Moreover, exclusion criteria for controls provided by the eye tissue banks were the presence of neurological and psychiatric disorders, human immunodeficiency virus infection, alcohol abuse, and diabetes.
All postmortem tissues were immersion‐fixed in neutral buffered formalin. Eyes and optic nerves were oriented for superior and temporal zones with tissue ink. The eyes were dissected at the horizontal meridian producing 2 calottes each containing the nasal and temporal retina at the level horizontally bisecting the papillomacular bundle. A ribbon of tissue from the superior half was embedded into paraffin and serially sectioned at 5 μm. Sections were immunostained for melanopsin (rabbit antihuman melanopsin polyclonal antibody, code No. 5J68, characterized in detail by Hannibal et al47, 48 using an indirect immunoperoxidase technique with diaminobenzidine (DAB) as the substrate/chromogen.36 The C‐terminal antihuman melanopsin antibody was raised in‐house in a rabbit against a fusion protein representing the C‐terminal part of human melanopsin (amino acid sequence of the fusion protein was MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDHPFTHPKYRVAIAQHLPCLGVLLGVSRRHSRPYPSYRSTHRSTLISHTSNLSWISIRRRQESLGSESEVGWTHMEAAAVWGAAQQANGRSLYQGLEDLEAKAPPRPQGHEAETPGKTKGLIPSQDPRM) as previously detailed.47, 48 The C‐terminal antimelanopsin antibody (diluted 1:10,000) was characterized by preabsorption of the antibody with the immunization material dialyzed against phosphate‐buffered saline (PBS) with 0.1% Tween, which abolished all staining, as previously reported.47, 48 The rabbit antihuman melanopsin antiserum recognized human and monkey but not rodent melanopsin. An N‐terminal antimelanopsin antibody (a kind gift from Dr K.‐W. Yau, Johns Hopkins University; code hNA diluted 1:500), which was raised in rabbit against a peptide consisting of 19 amino acid residues, MNPPSGPRVPPSPTQEPSC, from the N‐terminus of the conceptually translated human melanopsin protein (National Center for Biotechnology Information accession number AAF24978),49 was used to verify that the C– and N‐terminal melanopsin antibodies stained identically. Identical staining was obtained on the same section using retinal sections from either human (unpublished data) or monkey.48
We analyzed 1 eye from each postmortem subject for mRGC number and morphology and its optic nerve for total axonal counts. Melanopsin RGCs were identified and manually counted and further confirmed and photographed at ×1,000 magnification. Counting has been performed based on a modified version of a previously published protocol.36 Briefly, we counted only immunoreactive mRGCs containing a prominent nucleus (central profile counting).50 Furthermore, to avoid counting the same nucleus twice, we applied a “section separation” approach by counting central profiles for every 5th section.50 Central profile counting, coupled with section separation, may minimize counting errors,50 but does not eliminate the problem of overcounting large objects embedded in thin sections.51, 52 Thus, we used Abercrombie's equation to correct the immunoreactive mRGC number, multiplying the central profile counts by T/(T + h), where T is the section thickness (5 µm) and h is the mean diameter of the nucleus.52
To obtain the mean diameter of the mRGC nuclei, we first performed a simple random sampling of 2 immunoreactive mRGCs from the 5 immunostained slides available for each patient in the control and AD groups. Two of the 5 numbered slides were selected randomly, and the first cell identified with a nucleus was chosen for measurement. There were 13 controls and 14 AD patients. Thus, we measured 26 nuclei in the control group and 28 nuclei in the AD group at a magnification of ×1,000 on a bright‐field microscope. The shortest diameter of mRGC nuclei was considered for calculations.
To perform the measurements of the nuclei from immunoreactive mRGCs, we used a SPOT Imaging Solutions software program, version 4.6 (Diagnostic Instruments, Sterling Heights, MI), which was calibrated at ×1,000 with a Bausch + Lomb (Rochester, NY) stage micrometer. The mRGC nuclei were observed on an Axioskop microscope Carl Zeiss Microscopy LLC, Thornwood, NY, USA and photographed with a SPOT RTke digital camera, and the images were saved on a computer.
Finally, mRGC density was obtained dividing the Abercrombie's corrected number of mRGCs by the retinal surface sampled (retinal length × section thickness).
We computed separately mRGCs located in the inner nuclear layer (INL) and in the ganglion cell layer (GCL), and their ratio was calculated to assess the difference in distribution across groups.
Optic nerves were dissected into cross‐sectional profiles 3mm posterior to the globe, postfixed in 3% phosphate‐buffered glutaraldehyde, processed, embedded into plastic blocks, and cut on an ultramicrotome at 1 µm. Tissue sections were placed on glass slides with a drop of water, dried using a hot plate, cooled, then stained with para‐phenylenediamine (PPD) to label the myelin ring of the entire population of RGC axons (including mRGCs). Axons were manually counted on images acquired at ×1,000.36 For total axon counts, each nerve cross‐section was partitioned into 4 regions (temporal, nasal, superior, inferior). All light microscopy (LM) photos of eyes (retinas) and nerves (axons) were acquired with a Spot II digital camera (Diagnostic Instruments) and saved on a computer.
To grade the severity of brain pathology of AD patients, we used the published ABC score.53 However, we were able to compute only the B and the C subscores using the silver stain for the detection of neurofibrillary tangles (B) and the thioflavin stain for the detection of neuritic plaques (C). The brain regions analyzed were: hippocampus CA‐1 (uncus and lateral geniculate body), entorhinal cortex, middle frontal, superior/middle temporal, inferior parietal, primary visual, and visual associative cortex.
For each subject, we retrieved the density and total number of mRGCs and the total axon number in optic nerve cross‐sections. These data were used for comparisons between AD patients and controls, and for correlation with clinical and neuropathological data (see Statistical Analysis).
Morphological Analysis of mRGCs in Flat‐Mounted Retinal Preparations from AD Patients and Controls
Flat‐mounted retinas from 3 controls and 4 age‐matched AD patients, all belonging to the same postmortem cohort, were treated by antigen retrieval solution (ChemMate; Dako, Carpinteria, CA; code No. S2367 in distilled water, pH 9) at 80 °C for 1½ to 2 hours before processing for IHC using the antimelanopsin antibody (code No. 5J68). IHC detection for melanopsin on flat‐mounted retinas was performed as previously described.47, 48 Briefly, after incubation of the primary antibodies for 72 to 84 hours at 4 °C (diluted 1:10,000), melanopsin was visualized using the Dako Envision kit (code No. K4002, diluted 1:2) and tyramide‐conjugated Alexa 488 (Molecular Probes, Eugene, OR, USA). Images were obtained using an iMIC confocal microscope (FEI, Till Photonics, Munich, Germany) equipped with appropriate filter settings for detecting 4′,6‐diamidino‐2‐phenylindole (DAPI) and CY2/Alexa 488. For a more detailed visualization of the melanopsin immunoreactive network of outer and inner stratifying dendritic processes, Z‐stacks with a focal depth of 30 to 40 μm were generated and the Z‐stacks of images were stitched using the LA‐stitch module of Fiji/ImageJ, version 1.47n, 64 bit. Images for 3‐dimensional (3D) analysis were obtained by the spinning disk confocal part of the microscope. Z‐stacks of typically 60 to 70 images separated at the Z‐level by 0.5 µm were deconvoluted in AutoQuant X, version 3.02 (Media Cybernetics, Rockville, MD, USA), and the localization of dendritic processes and cell bodies was further analyzed by IMARIS, version 7.6.4 (Bitplane Scientific Software, Zurich, Switzerland) using the filament tracer module.48 After deconvolution, an autoNetwork was created for each cell analyzed. Thereafter, the lowest (L) and highest (H) values of dendritic diameters were defined (L = 0.323 µm, H = 16.15 µm). We identified dendrites belonging to the analyzed cells by manual tracer, the measures obtained were exported into a spreadsheet, and values from each cell were computed as a mean value.
Aβ Immunohistochemistry and Aβ‐Melanopsin Colabeling for mRGCs in Flat‐Mounted Retinas from AD Patients and Controls
Flat‐mounted retinas from 5 controls and 5 age‐matched AD cases, all belonging to the same postmortem cohort, were washed in ×1 PBS and treated with target retrieval solution (S1699, Dako), pH 6.1, at 96 °C for 1 hour. Tissues were washed in ×1 PBS, treated with 3% hydrogen peroxide for 12 minutes, and washed again in ×1 PBS. Thereafter, retinas were immunostained using the Vectastain ABC kit (PK‐610; Vector Laboratories, Burlingame, CA); first treated with blocking solution and permeabilized with 0.1% Triton X‐100 for 30 minutes at room temperature. Tissues were then incubated with primary anti‐Aβ monoclonal antibodies (6E10, recognizing the 1‐16aa N‐terminal or 12F4, recognizing the 42 aa C‐terminal) diluted 1:100 in PBS containing 10% permeabilization/blocking solution for 72 hours at 4 °C. Aβ immunoreactivity was detected by standard immunoperoxidase methodology using the DAB + Substrate Chromogen System (K3467, Dako). Bright‐field images were acquired using a Axio Imager Z1 fluorescence microscope equipped with ApoTome (Carl Zeiss Microimaging), with an AxioCam HRc camera. Images were repeatedly captured at several focal planes in the same area of the retinas (superior–temporal quadrant). Controls were processed using the same protocol with the omission of the primary antibody to assess nonspecific labeling.
