

Abstract
Sugar phosphates play an important role in metabolism and signaling, but also as constituents of macromolecular structures. Selective phosphorylation of sugars is chemically difficult, particularly at the anomeric center. We report phosphatase‐catalyzed diastereoselective “anomeric” phosphorylation of various aldose substrates with α‐d‐glucose 1‐phosphate, derived from phosphorylase‐catalyzed conversion of sucrose and inorganic phosphate, as the phosphoryl donor. Simultaneous and sequential two‐step transformations by the phosphorylase–phosphatase combination catalyst yielded glycosyl phosphates of defined anomeric configuration in yields of up to 70 % based on the phosphate applied to the reaction. An efficient enzyme‐assisted purification of the glycosyl phosphate products from reaction mixtures was established.
Keywords: biocatalysis, phosphatases, phosphorylases, phosphorylation, sugar phosphates
Phosphorylated carbohydrates constitute an important class of biomolecules.1 They are intermediates in metabolism and play central roles in signaling. Macromolecular structures, including the genome, are also built from sugar phosphates.1 Technological uses include applications in the food, cosmetic and detergent sectors.2, 3 Moreover they are used as precursors for the synthesis of nucleotide‐activated sugars2d, 4a and as intermediates in rare sugar synthesis.2e,f Therefore, convenient synthetic routes to sugar phosphates is of importance across disciplines, and selective phosphorylation of simple sugar substrates presents a strong option. However, the inherent structural complexity of sugars makes selective insertion of a phosphoryl group difficult.
During synthesis of glycosyl phosphates, stereocontrol at the anomeric center is a problem that requires special attention. Chemical methodologies normally require multiple steps,4 even if hydroxy‐protecting groups on the sugar are avoided.5 Most of these syntheses involve reactions with moderate yields and are limited to the formation of only a few different glycosyl phosphates. A number of glycosyl phosphates have been obtained effectively from the corresponding β‐glycosylsulfonohydrazides, which in turn were derived directly from free sugar hemiacetals. However, diastereoselectivity was only sufficient to obtain products enriched in the α‐anomer.5 Nucleoside triphosphate (NTP)‐dependent sugar 1‐kinases, which catalyze phosphorylation of the anomeric hydroxy group with precise α‐selectivity, have proven useful to overcome issues of stereocontrol.6 However relatively narrow substrate specificity (e.g. d‐Gal,7a l‐Fuc,7b d‐GalNAc/d‐GlcNAc,7c d‐GlcUA,7d d‐GalUA,7e l‐Ara7f) has essentially restricted the synthetic use of these enzymes to the phosphorylation of physiological sugar substrate(s) or close structural analogues. Only recently have sugar 1‐kinases with more relaxed substrate spectrum been discovered8 or alternatively derived through protein engineering.9 Chen and co‐workers applied d‐GalNAc/d‐GlcNAc kinase and d‐Gal kinase for the synthesis of α‐glycosyl phosphates of d‐Gal, d‐Glc, and d‐Man, based on their respective substrate preferences. Derivatives of the three sugars with the 2‐OH group substituted with H, F, NH2, NHAc, or N3 were also phosphorylated.10
Important advances made with “promiscuous” sugar 1‐kinases notwithstanding, there still exists gap in the scope of sugar substrates that can be phosphorylated by these enzymes with good activity. Moreover, it is not economical to use NTP phosphoryl donors in stoichiometric amounts, and despite notable developments in scaling up kinase‐catalyzed sugar phosphorylations to the gram scale,11 the requirement for their (enzymatic) regeneration adds complexity to the overall transformation.11, 12
Therefore, an alternative method of biocatalytic phosphorylation that combines the key feature of stereoselectivity with the additional advantages of broad substrate acceptance would be very useful, especially when the preparation of α‐glycosyl phosphates as commodity chemicals is considered. The use of a phosphoryl‐group donor more convenient than NTP could further improve the procedure. We herein describe a phosphorylase–phosphatase combination catalyst for the anomeric‐center phosphorylation of aldose sugars from inorganic phosphate (Scheme 1).
Scheme 1.