For fluorescent costaining of melanopsin and Aβ antibodies, on the same flat‐mount retina tissues that were previously stained with horseradish peroxidase, the antibodies from the first enzymatic staining were eluted with glycine sodium dodecyl sulfate pH 2 buffer for 45 minutes on a platform shaker in an incubator at 50 °C. Next, the tissues were washed extensively in PBS (see details in Pirici et al54 and treated with antigen retrieval solution, as described above. Tissues were then washed in PBS and blocked for 1 hour with blocking serum containing 20% donkey serum and 0.1% Triton X‐100 (Sigma‐Aldrich, Saint Louis, MO). Tissues were then stained for 72 hours at 4 °C with combinations of the primary antibodies in PBS with 2% blocking solution: mouse antihuman Aβ monoclonal antibody, clone 6E10 (1:100; Covance, Emeryville, CA) recognizing the amino acid 1‐16, and the rabbit antihuman melanopsin antibody (code No. 5J58; 1:2,000). Secondary polyclonal antibodies donkey antimouse, and antirabbit (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA) conjugated to Cy‐3, Cy‐5, or DyLight 649, were incubated for 1 hour at 37 °C. Subsequently, the tissues were washed in PBS and mounted with Prolong mounting media containing DAPI (Thermo‐Fisher Scientific/Molecular Probes, Grand Island, NY). Microscopic analysis was performed by ApoTome equipped Carl Zeiss Z1 fluorescence microscope.
Statistical Analysis
Normal distribution of data was verified by Kolmogorov–Smirnov test.
Comparisons between groups for OCT, actigraphic, and postmortem data were computed by means of analysis of covariance (ANCOVA) with age and gender as covariates. When appropriate, a group × age interaction was evaluated in the ANCOVA model. Significance was assumed for p < 0.05. Corrections for multiple comparisons were applied with the Bonferroni method. The adjusted critical p value is reported in the legends of Tables 1 and 2. We also calculated for each comparison the minimum detectable effect size assuming an 80% power and α = 0.05 (dES80%)55 and compared it to the observed effect size (expressed as Cohen f).
Table 1.
Optical Coherence Tomography Results in AD Patients and Controls
| Characteristic | AD, n = 21 | Controls, n = 74 | ANCOVA |
|---|---|---|---|
| Gender | 11 M, 10 F | 31 M, 43 F | |
| Age | 71.2 ± 10.5 (51–84) | 69.1 ± 8.1 (52–85) | |
| Disease duration, yr | 5.2 ± 2.5 (1–9) | ||
| MMSE | 18.3 ± 3.6 (13.5–26.1) | ||
| RNFL average, μm | 88.8 ± 8.4 (71–104) | 95.8 ± 12.3 (68–130) | *p = 0.038,a f = 0.2196 |
| RNFL‐T, μm | 67.2 ± 10.4 (49–84) | 69.3 ± 10.5 (41–101) | p = 0.762, f = 0.0316 |
| RNFL‐S, μm | 104.4 ± 13.4 (83–124) | 117.1 ± 17.6 (81–152) | *p = 0.006,a f = 0.2949 |
| RNFL‐N, μm | 66.6 ± 13.9 (37–86) | 74.3 ± 15.3 (48–133) | p = 0.081, f = 0.1759 |
| RNFL‐I, μm | 116.5 ± 15.5 (91–158) | 122.4 ± 18.1 (85–154) | p = 0.303, f = 0.1102 |
The critical probability value for multiple comparisons (n = 4) corrected with Bonferroni method is 0.013.
AD = Alzheimer disease; ANCOVA = analysis of covariance; f = Cohen f (detectable effect size with 80% power is 0.3075); F = female; I = inferior; M = male; MMSE = Mini‐Mental State Examination score; N = nasal; RNFL = retinal nerve fiber layer thickness; S = superior; T = temporal.
Table 2.
Demographic and Clinical Data of AD Patients and Controls Included for Actigraphic Studies
| Subject Group | Characteristic | Min | Max | Mean | SD |
|---|---|---|---|---|---|
| AD, n = 16 | DISDUR, yr | 1.00 | 9.00 | 4.8 | 2.5 |
| MMSE | 13.4 | 26.1 | 18.9 | 3.8 | |
| Age, yr | 52 | 84 | 70.25 | 10.2 | |
| Controls, n = 10 | Age, yr | 54 | 80 | 65.8 | 7.5 |
AD = Alzheimer disease; DISDUR = disease duration; Max = maximum; Min = minimum; MMSE = Mini‐Mental State Examination score; SD = standard deviation.
For in vivo studies, bivariate correlation analysis between experimental measurements (OCT or actigraphic data) and clinical data (age, age at onset, disease duration, and MMSE) was performed using Pearson coefficient. We also evaluated the interaction between age and disease severity by means of an age × disease severity interaction model (general linear model).
For postmortem studies, bivariate correlation analysis between histological (mRGC density, axonal counts, Braak score, severity of brain pathology) and clinical data (age, disease duration, MMSE score) was performed using Pearson coefficient. We also calculated the ratio of mRGCs to total RGCs. To do this, given the very different order of magnitude of the values for mRGC and RGC counts, we normalized (transforming into T scores) both axonal and mRGC counts. We thus compared the obtained ratio (T‐score‐mRGC/T‐score‐RGC = mRGC/RGC ratio) between AD and controls by means of ANCOVA (with age and gender as covariates).
Statistical analysis was performed using Statistical Package for Social Sciences (version 22.0; IBM, Armonk, NY).
Results
OCT Evaluation Demonstrated Age‐Dependent Loss of Retinal Nerve Fibers in AD Patients and Controls
We recruited 21 AD patients and 74 age‐matched controls. Seven AD eyes were excluded due to previously established criteria (see Subjects and Methods) or because the OCT was technically unreliable. Overall, we included 21 and 74 eyes, randomly selected, for AD and age‐matched control cohorts, respectively. Three AD patients had nonspecific visual complains (blurred vision), whereas the remaining 18 AD patients did not present visual symptoms. Demographic, clinical, and OCT findings of AD patients and controls are summarized in Table 1.
The average RNFL thickness was reduced in AD patients compared with controls (p = 0.038; Fig 1A and Table 1). Considering the 4 quadrants separately, the superior quadrant was significantly thinner (p = 0.006) in AD patients compared to controls (see Table 1). Average RNFL thickness was significantly correlated with age (p < 0.001; see Fig 1B). The group × age interaction model failed to reveal a significant difference between AD and controls for the RNFL thickness age‐related decline. RNFL thickness measurements did not significantly correlate with MMSE score, age at onset, or disease duration. Moreover, we failed to demonstrate a significant interaction between age and disease severity on RNFL thickness measurements in AD.
Figure 1.
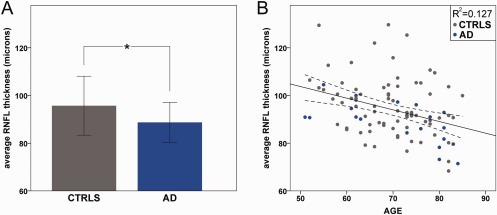
Optical coherence tomography results in controls (CTRLS) and Alzheimer disease (AD) patients. (A) Average retinal nerve fiber layer (RNFL) thickness results are reported for CTRLS and AD patients (*significant difference, p = 0.038). (B) Correlations between average RNFL thickness and age in years is reported (p < 0.001, R 2 = 0.127).
Overall, AD patients demonstrated an age‐related loss of retinal fibers involving predominantly the superior sector.
Actigraphic Evaluation of Rest–Activity Rhythm Revealed Variable Degrees of Circadian Rhythm Dysfunction and Sleep Efficiency Reduction in AD Patients
Actigraphic monitoring of rest–activity rhythm was performed in 16 AD patients and 10 controls, a subgroup of the cohort investigated by OCT. Good compliance was ensured insofar as bad hours represented only 0.6% of the total recording time for AD patients and 0.3% for controls (12 hours for AD and 4 hours for controls). Clinical and demographic data of patients and controls are summarized in Table 2. All but 4 AD patients were on therapy for dementia consisting either of donepezil (n = 4), rivastigmine (n = 2), memantine (n = 4), galantamine (n = 2), or both galantamine and rivastigmine (n = 1).
There was a large variability of actigraphic profiles among AD patients. Some AD patients resembled controls, whereas others showed markedly disturbed circadian rest–activity rhythms (Fig 2). Comparisons between groups of nonparametric actigraphic measurements are reported in Figure 2D–F and Table 3. RA was slightly reduced in AD patients (0.6 ± 0.13) compared to controls (0.7 ± 0.1), indicating a reduced variability of the activity level across the 24‐hour period; M10 was 258.9 ± 36.07 in AD and 282.59 ± 15.25 in controls, indicating that AD patients were less active during the wake period. L5 was 67 ± 27.54 in AD and 50.54 ± 17.17 in controls, indicating that AD patients were more active during the night period. However, these differences between AD and controls in terms of circadian parameters were not significant after Bonferroni correction for multiple comparisons.
Figure 2.
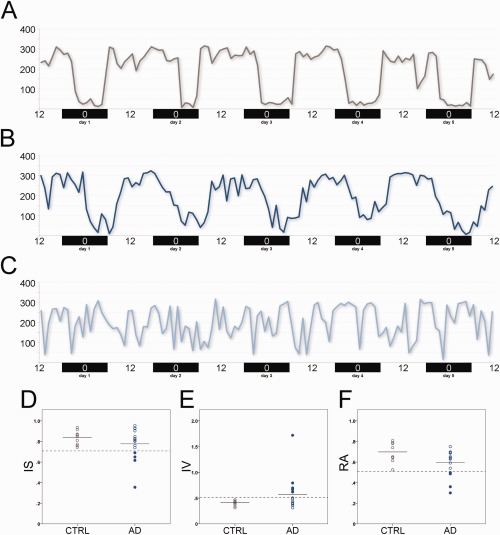
Actigraphic results in controls and Alzheimer disease (AD) patients. (A–C) Examples of actigraphic profiles are shown for a control (A), an AD patient with a profile resembling a control (B), and an AD patient with a severe disruption of the rest–activity circadian rhythm (C). (D–F) Scatter plots of (D) interdaily stability (IS), (E) intradaily variability (IV), and (F) relative amplitude (RA) for controls (gray circles) and AD patients (blue circles) are provided. Filled blue circles represent individual AD patients with values >2 standard deviations from the mean of controls (“circadian‐impaired” individuals).
Table 3.
Actigraphic Results for AD Patients and Controls
| Nonparametric measures | AD, n = 16 | Controls, n = 10 | ANCOVA |
|---|---|---|---|
| IS | 0.78 ± 0.15 | 0.84 ± 0.07 | p = 0.126, f = 0.3389 |
| IV | 0.57 ± 0.34 | 0.41 ± 0.05 | p = 0.198, f = 0.2827 |
| RA | 0.6 ± 0.13 | 0.7 ± 0.1 | p = 0.041,a f = 0.4622 |
| M10 | 258.9 ± 36.07 | 282.59 ± 15.25 | p = 0.036,a f = 0.4749 |
| L5 | 67 ± 27.54 | 50.54 ± 17.17 | p = 0.122, f = 0.3425 |
AD = Alzheimer disease; ANCOVA = analysis of covariance; f = Cohen f (detectable effect size with 80% power is 0.5749); IS = interdaily stability; IV = intradaily variability; L5 = least active 5‐hour period; M10 = most active 10‐hour period; RA = relative amplitude.
The critical probability value for multiple comparisons (n = 5) corrected with Bonferroni method is 0.01.
We also found a significant reduction of sleep efficiency in AD patients (75.4% ± 9.58%) compared with controls (90.2% ± 6.1%, p = 0.001). Patients and controls did not significantly differ for subjective sleepiness as evaluated by ESS, sleep quality as evaluated by PSQI, and risk of sleep apnea as evaluated by Berlin Questionnaire.
Furthermore, none of the actigraphic data assessing rest–activity circadian rhythms significantly correlated with clinical parameters (age, age at onset, disease duration, and MMSE score) or with OCT data.
Individual data for IS, IV, and RA of rest–activity circadian rhythm results are shown in Figure 2D–F. Overall, the graphic representation of single individuals showed that AD patients demonstrated higher variability, with scattered values, compared with controls, highlighting that only a subgroup of them are “circadian impaired.” The circadian impaired patients were defined as cases with circadian values >2 standard deviations (SD) from the mean of controls, as previously proposed.3
Overall, our in vivo studies documented that in AD there is an age‐dependent optic neuropathy and a rest–activity circadian dysfunction affecting a subgroup of patients. However, considering that mRGCs are only 1 to 2% of the total RGCs, their possible loss in the context of reduced RNFL thickness, as a contributor to circadian dysfunction, is overshadowed in the OCT results. Thus, we directly investigated these cells in postmortem ocular specimens (retina and optic nerve; see next section).
Postmortem Studies Confirmed the Age‐Related Loss of Axons and Revealed a Loss of mRGCs in AD Patients
We investigated horizontal retinal sections stained with an antibody targeting melanopsin to identify and count immunoreactive mRGCs from 17 AD patients and 13 controls, adopting the same protocol used in a previous study.36 Demographic, clinical, and neuropathological information for controls are reported in Table 4.
Table 4.
Summary of the Postmortem Control Cohort
| ID | Age at Death, yr | Gender | Time at Death | Postmortem Time | Cause of Death | NP Report |
|---|---|---|---|---|---|---|
| C1 | 54 | M | 12:55 | 10:28 | Anoxic brain injury secondary to acute myocardial infarction | n.a. |
| C2 | 58 | M | 9:10 | 7:20 | Upper gastrointestinal bleed secondary to esophageal cancer | n.a. |
| C3 | 60 | M | 21:15 | 7:20 | Metastatic lung cancer | n.a. |
| C4 | 64 | F | 19:40 | 11:03 | Cardiogenic shock secondary to congestive heart failure | n.a. |
| C5 | 70 | F | 10:29 | 10:46 | End‐stage chronic obstructive pulmonary disease | n.a. |
| C6 | 74 | M | 5:59 | 7:11 | Ruptured brain aneurysm | n.a. |
| C7 | 80 | M | 11:10 | 18:29 | Respiratory failure secondary to postoperative sequelae | n.a. |
| C8 | 80 | M | 16:30 | 13:12 | Sudden respiratory failure secondary to pneumonia | n.a. |
| C9 | 85 | M | 10:10 | 11:26 | Gastrointestinal bleeding | n.a. |
| C10 | 93 | M | 4:30 | 6:30 | Subarachnoid hemorrhage | Subarachnoid hemorrhage; exclusion of AD diagnosis |
| C11 | 93 | M | 23:45 | 3:45 | n.a. | Amyloid angiopathy; exclusion of AD diagnosis |
| C12 | 95 | F | 23:45 | 3:15 | n.a. | Normal brain (Braak score I); amyloid angiopathy in meningeal vessels |
| C13 | 105 | F | 5:30 | 6:00 | Cardiopulmonary arrest | Cerebrum infarct, severe arteriosclerosis; exclusion of AD diagnosis |
C = control; F = female; M = male; n.a. = not available; NP = neuropathological.
Histological examination revealed the presence of age‐related macular degeneration in a 98‐year‐old AD patient and proliferative diabetic retinopathy in a 70‐year‐old AD case. Based on these comorbidities, we excluded these 2 cases from the subsequent analysis. Moreover, LM evaluation of optic nerve cross‐sections revealed a massive regional loss of axons compatible with nonarteritic ischemic optic neuropathy in 1 AD case (age = 86 years), which was also excluded from further analysis. All the included AD cases (n = 14) showed preserved retinal anatomy. The main clinical premortem and neuropathological features of the AD cases included are summarized in Table 5.
Table 5.
Summary of the Postmortem AD Cohort
| ID | Age at Death, yr | Gender | Time at Death | Postmortem Time | Premortem dx | MMSE | CDR | DISDUR | Braak Stage |
|---|---|---|---|---|---|---|---|---|---|
| A1 | 51 | F | 10:59 | 6 hours | Familial AD (T245P PS1 mutation) | n.a. | n.a. | n.a. | VI |
| A2 | 62 | M | 21:25 | 5:50 | Dementia | 26 (59 years) | n.a. | 4 | V |
| A3 | 64 | F | 1:17 | 3:23 | AD possible | 3 (64 years) | 3 (61 years) | 6 | VI |
| A4 | 71 | M | 00:01 | 13:40 | FTD | n.a. | n.a. | 14 | VI |
| A5 | 75 | F | 20:00 | 5:30 | AD | 21 (72 years) | n.a. | 7 | V |
| A6 | 80 | M | 14:48 | 6:12 | FTD | Unable to complete | 4 | n.a. | V |
| A7 | 80 | M | 18:10 | 6:50 | AD | 7 (80 years) | 3 (80 years) | 7 | V–VI |
| A8 | 81 | M | 12:15 | 4 hours | AD probable | Unable to complete | 3 (81 years) | 11 | VI |
| A9 | 83 | M | 23:20 | 13 hours | AD probable | 14 (78 years) | n.a. | 7 | VI |
| A10 | 86 | M | 6:45 | 9:15 | AD probable | 2 (84 years) | 3 (84 years) | 6 | VI |
| A11 | 95 | F | 7:25 | 6 hours | AD | 17 (93 years) | 3 (93 years) | 18 | V |
| A12 | 95 | F | 13:30 | 6 hours | AD | n.a. | 3 (92 years) | 11 | V |
| A13 | 96 | F | 16:17 | 5:13 | AD possible plus LBD | 0 (95 years) | 4 (95 years) | 10 | V |
| A14 | 98 | F | 7:30 | 7.5 hours | AD possible | n.a. | 2 (96 years) | n.a. | V |
AD = Alzheimer disease; CDR = Clinical Dementia Rating score; DISDUR = disease duration; dx = diagnosis; F = female; FTD = frontotemporal dementia; LBD = Lewy body disease; M = male; MMSE = Mini‐Mental State Examination score; n.a. = not available.