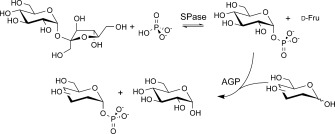
Diastereoselective synthesis of glycosyl phosphates by a sucrose phosphorylase (SPase) and glucose 1‐phosphatase (AGP) combination catalyst.
The overall two‐step conversion proceeded via the activated phosphoryl donor α‐d‐glucose 1‐phosphate (αGlc 1‐P), which was derived from sucrose and phosphate in a thermodynamically favored reaction catalyzed by sucrose phosphorylase (from Leuconostoc mesenteroides; SPase; EC 2.4.1.7).13 αGlc 1‐P was then utilized directly in a selective transphosphorylation, in which the sugar acceptor was reacted in the presence of a suitable phosphomonoester hydrolase (glucose 1‐phosphatase (AGP); EC 3.1.3.10). The large number of phosphatases encoded by the genome and the often strongly overlapping substrate specificities of these enzymes14 makes the dedicated selection of candidate biocatalysts a challenging task.
Expedient synthesis of several aldohexose phosphates by phosphatase‐catalyzed transphosphorylation from pyrophosphate has been reported.15 However, the phosphorylation was always at C6 and never at the anomeric center. αGlc 1‐P is hydrolyzed by various phosphatases but until recently,16 its use as a phosphoryl donor for enzymatic synthesis was not considered. We speculated that phosphatases active in the cleavage of αGlc 1‐P might be also efficient and specific in synthesizing phosphomonoesters with the anomeric sugar hydroxy group. Three sugar phosphate phosphatases from Escherichia coli (AGP, HAD4, HAD13; see the Supporting Information) that have been reported to use αGlc 1‐P as their preferred substrate for hydrolysis14, 17 were tested for phosphorylation of different sugars (see below, 100–200 mm) in the presence of αGlc 1‐P (20 mm). Reactions were followed by monitoring αGlc 1‐P consumption, phosphate release, and formation of the phosphorylated product(s) (see Methods in the Supporting Information). HAD4 and HAD13 converted αGlc 1‐P exclusively through hydrolysis and thus proved useless for synthesis. By contrast, AGP was active with various acceptors (e.g. d‐Man) and utilized a substantial portion (≥50 %) of the offered donor substrate for sugar phosphate synthesis. Preliminary product analysis of the AGP reaction with d‐Man revealed that the phosphorylation product was not d‐mannose 6‐phosphate (Man 6‐P). This indicates that AGP might exhibit a selectivity strongly divergent from the phosphatases described in the literature.15
AGP is a member of the histidine acid phosphatase protein family.18 Its proposed mechanism involves a double nucleophilic substitution and proceeds via a covalent phosphohistidine enzyme intermediate.17 Transphosphorylation is explained by partitioning of the phosphorylated AGP between reactions with the acceptor and water, as shown in Scheme 2.16 Sugar phosphate synthesis by AGP therefore occurs under kinetic control and its efficiency is linked to key enzyme catalytic properties: stereoselectivity during enzyme dephosphorylation by the sugar acceptor; low hydrolysis of the phosphoenzyme intermediate; and low hydrolysis of the synthesis product.16
Scheme 2.
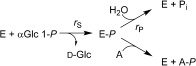
Kinetic scheme of AGP‐catalyzed transphosphorylation. A is the sugar acceptor and Pi is phosphate.
Purified AGP (0.2 U mL−1) was applied to the phosphorylation (37 °C; pH 7.0) of different monosaccharides (200 mm; d‐Man, d‐Gal, d‐GlcNAc, d‐GlcNH2, d‐GalNH2, d‐GlcUA, l‐Fuc, d‐Xyl, d‐Ara, and l‐Ara) from αGlc 1‐P (20 mm). Xylitol (Xol) was also used. Reaction time courses are shown in Figure S2 in the Supporting Information. Except for d‐GlcUA and d‐GalNH2, which were not accepted by AGP, all other compounds were active. Extensive 1D and 2D NMR analyses (1H, 31P) conducted directly on the reaction mixtures and on recovered compounds revealed the identity of the phosphoryl transfer products (Figures S3–S7, Tables S2, S3). AGP phosphorylated d‐Man, d‐GlcNAc, d‐Gal, and l‐Fuc at the anomeric hydroxy group, and it did so with absolute axial selectivity to yield the corresponding α‐d‐ or β‐l‐glycosyl phosphate products (Figures S3–S6). Interestingly, d‐GlcNH2 was phosphorylated primarily at the 6‐OH (Figure S7). Xol, d‐Xyl, d‐Ara, and l‐Ara were also phosphorylated.