The qualitative evaluation of the melanopsin‐stained AD retinal sections at the LM level showed that the retinal layers were preserved as compared to age‐matched controls. We observed immunoreactive mRGCs located in both the GCL and the INL, as previously reported.47 Representative examples of immunoreactive mRGCs and their dendrites in controls and AD patients are provided in Figure 3A–H. In AD patients, we frequently observed smaller somas and thinner dendrites. At higher magnification, AD immunoreactive mRGCs often displayed “patchy” melanopsin immunostaining in cell bodies (see Fig 3B, D) and thinning of dendrites, which, when seen between varicosities, produced a “beading” effect to its appearance that we termed “focal attenuations” (see Fig 3F, H).
Figure 3.
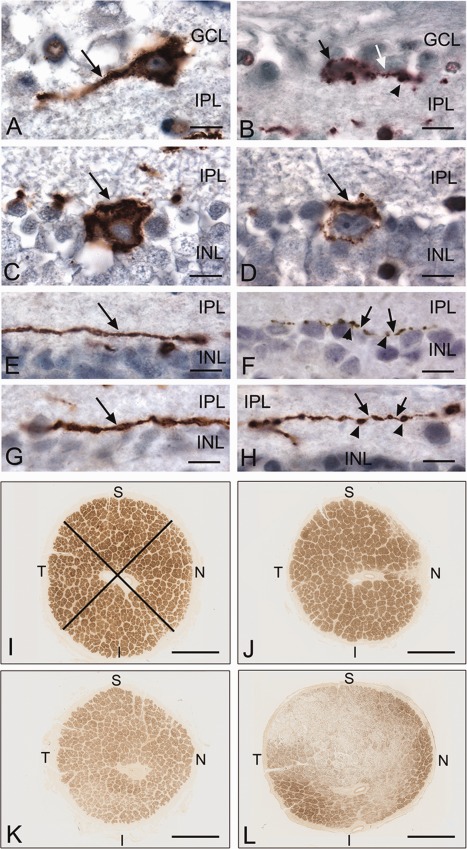
Melanopsin retinal ganglion cells, their dendrites, and optic nerve cross‐sections from controls and Alzheimer disease (AD) patients. Light micrographs of paraffin‐embedded retinas, immunoperoxidase stained for melanopsin with diaminobenzidine (brown color; A–H), and plastic‐embedded optic nerve cross‐sectional profiles, stained with paraphenylenediamine (brown color) for myelin (I–L). (A) Control retina with a single melanopsin retinal ganglion cell (mRGC) in the ganglion cell layer (GCL). Note homogeneous staining of cell body and dendrite (arrow). (B) AD retina with a single mRGC in the GCL. Note patchy staining of melanopsin in the cell body (black arrow). A single dendrite can be seen with a focal attenuation (white arrow) and varicosity (arrowhead). (C) Control retina with a single mRGC in the inner nuclear layer (INL). Note homogeneous staining of the cell body (arrow). (D) AD retina with a single mRGC in the INL. Note patchy staining of melanopsin (arrow). (E, G) Control retinas with a single immunostained dendrite (arrow) for melanopsin. Note caliber thickness. (F, H) AD retinas with a single immunostained dendrite for melanopsin. Note thinning or focal attenuations (arrows) of dendrites between varicosities (arrowheads). (I) Control optic nerve (1.18 million axons) with normal staining of axon bundles. The crossed lines delineate an example of sectorial sampling in quadrants for axon counting. (J–L) Examples of AD optic nerves with mild (J; A3, 822,000 axons), moderate (K; A14, 706,000 axons), and severe (L; A6, 306,000 axons) axonal depletion. I = inferior; INL = inner nuclear layer; IPL = inner plexiform layer; N = nasal; S = superior; T = temporal. Scale bars: A–H, 10 µm; I–L, 1mm.
Analysis of optic nerve cross‐sections showed, in some AD patients, evidence of optic neuropathy with different degrees of axonal loss, corroborating previous reports.9, 10, 11 Examples of optic nerve cross‐sectional profiles from controls and AD cases with different degrees of axonal loss are provided in Figure 3I–L. Interestingly, the axonal loss predominantly affected the larger fibers in the superior quadrants, and, to a lesser extent, the nasal and inferior quadrants, whereas the temporal quadrants, containing most of the smaller fibers, were largely spared (see Fig 3K, L).
Overall, quantitative and statistical analyses for axon and immunoreactive mRGC counts were performed on 14 AD patients (age = 79.8 ± 14.1 years; 7 males) and 13 age‐matched controls (age = 77.8 ± 16.1 years; 9 males). Analysis of axonal counts showed a reduction in AD patients compared to controls (Fig 4A), which did not reach statistical significance (p = 0.346, f = 0.2929, dES80% = 0.5630). Considering the different sectors (superior, temporal, nasal, and inferior), although a greater reduction was apparent for the superior quadrant in AD, this was not significant compared to controls (data not shown).
Figure 4.
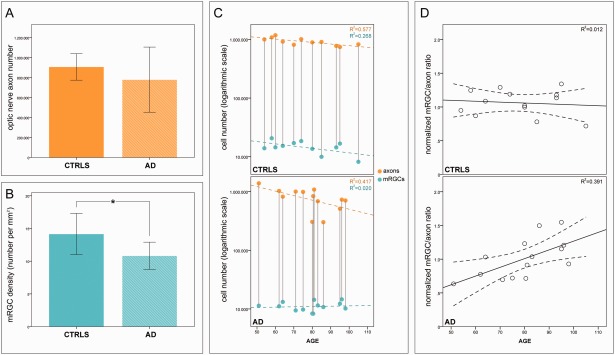
Melanopsin retinal ganglion cell (mRGC) density and axon number in controls and Alzheimer disease (AD) patients. (A) Mean and standard deviation of axon counts in controls and AD. (B) Mean and standard deviation of mRGC density in controls and AD patients (*significant difference, p = 0.003). (C) Correlation of axon count with age in years in controls (p = 0.001, R 2 = 0.577; orange circles, upper panel), correlation of mRGC number with age in years in controls (p = 0.035, R 2 = 0.268; cyan circles, upper panel), correlation of axon count with age in years in AD (p = 0.006, R 2 = 0.417; orange circles, lower panel), and correlation of mRGC number with age in years in AD (p = 0.313, R 2 = 0.020; cyan circles, lower panel). (D) Correlations between the standardized mRGC/RGC ratio and age in years are shown for controls (R 2 = 0.012; upper panel) and AD (R 2 = 0.391; lower panel).
The immunoreactive mRGCs were equally distributed between the GCL and the INL in both AD patients and controls, and there were no differences between groups in terms of their distribution. The mRGC density was significantly reduced in AD patients (mean = 10.824 ± 2.079) compared with controls (mean = 14.153 ± 3.149; p = 0.003, f = 0.6844, dES80% = 0.5630; see Fig 4B). Melanopsin RGC number was significantly correlated with age in controls (p = 0.035; r = −0.517; see Fig 4C, upper panel) but not in AD patients (p = 0.313; r = 0.143; see Fig 4C, lower panel).
Axon number significantly correlated with age in both controls (p = 0.001, r = −0.760) and AD patients (p = 0.006, r = −0.646; see Fig 4C). Remarkably, in the 5th to 7th decades, the reduction of mRGC number in AD patients occurred without a concomitant loss of regular RGCs, as reflected by axonal counts (see Fig 4C, lower panel).
The relation between the normalized mRGC/RGC ratio and age is shown in Figure 4D. We found a significant principal effect of the group (p = 0.021), whereas no significant effect of age (p = 0.082) or gender (p = 0.718) was evident. A significant group × age interaction (p = 0.026) indicated that the age‐related variation is different between AD and controls. This analysis highlights that mRGC loss in AD is evident also at an age where AD patients have a normal RGC count (see Fig. 4D).
Finally, there was no significant correlation between mRGC density and axonal counts with either disease duration, MMSE score, Braak score, or severity of AD pathology, as assessed by B and C scores (see Subjects and Methods).
In summary, we demonstrated mRGC loss in AD compared with controls, which is evident before the occurrence of the age‐related axonal loss.