The efficiency with which each sugar acceptor was phosphorylated was evaluated by using the rate ratio of αGlc 1‐P consumption (r S) and phosphate release (r P) as the key parameter (Scheme 2). Under hydrolysis conditions, r S/r P has a value of unity. Phosphoryl transfer to the acceptor is indicated by an r S/r P value exceeding unity, and Figure 1 shows the different acceptors ranked according to r S/r P .
Figure 1.
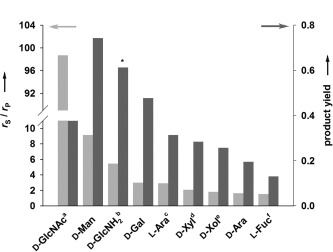
AGP‐catalyzed phosphorylation of different acceptor substrates (200 mm) from αGlc 1‐P (20 mm). Product yield was determined at a αGlc 1‐P conversion of ≥90 %, except for [a] 38 %, [b] 81 %, [c] 69 %, [d] 86 %, [e] 89 % and [f] 75 %. The asterisk (*) indicates the total amount of sugar 1‐phosphate products (GlcNH2 1‐P, GlcNH2 6‐P), which was used to calculate the yield.
As expected from Scheme 2, the r S/r P and the yield of phosphorylated product were both dependent on the amount of acceptor used in the reaction (Figure S8). Given that working at a low donor/acceptor ratio is not practical synthetically, it is convenient that donor and acceptor were effectively co‐utilized at high substrate concentrations. This is shown in Figure 2 (and the associated Figure S9) for the example of αMan 1‐P synthesis. Up to 400 mm (ca. 100 g L−1) of phosphorylated product was obtained from approximately 1 m each of αGlc 1‐P and d‐Man.
Figure 2.
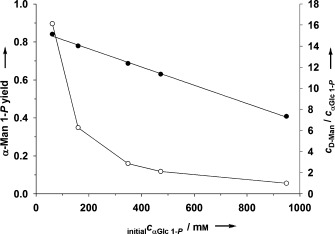
AGP‐catalyzed synthesis of αMan 1‐P at different concentrations of αGlc 1‐P donor and d‐Man acceptor. (○) αMan 1‐P yield, (•) c D‐Man/c αGlc 1‐P . The yield of αMan 1‐P was determined after 120 min. αGlc 1‐P conversion decreased from 95 % to 52 % with increasing initial concentration of the donor substrate.
High substrate concentrations were also useful to achieve control over two side reactions in the overall transformation, namely secondary hydrolysis of the phosphorylated sugar synthesized and formation of d‐glucose 6‐phosphate (Glc 6‐P) from the d‐Glc released through αGlc 1‐P conversion. Generally, the products of phosphoryl transfer by AGP exhibited adequate kinetic stability to enable their convenient production (Figure S2). However, the concentrations of αGal 1‐P, αMan 1‐P, and β‐l‐Fuc 1‐P decreased at extended incubation times, so timely stopping of the reaction was important (Figure S2). This was confirmed by in situ 31P NMR monitoring of the enzymatic αMan 1‐P synthesis (Figure S10). Up to the optimum reaction time (approximately 300 min), target product was formed in large excess over hydrolysis and alternative transphosphorylation products. Product titer and purity decreased afterwards. It could also be shown (Figure S9) that the degradation of αMan 1‐P was strongly suppressed at high substrate concentrations (≥400 mm). Concerning C6 phosphorylation of d‐Glc, the relative amount of Glc 6‐P in the total transphosphorylation product was dependent on r S/r P and was typically well below 10 % for the good acceptors (r S/r P≥3; Figure S11). Use of the acceptor in suitable excess over the donor was effective in minimizing Glc 6‐P formation.
Next, sugar phosphate synthesis was examined by using the combination catalytic cascade from Scheme 1. Conversions were performed with the phosphorylase (SPase) and phosphatase (AGP) reactions run sequentially or simultaneously in one pot. d‐Man, d‐Gal, and d‐GlcNAc (1 m) were phosphorylated from sucrose and phosphate (100 mm each; Figure 3 and Figure S12). Based on the phosphate applied to the enzymatic conversion, the yield of total phosphorylated product was around 50–70 % depending on the phosphate acceptor substrate used (Figure 3 and Figure S12). Time courses for αMan 1‐P synthesis are depicted in Figure 3. The sequential‐mode reaction (Figure 3 a) involved equilibrium controlled synthesis of αGlc 1‐P (150 mm; ca. 80 % substrate conversion) followed by phosphorylation of d‐Man after the addition of AGP and acceptor. αGlc 1‐P was used up completely in the reaction, and roughly the equivalent amount of αMan 1‐P was synthesized. Formation of Glc 6‐P was low (≤6 %) and phosphorylated d‐Fru16 was below the detection limit. Phosphorylated sucrose was not observed. The concentration of free phosphate decreased slightly during the AGP reaction in Figure 3 a, thus indicating that SPase‐catalyzed conversion of sucrose continued owing to gradual depletion of the αGlc 1‐P.
Figure 3.
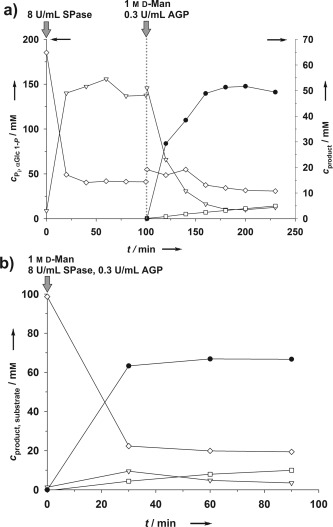
Synthesis of αMan 1‐P (•) by using the phosphorylase–phosphatase combination catalyst in sequential (a) and simultaneous (b) reaction modes. Glc 1‐P (▿), Glc 6‐P (□), and phosphate (◊) are also indicated.
Interestingly, the simultaneous reaction of SPase and AGP was also effective, giving rapid phosphorylation of d‐Man (Figure 3 b) in a yield comparable to that of the sequential reaction. To prevent αMan 1‐P degradation, as well as gradual accumulation of Glc 6‐P, control of the reaction time was important in both reaction modes. The activity ratio of phosphorylase and phosphatase was also important: phosphorylase had to be present in suitable excess (≥20‐fold) over AGP. Because of shortened overall reaction time, the space–time yield of αMan 1‐P in simultaneous mode exceeded that of the sequential mode by a factor of 5 or more (Figure 3).
Separation of the glycosyl phosphate product from minor amounts of other sugar phosphates (e.g., Glc 6‐P) present in the reaction mixture was an important task for product isolation. An efficient procedure was developed in which selective enzymatic hydrolysis of undesired sugar phosphates constitutes the key step. The phosphatase BT4131 from Bacteroides thetaiotaomicron was selected. Kinetic studies in the literature19 and “competition experiments” performed herein (Figure S13) suggest that this enzyme effectively discriminates between sugar phosphates phosphorylated at the primary and the anomeric hydroxy groups. Indeed, complete removal of phosphorylated byproducts from the reaction mixture was achieved, as shown for αMan 1‐P, αGlcNAc 1‐P, and αGal 1‐P production. Authentic glycosyl phosphates (Figures S14–S19, Tables S4, S5) were obtained after anion‐exchange chromatography and precipitation as a barium salt. From a single small‐scale reaction (1.5 mL), around 40 mg product was recovered in useful purity (Table S5).
In summary, the diastereoselective synthesis of glycosyl phosphates by phosphatase‐catalyzed transphosphorylation of unprotected aldose sugars is reported. Targeted identification of the enzyme was key, and AGP is a unique phosphatase for glycosyl phosphate synthesis. A two‐step cascade reaction based on a phosphorylase–phosphatase combination catalyst was developed to enable sugar phosphorylation at the anomeric center from inorganic phosphate and sucrose via a αGlc 1‐P intermediate. Various structurally defined glycosyl phosphates were thus obtained in good yields of up to 70 % (based on the phosphate or αGlc 1‐P applied to the reaction). An enzyme‐assisted work‐up method was developed to isolate glycosyl phosphates from the reaction mixtures. This method provides a promising new route to defined glycosyl phosphates as fine or bulk chemicals and complements the reported kinase‐catalyzed syntheses of valuable glycosyl phosphate products.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
Financial support from the Austrian Science Fund FWF (FWF projects L‐586‐B03 and W901‐B05 to B.N.) is gratefully acknowledged. Prof. Hansjörg Weber (Institute of Organic Chemistry, TU Graz) performed NMR analyses of isolated glycosyl phosphates. Prof. Karen N. Allen (Department of Chemistry, Boston, United States) kindly provided the plasmid pET‐3A‐042 encoding the phosphatase BT4131.