Analysis of Flat‐Mounted Retinas Demonstrated Morphological Abnormalities of mRGCs and Reduction of Their Dendritic Size in AD
We also studied flat‐mounted retinal preparations from 4 AD patients (mean age = 72.25 years) and 3 controls (mean age = 75 years) from the same case series used in postmortem studies. In controls, melanopsin was distributed, as previously described,47 along the membranes of the soma, axons, and dendrites (Fig 5). Melanopsin immunoreactive processes were found in 2 distinct dendritic mosaics—one with dendrites stratified in the inner plexiform layer (IPL) close to the border of the GCL and one with dendrites stratified in the outer IPL close to the border of the INL. Inner stratifying cells had somas exclusively located in the GCL, whereas outer stratifying cell bodies were often displaced to the INL. Melanopsin was found in the membrane of the dendritic processes and in the relatively large “boutons en passant” characteristic of the mRGCs. In AD patients, melanopsin distribution was similar to controls, although several areas showed sparse or lacking immunoreactivity probably due to insufficient immunostaining (decreased penetration of primary or secondary antibody into the vitreous body). Such areas were also found in controls. However, in AD patients the dendritic processes looked thinner and the boutons en passant were smaller or missing. Furthermore, melanopsin immunoreactivity was patchy in the membrane of AD cells compared to what seen in controls.
Figure 5.
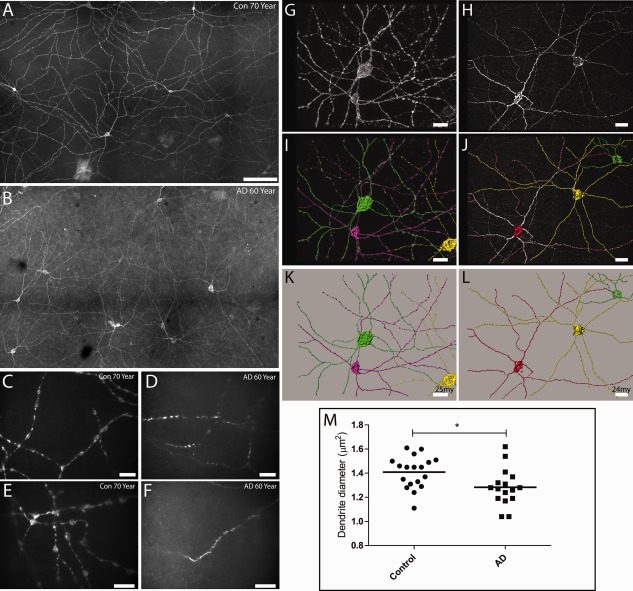
Flat‐mounted retinas and dendritic process analysis in Alzheimer disease (AD) patients and controls (Con). (A–F) Melanopsin immunoreactivity in control retinas (A, C, E) and in AD patients (B, D, F). At low magnification, melanopsin staining is present in a subpopulation of ganglion cells and displaced ganglion cells forming a dendritic network. The dendritic network of melanopsin processes in control retinas is characterized by long processes with a large “boutons en passant” (A, C, E). In contrast, melanopsin retinal ganglion cells (mRGCs) in AD patients have an abnormal morphology in which dendritic processes have a smaller diameter with a lack of “boutons en passant” (B, D, F). (G–L) Morphological analysis of control (G, I, K) and AD mRGCs (H, J, L). Stacks of images of mRGCs were generated, and a 3‐dimensional reconstruction was performed (see Subjects and Methods), followed by analysis of the processes using the filament trace module in Imaris (Bitplane). In G, I, and K, 3 cells from a control (green, purple, and yellow) and their dendritic processes are visualized. (G) The raw picture after deconvolution and (I) after tracing of the processes are shown. (K) Only the traced cells identified by the program are shown. Similarly, in H, J, and L, 3 mRGCs from AD an patient are shown. (M) The results of the total analysis of 18 cells from 3 controls (C3: 60 years old, 6 cells; C5: 70 years old, 8 cells; C12: 95 years old, 4 cells) and 16 cells from 4 AD patients (A1: 51 years old, 5 cells; A2: 62 years old, 5 cells; A6: 80 years old, 3 cells; A13: 96 years old, 3 cells) are shown, demonstrating a significant reduction of dendrite diameter in AD patients (*p = 0.003). Scale bars: A, B, 100 µm; C, D, 50 µm; E, F, 20 µm; G, I, K, 25 µm; H, J, L, 24 µm.
We analyzed a total of 18 cells from controls and 16 cells from AD patients in greater detail, measuring the mean diameter of dendritic processes after a 3D reconstruction of each cell using a specific software module applied to the Z‐stack of images (see Fig 5G–L). This analysis demonstrated that the dendrites of AD patients had a significantly smaller diameter compared to controls (p = 0.003; see Fig 5M).
In summary, these findings confirm the qualitative observations in the sagittal retinal sections and demonstrate that AD patients also display abnormal mRGC morphology.
Analysis of Flat‐Mounted Retinas Revealed Consistent Amyloid Deposition Affecting mRGCs in AD
We further investigated whether the observed abnormalities of immunoreactive mRGC and RGC degeneration in AD patients were associated with retinal evidence of Aβ accumulation. Thus, we studied the superior hemispheres flat‐mounted retinas in 5 definite AD patients (mean age = 73.4 years) and 5 age‐matched controls (mean age = 74.2 years) belonging to the original set studied for mRGC counts, randomly including 1 subject for each decade (Fig 6). We performed immunolabeling with antihuman Aβ monoclonal antibodies recognizing diverse epitopes within the N′ and C′ terminus of the Aβ sequence (6E10 and 12F4). Representative microscopic images from corresponding retinal locations demonstrate the overall Aβ burden in AD patients compared with age‐matched controls. Whereas no or minimal Aβ‐immunoreactivity was detected in control retinas, a substantial Aβ deposition was evident in all AD retinas. A few sparse Aβ deposits were also detected in the 95‐year‐old control retina, distributed in a few retinal locations (see Fig 6E and E1 inset, image at higher magnification). This is comparable with the neuropathological report of this individual indicating the presence of similar sparse mature and immature plaques in the occipital cortex (data not shown).
Figure 6.
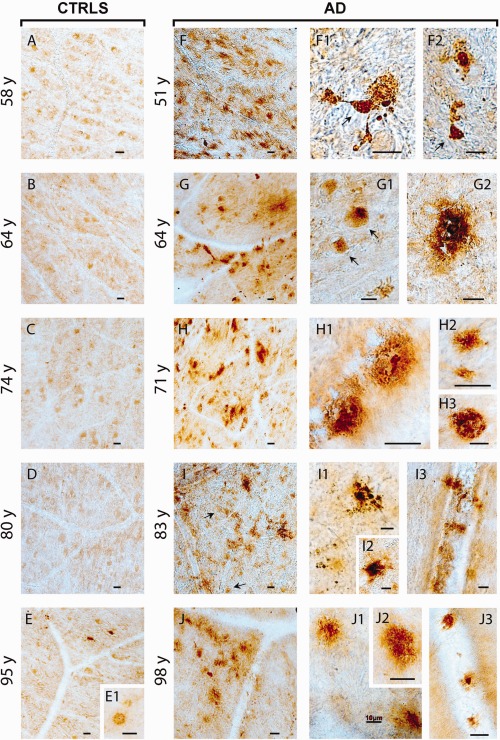
Amyloid‐β (Aβ) deposition in the superior retinas of Alzheimer disease (AD) patients. Examination of Aβ immunoreactivity in flat‐mounted retinas from 5 definite AD cases (range = 51–98 years; A1: 51 years; A3: 64 years; A4: 71 years; A9: 83 years; A14: 98 years) and age‐/gender‐matched controls (range = 58–95 years; C2: 58 years; C4: 64 years; C6: 74 years; C8: 80 years; C12: 95 years) using anti‐human Aβ monoclonal antibodies and standard peroxidase‐based techniques is shown. (A–J) Representative micrographs from corresponding superior temporal quadrant locations in the retina exhibit no to minimal Aβ immunoreactivity in healthy control retinas (A–E) and substantial Aβ burden in AD retinas (F–J; dark brown). Retinal Aβ plaques in AD patients are frequently found in clusters (F–J, left). Higher‐magnification images demonstrate variable morphology of extracellular Aβ depositions (F1–J3), including immature plaques (H2, I3, J1, J2), classical mature plaques with central dense Aβ core and radiating fibrillar arms (F2, G2, H1, I2, J3), and compact plaques composed of multiple dense cores (H3, I1). Diffuse plaques are typically found in postmortem retinas of AD patients (I3) but can occasionally be detected in aged healthy control retina (E1). Intracellular Aβ immunoreactivity is indicated with black arrows (F1, F2, G1, I). Furthermore, retinal Aβ deposits are also detected along blood vessels (I3, J3; blood vessel structures can be seen as transparent–lighter lane or tube shapes containing erythrocytes). Scale bars = 20 µm unless otherwise indicated.
In AD retinas, Aβ plaques were frequently found in clusters, and higher magnification revealed the diverse morphology of Aβ depositions (see Fig 6). Intracellular Aβ immunoreactivity was abundant in the 51‐, 64‐, and 83‐year‐old AD patients. Extracellular abluminal Aβ deposits, with diffuse, immature and mature structures, were the most prevalent in AD retinas. In all AD patients we detected Aβ deposits along the blood vessels (perivascular), with some cases having extensive accumulation. Similar to the brain, we found classical plaque structures, displaying a central dense Aβ core and radiating fibrillar arms, as well as “compact” plaques composed of multiple dense‐core globular Aβ deposits with no apparent radiating fibrils.
We further assessed the possible coexistence of Aβ deposition in and around immunoreactive mRGCs in the same set of flat‐mounted retinas from AD patients (n = 5) and controls (n = 5; Fig 7). Our analysis indicated absent Aβ‐immunoreactivity in and around intact immunoreactive mRGCs in controls. On the contrary, retinal Aβ deposition in AD patients was consistently found inside immunoreactive mRGCs soma (arrowheads) and extracellular/abluminal areas. The immunoreactive mRGCs in AD retinas had abnormal morphology associated with intracellular and extracellular Aβ. In AD retinas, we found that Aβ colocalized also in the neurites of immunoreactive mRGCs.
Figure 7.
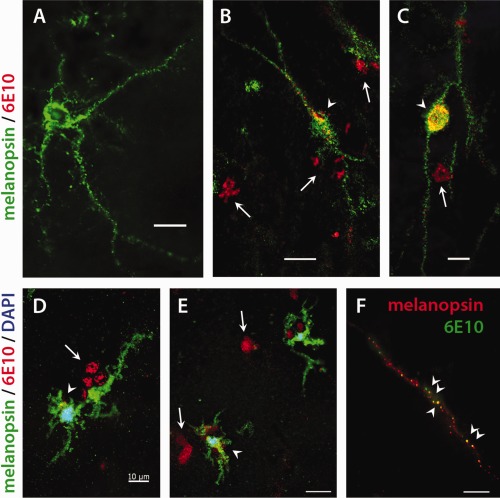
Colocalization of amyloid‐β (Aβ) deposition and melanopsin retinal ganglion cells (mRGCs) in flat‐mounted retinas of Alzheimer disease (AD) patients. (A–F) Fluorescent immunohistochemistry analysis of flat‐mounted retinas isolated from AD patients (n = 5; A1: 51 years; A3: 64 years; A4: 71 years; A9: 83 years; A14: 98 years) and age‐matched controls (n = 5; C2: 58 years; C4: 64 years; C6: 74 years; C8: 80 years; C12: 95 years) in different age groups (range = 51–98 years) colabeled with anti‐Aβ (6E10) and antimelanopsin antibodies. (A) Representative micrograph from control individual demonstrates an intact melanopsin‐containing RGC having long dendrites without presence of Aβ. (B, C) Representative micrographs from AD patients demonstrating intracellular Aβ deposition in mRGCs (arrowheads) as well as extracellular Aβ (arrows). (D, E) The mRGCs exhibited fewer cell processes. Abnormal morphology was frequently found in AD patients; mRGCs showed loss of dendritic arborization in the presence of intra‐ and extracellular Aβ. (F) An example of an mRGC neurite from AD retina showing colocalization with Aβ (arrowheads). Scale bars = 20 µm unless otherwise indicated. DAPI = 4′,6‐diamidino‐2‐phenylindole.
Discussion
The present study demonstrates, for the first time, loss of immunoreactive mRGCs in AD patients compared to controls. The remaining mRGCs in AD patients displayed morphological abnormalities, in particular, a reduction of dendritic diameter suggesting a possible impairment of function. Additionally, we showed an age‐related loss of total RGCs, as reflected by axonal counts, in optic nerves, confirmed in vivo by OCT studies. Remarkably, the mRGC loss in AD occurred even with a completely normal RGC count, suggesting that mRGCs are specifically affected in AD. Furthermore, the AD hallmark of amyloid pathology characterized postmortem AD retinas, affecting specifically mRGCs. Finally, we documented in vivo the presence of variable degrees of rest–activity circadian dysfunction in mild‐moderate AD. Thus, we here propose that mRGC loss is a plausible contributing factor to circadian dysfunction in AD.
The pattern of axonal loss documented by OCT corroborated a similar trend in postmortem studies and indicates a preferential involvement of the superior quadrant. This is consistent with previous OCT findings.13, 15, 16, 17, 18, 20 The preferential loss of fibers in the superior quadrant is also compatible with the predominant inferior visual field defect documented in AD patients.56 It is intriguing that the superior sector contains the largest fibers of the optic nerve and previous postmortem studies documented a preferential loss of the larger axons, suggesting early involvement of the magnocellular RGCs in AD.11 These findings, including the absence of large caliber fibers in the nerve fiber layer spectra, have also been reported in glaucoma.57, 58 Thus, AD optic neuropathy may share some similarities with glaucoma, which also has higher prevalence in AD patients.59 Interestingly, this pattern of axonal loss also fits with the demonstration of prominent Aβ deposition in the superior–temporal quadrant in flat‐mounted retinas (Koronyo et al: this study and unpublished results).
Our OCT findings and optic nerve axonal counts confirmed the age‐related decline of total RGCs, as reflected by loss of axons in the optic nerve, previously documented by histological60 and OCT studies.61
The circadian rest–activity assessment of AD patients revealed a wide variability for all the circadian parameters analyzed. This could be due to the intrinsic nature of pathology that produces variable severity of circadian derangement in this disease, as already reported in other case series.3 In our series of mild–moderate AD patients, although we failed to detect a significant difference in terms of circadian parameters (IS, IV, and RA) between AD and controls, there was a subgroup of AD patients with pathological circadian values (>2 SD from the mean of controls). Furthermore, AD patients tended to be less active during the day and more active during the night. The circadian measures did not correlate with dementia severity. These results are concordant with those reported by others.3, 62 The lack of significant differences compared to controls for circadian parameters may be due to the inclusion in our study of mild–moderate AD cases. By contrast, most of the previous studies, documenting a more severe circadian dysfunction in AD, included moderate or severe patients,63, 64, 65 and did not correct the results for multiple comparisons, whereas actigraphic studies investigating mild–moderate AD patients failed to reveal significant abnormalities.3, 64 Differently, laboratory‐based polysomnography studies showed significantly reduced sleep efficiency and slow wave sleep amount, even in the mild cases, as in our case series.66, 67 Selection of mild–moderate cases was constrained by the need to have the necessary compliance for reliable OCT studies. A previous study by Hatfield and coauthors, in a 1‐year follow‐up of mild–moderate AD patients, showed that the actigraphic pattern did not deteriorate over time, even in the presence of a significant decline of cognitive functions.3 This suggests that cognitive and circadian abnormalities may have different rates of progression, which could reflect different pathophysiological mechanisms, as suggested in previous studies.68 Moreover, the patients with the worst outcome had a significantly lower stability of circadian parameters at baseline evaluation.3 Thus, the presence of significant circadian dysfunction in the early stages of disease may be a predictive factor of poor outcome in AD patients.
In our series, only a subgroup of AD patients had a significant alteration of the circadian parameters. Melanopsin RGCs represent only a small subgroup of RGCs (about 1–2%), and it is difficult to infer from the OCT results a direct contribution of these cells to circadian dysfunction. Further investigation of mRGCs in these patients was hampered by a lack of reliable approaches in vivo. One possibility could have been the assessment of mRGC‐driven pupil response.69 However, the presence of concomitant cholinergic dysfunction in AD may represent a confounding factor in the interpretation of pupil response data.70 Moreover, different mRGC subtypes may regulate pupil response and circadian photoentrainment,31 and so the information obtained by pupil recordings may not describe the contribution of mRGCs to circadian rhythm regulation.
Therefore, to objectively and reliably evaluate the presence of mRGC pathology in AD we looked at postmortem AD retinas. Our postmortem studies demonstrated that, in addition to the total RGC loss, which correlated with aging, there was mRGC loss in AD cases that was present even in younger patients with a normal RGC count. This suggests that, in AD, the mRGC subpopulation of RGCs may be more directly sensitive to AD neuropathological mechanisms, in addition to the more generalized age‐related loss of total RGCs. This is further supported by the observed morphological changes of spared mRGCs, which were also abnormal, affecting the dendrites and their arborization patterns, as well as by their smaller cell bodies and patchy distribution of the melanopsin photopigment within their cytoplasm and in the membrane of the mRGC somata.
Furthermore, we documented in AD flat‐mounted retinas the presence of Aβ deposition with classical amyloid plaques, as previously described.21 Interestingly, a more recent study also documented phosphorylated‐tau deposition in AD human retinas.71 Importantly, Aβ deposits were frequently found inside and around mRGCs. These mRGCs exhibited abnormal morphology with substantial loss of axons and dendritic arborization. The presence of amyloid pathology in AD retinas, specifically in degenerating immunoreactive mRGCs, adds further evidence that mRGC may be primarily affected by Aβ pathology in AD.