References
- 1.
- 1a. Westheimer F. H., Science 1987, 235, 1173–1178; [DOI] [PubMed] [Google Scholar]
- 1b. Kamerlin S. C., Sharma P. K., Prasad R. B., Warshel A., Q. Rev. Biophys. 2013, 46, 1–132; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Nikolaev A. V., Botvinko I. V., Ross A. J., Carbohydr. Res. 2007, 342, 297–344. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Auriol D., Lefevre F., Nalin R., Redziniak G., Cosmetic and Pharmaceutical Composition Comprising N‐Acetylglucosamine‐6‐Phosphate, 2011, US 20130012475;
- 2b. Woodyer R., Taylor P., Demirjian D., Activated Sugars, 2011, US 20140024082 A1;
- 2c. de Groeve M. R., Depreitere V., Desmet T., Soetaert W., Biotechnol. Lett. 2009, 31, 1873–1877; [DOI] [PubMed] [Google Scholar]
- 2d. Bülter T., Elling L., Glycoconjugate J. 1999, 16, 147–159; [DOI] [PubMed] [Google Scholar]
- 2e. Wen L., Huang K., Wei M., Meisner J., Liu Y., Garner K., Zang L., Wang X., Li X., Fang J., Zhang H., Wang P. G., Angew. Chem. Int. Ed. 2015, 54, 12654–12658; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12845–12849; [Google Scholar]
- 2f. Clapés P., Fessner W.‐D., Sprenger G. A., Samland A. K., Curr. Opin. Chem. Biol. 2010, 14, 154–167. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Woo‐Jung S., Kim B.‐Y., Bang W.‐G., J. Microbiol. Biotechnol. 2007, 17, 769–773; [PubMed] [Google Scholar]
- 3b. Ishikawa K., Mihara Y., Shimba N., Ohtsu N., Kawasaki H., Suzuki E., Asano Y., Protein Eng. 2002, 15, 539–543. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Wagner G. K., Pesnot T., Field R. A., Nat. Prod. Rep. 2009, 26, 1172–1194; [DOI] [PubMed] [Google Scholar]
- 4b. Pale P., Whitesides G. M., J. Am. Chem. Soc. 1991, 56, 4547–4549; [Google Scholar]
- 4c. Sim M. M., Kondo H., Wong C.‐H., J. Am. Chem. Soc. 1993, 115, 2260–2267; [Google Scholar]
- 4d. Arlt M., Hindsgaul O., J. Org. Chem. 1995, 60, 14–15; [Google Scholar]
- 4e. Dohi H., et al., Vincent S. P., Chem. Eur. J. 2008, 14, 9530–9539; [DOI] [PubMed] [Google Scholar]
- 4f. Plante O. J., Andrade R. B., Seeberger P. H., Org. Lett. 1999, 1, 211–214; [DOI] [PubMed] [Google Scholar]
- 4g. Patel M. K., Davis B. J., Org. Lett. 2013, 15, 346–349; [DOI] [PubMed] [Google Scholar]
- 4h. Timmons S. C., Jakeman D. L., Carbohydr. Res. 2008, 343, 865–874; [DOI] [PubMed] [Google Scholar]
- 4i. Boons G.‐Y., Burton A., Wyatt P., Synlett 1996, 310–312. [Google Scholar]
- 5. Edgar L. J. G., Dasgupta S., Nitz M., Org. Lett. 2012, 14, 4226–4229. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Yang J., et al., Org. Lett. 2003, 5, 2223–2226; [DOI] [PubMed] [Google Scholar]
- 6b. Park S. H., Pastusza I., Drake R., Elbein A. D., J. Biol. Chem. 1998, 273, 5685–5691; [DOI] [PubMed] [Google Scholar]