An alternative explanation for the specific loss of mRGCs in AD may be retrograde axonal degeneration deriving from primary pathology of the SCN. Neuronal loss and classical AD neuropathology have been shown in the SCN.2, 5, 6, 7 A recent longitudinal study also demonstrated a significant loss of neurons in the hypothalamic ventrolateral preoptic nucleus correlated with sleep fragmentation, as documented by actigraphic recordings.72 Thus, the significant loss of mRGCs and the possible dysfunction of remaining mRGCs, in conjunction with other AD‐related hypothalamic pathology, may all contribute to the circadian rhythm and sleep abnormalities in AD patients, and it may be difficult to distinguish the individual contribution of the retinal and hypothalamic damage.
The current study has some obvious limitations. First, this is a cross‐sectional study of 2 different cohorts for in vivo and postmortem studies, where a direct correlation between circadian dysfunction and mRGC loss cannot be established. The 2 cohorts were enrolled with different inclusion criteria, and we cannot exclude that this may introduce a selection bias. Additionally, we recognize the relatively small sample size of the in vivo studies, the lack of an in vivo quantitative measure of mRGC function, and the lack of access to postmortem hypothalamic material. Furthermore, in our postmortem studies, we relied on immunoreactive mRGC counts using melanopsin staining, and there is the remote possibility that the AD degenerative process might reduce melanopsin expression. Finally, neuropathological data were not available for all controls, but dementia was an exclusion criterion for controls.
Overall, our study confirms the presence of optic neuropathy and hallmarks of AD pathology in the retina and shows, for the first time, the additional specific AD‐related pathology affecting mRGCs in AD. The mRGC loss may provide the anatomical basis for a contributing factor to the pathogenesis of circadian rhythm dysfunction in AD. The understanding of the mRGC–hypothalamic contribution to circadian dysfunction in AD is particularly important for the development of future strategies aimed at counteracting circadian derangement in AD patients, such as a specifically designed light therapy.73 This is relevant considering that there is evidence that circadian dysfunction is a strong predictor of dementia in the elderly74 and robust circadian photoentrainment may prime longevity as well as quality of life.75
Author Contributions
C.L.M., F.N.R.‐C., and Y.K. contributed equally to the study. Conception and design of the study: C.L.M., F.N.R.‐C., Y.K., A.A.S., V.C.; acquisition and analysis of data: C.L.M., F.N.R.‐C., Y.K., J.H., R.G., G.C., L.S., B.X.P., K.R.T., P.B., F.P., P.A., M.C., A.P., H.C., R.L., A.B., M.K.‐H., A.A.S., V.C.; drafting the manuscript or figures: C.L.M., F.N.R.‐C., Y.K., J.H., G.C., M.K.‐H., A.A.S., V.C.
Potential Conflicts of Interest
Nothing to report.
Dedication
This work is dedicated to the memory of Professor Pasquale Montagna, who was a pioneer in promoting this study.
Acknowledgment
This work was supported by the Gino Galletti Foundation (V.C.); Research to Prevent Blindness, the International Foundation for Optic Nerve Diseases (IFOND), and NIH National Eye Institute grant EY03040 (F.N.R.‐C., A.A.S.); NIH National Institute on Aging grant P50‐AG05142 (USC ADRC Neuropathology Core Grant; A.A.S.); NIH National Institute on Aging grant R41 AG044897‐01 (M.K.‐H.); the Marciano Family Foundation and Cheryl and Haim Saban Family Foundation (M.K.‐H.); and the Danish Biotechnology Center for Cellular Communication (J.H.).
We thank Drs. F. Pizza and G. Plazzi for their support in the actigraphic studies; Drs. C. Miller and P. Parchi for their advice on postmortem studies; Dr. C. Zenesini, J. Tran, and L. Dustin for statistical consultation; C. Church and J. Espinosa for administrative support; E. Barron and Mario M. Schunimann for technical support; A. Hansen for excellent technical assistance during the IHC staining of melanopsin; and the patients and their families for their gracious participation in this study.
References
- 1. Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 2010;362:329–344. [DOI] [PubMed] [Google Scholar]
- 2. Wu YH, Swaab DF. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer's disease. Sleep Med 2007;8:623–636. [DOI] [PubMed] [Google Scholar]
- 3. Hatfield CF, Herbert J, van Someren EJ, et al. Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home‐dwelling patients with early Alzheimer's dementia. Brain 2004;127:1061–1074. [DOI] [PubMed] [Google Scholar]
- 4. Dowling GA, Burr RL, Van Someren EJ, et al. Melatonin and bright‐light treatment for rest‐activity disruption in institutionalized patients with Alzheimer's disease. J Am Geriatr Soc 2008;56:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swaab DF, Fliers E, Partiman TS. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res 1985;342:37–44. [DOI] [PubMed] [Google Scholar]
- 6. Stopa EG, Volicer L, Kuo‐Leblanc V, et al. Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J Neuropathol Exp Neurol 1999;58:29–39. [DOI] [PubMed] [Google Scholar]
- 7. Harper DG, Stopa EG, Kuo‐Leblanc V, et al. Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain 2008;131:1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dowling GA, Hubbard EM, Mastick J, et al. Effect of morning bright light treatment for rest‐activity disruption in institutionalized patients with severe Alzheimer's disease. Int Psychogeriatr 2005;17:221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic‐nerve degeneration in Alzheimer's disease. N Engl J Med 1986;315:485–487. [DOI] [PubMed] [Google Scholar]
- 10. Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer's disease. Brain Res 1989;501:364–372. [DOI] [PubMed] [Google Scholar]
- 11. Sadun AA, Bassi CJ. Optic nerve damage in Alzheimer's disease. Ophthalmology 1990;97:9–17. [DOI] [PubMed] [Google Scholar]
- 12. Iseri PK, Altinaş O, Tokay T, Yüksel N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J Neuroophthalmol 2006;26:18–24. [DOI] [PubMed] [Google Scholar]
- 13. Berisha F, Feke GT, Trempe CL, et al. Retinal abnormalities in early Alzheimer's disease. Invest Ophthalmol Vis Sci 2007;48:2285–2289. [DOI] [PubMed] [Google Scholar]
- 14. Paquet C, Boissonnot M, Roger F, et al. Abnormal retinal thickness in patients with mild cognitive impairment and Alzheimer's disease. Neurosci Lett 2007;420:97–99. [DOI] [PubMed] [Google Scholar]
- 15. Lu Y, Li Z, Zhang X, et al. Retinal nerve fiber layer structure abnormalities in early Alzheimer's disease: evidence in optical coherence tomography. Neurosci Lett 2010;480:69–72. [DOI] [PubMed] [Google Scholar]
- 16. Kesler A, Vakhapova V, Korczyn AD, et al. Retinal thickness in patients with mild cognitive impairment and Alzheimer's disease. Clin Neurol Neurosurg 2011;113:523–526. [DOI] [PubMed] [Google Scholar]
- 17. Moschos MM, Markopoulos I, Chatziralli I, et al. Structural and functional impairment of the retina and optic nerve in Alzheimer's disease. Curr Alzheimer Res 2012;9:782–788. [DOI] [PubMed] [Google Scholar]
- 18. Kirbas S, Turkyilmaz K, Anlar O, et al. Retinal nerve fiber layer thickness in patients with Alzheimer disease. J Neuroophthalmol 2013;33:58–61. [DOI] [PubMed] [Google Scholar]
- 19. Marziani E, Pomati S, Ramolfo P, et al. Evaluation of retinal nerve fiber layer and ganglion cell layer thickness in Alzheimer's disease using spectral‐domain optical coherence tomography. Invest Ophthalmol Vis Sci 2013;54:5953–5958. [DOI] [PubMed] [Google Scholar]
- 20. Kromer R, Serbecic N, Hausner L, et al. Detection of retinal nerve fiber layer defects in Alzheimer's disease using SD‐OCT. Front Psychiatry 2014;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koronyo‐Hamaoui M, Koronyo Y, Ljubimov AV, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011;54(suppl 1):S204–S217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer's disease. Neuroreport 2011;22:623–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsai Y, Lu B, Ljubimov AV, et al. Ocular changes in TgF344‐AD rat model of Alzheimer's disease. Invest Ophthalmol Vis Sci 2014;55:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koronyo Y, Salumbides BC, Black KL, Koronyo‐Hamaoui M. Alzheimer's disease in the retina: imaging retinal Aβ plaques for early diagnosis and therapy assessment. Neurodegener Dis 2012;10:285–293. [DOI] [PubMed] [Google Scholar]
- 25. Ning A, Cui J, To E, et al. Amyloid‐beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci 2008;49:5136–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perez SE, Lumayag S, Kovacs B, et al. Beta‐amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest Ophthalmol Vis Sci 2009;50:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiu K, Chan TF, Wu A, et al. Neurodegeneration of the retina in mouse models of Alzheimer's disease: what can we learn from the retina? Age (Dordr) 2012;34:633–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hattar S, Liao HW, Takao M, et al. Melanopsin‐containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science 2002;295:1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science 2002;295:1070–1073. [DOI] [PubMed] [Google Scholar]
- 30. Sand A, Schmidt TM, Kofuji P. Diverse types of ganglion cell photoreceptors in the mammalian retina. Prog Retin Eye Res 2012;31:287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schmidt TM, Chen SK, Hattar S. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci 2011;34:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sadun AA, Schaechter JD, Smith LE. A retinohypothalamic pathway in man: light mediation of circadian rhythms. Brain Res 1984;302:371–377. [DOI] [PubMed] [Google Scholar]
- 33. Hannibal J. Neurotransmitters of the retino‐hypothalamic tract. Cell Tissue Res 2002;309:73–88. [DOI] [PubMed] [Google Scholar]
- 34. Hannibal J, Fahrenkrug J. Neuronal input pathways to the brain's biological clock and their functional significance. Adv Anat Embryol Cell Biol 2006;182:1–71. [PubMed] [Google Scholar]
- 35. Semo M, Peirson S, Lupi D, et al. Melanopsin retinal ganglion cells and the maintenance of circadian and pupillary responses to light in aged rodless/coneless (rd/rd cl) mice. Eur J Neurosci 2003;17:1793–1801. [DOI] [PubMed] [Google Scholar]
- 36. La Morgia C, Ross‐Cisneros FN, Sadun AA, et al. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain 2010;133:2426–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feigl B, Mattes D, Thomas R, Zele AJ. Intrinsically photosensitive (melanopsin) retinal ganglion cell function in glaucoma. Invest Ophthalmol Vis Sci 2011;52:4362–4367. [DOI] [PubMed] [Google Scholar]
- 38. La Morgia C, Ross‐Cisneros FN, Hannibal J, et al. Melanopsin‐expressing retinal ganglion cells: implications for human diseases. Vision Res 2011;51:296–302. [DOI] [PubMed] [Google Scholar]
- 39. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 40. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 41. Barboni P, Savini G, Valentino ML, et al. Retinal nerve fiber layer evaluation by optical coherence tomography in Leber's hereditary optic neuropathy. Ophthalmology 2005;112:120–126. [DOI] [PubMed] [Google Scholar]
- 42. Witting W, Kwa IH, Eikelenboom P, et al. Alterations in the circadian rest‐activity rhythm in aging and Alzheimer's disease. Biol Psychiatry 1990;27:563–572. [DOI] [PubMed] [Google Scholar]
- 43. Van Someren EJ, Swaab DF, Colenda CC, et al. Bright light therapy: improved sensitivity to its effects on rest‐activity rhythms in Alzheimer patients by application of nonparametric methods. Chronobiol Int 1999;16:505–518. [DOI] [PubMed] [Google Scholar]
- 44. Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997;56:1095–1097. [DOI] [PubMed] [Google Scholar]
- 45. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 46. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging 1995;16:271–278. [DOI] [PubMed] [Google Scholar]
- 47. Hannibal J, Hindersson P, Ostergaard J, et al. Melanopsin is expressed in PACAP‐containing retinal ganglion cells of the human retinohypothalamic tract. Invest Ophthalmol Vis Sci 2004;45:4202–4209. [DOI] [PubMed] [Google Scholar]
- 48. Hannibal J, Kankipati L, Strang CE, et al. Central projections of intrinsically photosensitive retinal ganglion cells in the macaque monkey. J Comp Neurol 2014;522:2231–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dacey DM, Liao HW, Peterson BB, et al. Melanopsin‐expressing ganglion cells in primate retina signal colour and irradiance and project to the LGN. Nature 2005;433:749–754. [DOI] [PubMed] [Google Scholar]
- 50. Coggeshall RE, Lekan HA. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol 1996;364:6–15; erratum 1996;369:162. [DOI] [PubMed] [Google Scholar]
- 51. Saper CB. Any way you cut it: a new journal policy for the use of unbiased counting methods. J Comp Neurol 1996;364:5. [DOI] [PubMed] [Google Scholar]
- 52. Guillery RW. On counting and counting errors. J Comp Neurol 2002;447:1–7. [DOI] [PubMed] [Google Scholar]
- 53. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging; Alzheimer's Association. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pirici D, Mogoanta L, Kumar‐Singh S, et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J Histochem Cytochem 2009;57:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 2007;39:175–191. [DOI] [PubMed] [Google Scholar]
- 56. Trick GL, Trick LR, Morris P, Wolf M. Visual field loss in senile dementia of the Alzheimer's type. Neurology 1995;45:68–74. [DOI] [PubMed] [Google Scholar]
- 57. Quigley HA, Dunkelberger GR, Green WR. Chronic human glaucoma causing selectively greater loss of large optic nerve fibers. Ophthalmology 1988;95:357–363. [DOI] [PubMed] [Google Scholar]
- 58. Sadun AA, Johnson B, Miao M. Axon caliber populations in the human optic nerve: changes with age and disease In: Ishikawa S, Mukuno K, Uga S, Van Dalen JTW, eds. Highlights in neuro‐ophthalmology: proceedings of the sixth meeting of the International Neuro‐Ophthalmology Society (INOS), Hakone, Japan, 8–14 June 1986. Amsterdam, the Netherlands: Aeolus, 1987:15–20. [Google Scholar]
- 59. Bayer AU, Ferrari F, Erb C. High occurrence rate of glaucoma among patients with Alzheimer's disease. Eur Neurol 2002;47:165–168. [DOI] [PubMed] [Google Scholar]
- 60. Johnson BM, Miao M, Sadun AA. Age‐related decline of human optic nerve axon populations. Age 1987;10:5–9. [Google Scholar]
- 61. Budenz DL, Anderson DR, Varma R, et al. Determinants of normal retinal nerve fiber layer thickness measured by Stratus OCT. Ophthalmology 2007;114:1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hooghiemstra AM, Eggermont LH, Scheltens P, et al. The Rest‐Activity Rhythm and Physical Activity in Early‐Onset Dementia. Alzheimer Dis Assoc Disord 2015;29:45–49. [DOI] [PubMed] [Google Scholar]
- 63. Satlin A, Volicer L, Stopa EG, Harper D. Circadian locomotor activity and core‐body temperature rhythms in Alzheimer's disease. Neurobiol Aging 1995;16:765–771. [DOI] [PubMed] [Google Scholar]
- 64. Van Someren EJ, Hagebeuk EE, Lijzenga C, et al. Circadian rest‐activity rhythm disturbances in Alzheimer's disease. Biol Psychiatry 1996;40:259–270. [DOI] [PubMed] [Google Scholar]
- 65. Volicer L, Harper DG, Manning BC, et al. Sundowning and circadian rhythms in Alzheimer's disease. Am J Psychiatry 2001;158:704–711. [DOI] [PubMed] [Google Scholar]
- 66. Prinz PN, Larsen LH, Moe KE, Vitiello MV. EEG markers of early Alzheimer's disease in computer selected tonic REM sleep. Electroencephalogr Clin Neurophysiol 1992;83:36–43. [DOI] [PubMed] [Google Scholar]
- 67. Moe KE, Vitiello MV, Larsen LH, Prinz PN. Symposium: Cognitive processes and sleep disturbances: sleep/wake patterns in Alzheimer's disease: relationships with cognition and function. J Sleep Res 1995;4:15–20. [DOI] [PubMed] [Google Scholar]
- 68. Wu YH, Fischer DF, Kalsbeek A, et al. Pineal clock gene oscillation is disturbed in Alzheimer's disease, due to functional disconnection from the “master clock”. FASEB J 2006;20:1874–1876. [DOI] [PubMed] [Google Scholar]
- 69. Moura AL, Nagy BV, La Morgia C, et al. The pupil light reflex in Leber's hereditary optic neuropathy: evidence for preservation of melanopsin‐expressing retinal ganglion cells. Invest Ophthalmol Vis Sci 2013;54:4471–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fotiou DF, Stergiou V, Tsiptsios D, et al. Cholinergic deficiency in Alzheimer's and Parkinson's disease: evaluation with pupillometry. Int J Psychophysiol 2009;73:143–149. [DOI] [PubMed] [Google Scholar]
- 71. Schön C, Hoffmann NA, Ochs SM, et al. Long‐term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLoS One 2012;7:e53547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lim AS, Ellison BA, Wang JL, et al. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain 2014;137:2847–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Forbes D, Blake CM, Thiessen EJ, et al. Light therapy for improving cognition, activities of daily living, sleep, challenging behaviour, and psychiatric disturbances in dementia. Cochrane Database Syst Rev 2014;2:CD003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tranah GJ, Blackwell T, Stone KL, et al; SOF Research Group . Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann Neurol 2011;70:722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Froy O. Circadian rhythms, aging, and life span in mammals. Physiology (Bethesda) 2011;26:225–235. [DOI] [PubMed] [Google Scholar]