- 6c. Nishimoto M., Kitaoka M., Appl. Environ. Microbiol. 2007, 73, 6444–6449; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Pieslinger A. M., Hoepflinger M. C., Tenhaken R., J. Biol. Chem. 2010, 285, 2902–2910; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Yang T., Bar‐Peled L., Gebhart L., Lee S. G., Bar‐Peled M., J. Biol. Chem. 2009, 284, 21526–21535; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Sherson S., Gy I., Medd J., Schmidt R., Dean C., Kreis M., Lecharny A., Cobbett C., Plant Mol. Biol. 1999, 39, 1003–1012. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Zou Y., Wang W., Cai L., Chen L., Xue M., Zhang X., Shen J., Chen M., Bioorg. Med. Chem. Lett. 2012, 22, 3540–3543; [DOI] [PubMed] [Google Scholar]
- 7b. Bourgeaux V., Piller F., Piller V., Bioorg. Med. Chem. Lett. 2005, 15, 5459–5462; [DOI] [PubMed] [Google Scholar]
- 7c. Chen M., et al., Carbohydr. Res. 2011, 346, 2421–2425; [DOI] [PubMed] [Google Scholar]
- 7d. Li L., Liu Y., Wang W., Cheng J., Zhao W., Wang P., Carbohydr. Res. 2012, 355, 35–39. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Li Y., et al., Molecules 2011, 16, 6396–6407; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Cai L., Guan W., Kitaoka M., Shen J., Xia C., Chen W., Wang P. G., Chem. Commun. 2009, 2944–2946. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hoffmeister D., Yang J., Liu L., Thorson J. S., Proc. Natl. Acad. Sci. USA 2003, 100, 13184–13189; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Hoffmeister D., Thorson J. S., ChemBioChem 2004, 5, 989–992; [DOI] [PubMed] [Google Scholar]
- 9c. Yang J., Fu X., Liao J., Liu L., Thorson J. S., Chem. Biol. 2005, 12, 657–664. [DOI] [PubMed] [Google Scholar]
- 10. Muthana M. M., Qu J., Li Y., Zhang L., Yu H., Ding L., Malekan H., Chen X., Chem. Commun. 2012, 48, 2728–2730. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y., Nishimoto M., Kitaoka M., Carbohydr. Res. 2015, 401, 1–4. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Berke W., Schüz H. J., Wandrey C., Morr M., Denda G., Kula M. R., Biotechnol. Bioeng. 1988, 32, 130–139; [DOI] [PubMed] [Google Scholar]
- 12b. Zhao H., van der Donk W. A., Curr. Opin. Biotechnol. 2003, 14, 583–589. [DOI] [PubMed] [Google Scholar]
- 13. Goedl C., Schwarz A., Minani A., Nidetzky B., J. Biotechnol. 2007, 129, 77–86. [DOI] [PubMed] [Google Scholar]
- 14. Kuznetsova E., et al., J. Biol. Chem. 2006, 281, 36149–36161. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. van Herk T., Hartog A. F., van der Burg A. M., Wever R., Adv. Synth. Catal. 2005, 347, 1155–1162; [Google Scholar]
- 15b. Tanaka N., Hasan Z., Hartog A. F., van Herk T., Wever R., Sanders R. J., Org. Biomol. Chem. 2003, 1, 2833–2839. [DOI] [PubMed] [Google Scholar]
- 16. Wildberger P., Pfeiffer M., Brecker L., Rechberger G. N., Birner‐Gruenberger R., Nidetzky B., Appl. Environ. Microbiol. 2015, 81, 1559–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee D. C., Cottrill M. A., Forsberg C. W., Jia Z., J. Biol. Chem. 2003, 278, 31412–31418. [DOI] [PubMed] [Google Scholar]
- 18. Chi H., Tiller G. E., Dasouki M. J., Romano P. R., Wang J., O’Keefe J., Puzas J. E., Rosier R. N., Reynolds P. R., Genomics 1999, 56, 324–336. [DOI] [PubMed] [Google Scholar]
- 19. Lu Z., Dunaway‐Mariano D., Allen K. N., Biochemistry 2005, 44, 8684–8696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information