

Abstract
To clarify molecular alterations in serrated pathway of colorectal cancer (CRC), we performed epigenetic and genetic analyses in sessile serrated adenoma/polyps (SSA/P), traditional serrated adenomas (TSAs) and high‐methylation CRC. The methylation levels of six Group‐1 and 14 Group‐2 markers, established in our previous studies, were analyzed quantitatively using pyrosequencing. Subsequently, we performed targeted exon sequencing analyses of 126 candidate driver genes and examined molecular alterations that are associated with cancer development. SSA/P showed high methylation levels of both Group‐1 and Group‐2 markers, frequent BRAF mutation and occurrence in proximal colon, which were features of high‐methylation CRC. But TSA showed low‐methylation levels of Group‐1 markers, less frequent BRAF mutation and occurrence at distal colon. SSA/P, but not TSA, is thus considered to be precursor of high‐methylation CRC. High‐methylation CRC had even higher methylation levels of some genes, e.g., MLH1, than SSA/P, and significant frequency of somatic mutations in nonsynonymous mutations (p < 0.0001) and insertion/deletions (p = 0.002). MLH1‐methylated SSA/P showed lower methylation level of MLH1 compared with high‐methylation CRC, and rarely accompanied silencing of MLH1 expression. The mutation frequencies were not different between MLH1‐methylated and MLH1‐unmethylated SSA/P, suggesting that MLH1 methylation might be insufficient in SSA/P to acquire a hypermutation phenotype. Mutations of mismatch repair genes, e.g., MSH3 and MSH6, and genes in PI3K, WNT, TGF‐β and BMP signaling (but not in TP53 signaling) were significantly involved in high‐methylation CRC compared with adenoma, suggesting importance of abrogation of these genes in serrated pathway.
Keywords: colorectal cancer (CRC), sessile serrated adenoma/polyp (SSA/P), traditional serrated adenoma (TSA), DNA methylation, gene mutation
Short abstract
What's new?
The serrated pathway of colorectal cancer (CRC) development is characterized by the presence of saw‐toothed colonic crypts and by a high‐methylation epigenotype. Not all serrated lesions, however, give rise to CRC. Here, only sessile serrated adenoma/polyps, a potentially malignant serrated polyp subtype, were identified as precursors of high‐methylation CRC. Moreover, MLH1 expression, silencing of which previously was linked to microsatellite instability in CRC, was found to be preserved in most serrated adenoma/polyp samples. The findings suggest that the acquisition of a hypermutation phenotype in serrated CRC likely depends on extensive MLH1 methylation and mutation of additional mismatch repair genes.
Abbreviations
- CIMP
CpG island methylator phenotype
- CRC
colorectal cancer; indel, insertion/deletion
- LST
laterally spreading tumor
- MMR
mismatch repair
- MSI
microsatellite instability
- SSA/P
sessile serrated adenoma/polyp
- TSA
traditional serrated adenoma
Introduction
Colorectal cancer (CRC) is caused by the stepwise accumulation of multiple genetic and epigenetic alterations.1 Recent genomic research has revealed that CRC can develop through various pathways, resulting in tumors with distinct molecular characteristics.2, 3, 4 Sporadic CRC can be divided into cancers with chromosomal instability and those with microsatellite instability (MSI), the latter of which is frequently associated with CpG island methylator phenotype (CIMP). The majority of sporadic CRC is thought to develop through the adenoma–carcinoma sequence,5 whereas the serrated pathway has been considered as an alternative pathway distinct from the adenoma–carcinoma sequence. The development of CRC through the serrated pathway was reported to involve MSI, CIMP and the mutation of BRAF,6, 7, 8 and occur predominantly in the proximal colon.8 CIMP‐mediated gene silencing of the mismatch repair (MMR) gene MLH1 has been reported to be a molecular basis for MSI,9, 10 and is thus thought to be closely associated with the serrated pathway of CRC development.11
Recent exome sequencing analyses have revealed the involvement of many somatically mutated genes, including ARID1A, SMAD4, FBXW7, TCF7L2 and FAM123B.12, 13, 14 According to a report by the Cancer Genome Atlas (TCGA) group, 16% of CRCs exhibited frequent gene mutations (e.g., BRAF, ACVR2A, MSH3 and MSH6), and those CRCs are known as hypermutated tumors and usually showed MSI‐high, CIMP(+) and MLH1 silencing.12
Meanwhile, we and other groups have stratified CRCs using comprehensive and quantitative DNA methylation data.2, 4, 15 We developed two groups of methylation markers to classify CRC clearly into three distinct epigenotypes: high‐, intermediate‐ and low‐methylation epigenotypes. High‐methylation cancer was strongly correlated with the presence of the BRAF mutation and MSI‐high, and is thus thought to develop through the serrated pathway, similar to hypermutated tumors.4 We also epigenotyped early colorectal lesions and reported that serrated adenoma exhibited a high‐methylation epigenotype, whereas conventional adenoma was classified into intermediate‐ and low‐methylation epigenotypes. For flat, early colorectal lesions, so‐called laterally spreading tumors (LST), granular‐type LST showed intermediate‐methylation epigenotype, whereas nongranular LST showed low‐methylation epigenotype.3, 16 Importantly, no difference in the methylation level of each gene was observed between intermediate‐methylation adenoma and intermediate‐methylation cancer, suggesting that DNA methylation accumulation is mostly completed by the adenoma stage, at least in the development of intermediate CRC.4, 16
Serrated polyps, in contrast to conventional adenomas, present with a saw‐toothed appearance of the colonic crypts.17 According to the 2010 WHO classification schema,18 serrated polyps can be categorized into three subtypes: hyperplastic polyps, sessile serrated adenoma/polyps (SSA/P) and traditional serrated adenomas (TSAs). Although most hyperplastic polyps are non‐neoplastic lesions without malignant potential, both SSA/P and TSA can potentially develop into malignant CRC.19 Recent studies have revealed that SSA/P is likely to occur in the proximal colon and, to exhibit a high frequency of BRAF mutation and CIMP20, 21, 22, 23; thus, clinical and genetic characteristics of SSA/P resemble those of CRC with MSI. TSAs also reportedly have a relatively high frequency of CIMP and BRAF mutation, but they tend to occur in the distal colon and to exhibit KRAS mutation.20
To clarify the involvement of epigenetic and genetic alterations in the serrated pathway of CRC, we performed a quantitative methylation analysis of our two groups of methylation markers to determine the epigenotype of serrated tumors (45 SSA/P and 14 TSA samples); we then compared the results with those for other colorectal tumors, including LST and high‐methylation cancer. SSA/P showed high‐methylation epigenotype and was strongly associated with BRAF mutation, whereas TSA clearly showed intermediate‐methylation epigenotype and rather associated with KRAS mutation, suggesting that SSA/P, but not TSA, can be considered as a precursor of high‐methylation cancer. We subsequently performed targeted exon sequencing to evaluate 126 candidate CRC driver genes to gain insight into how these genes may modulate the genesis and progression of these tumors. Whereas DNA methylation accumulation in the serrated pathways has been investigated,24 the novelty of this study is the extensive mutational profiling of SSA/P and TSA. Our results will contribute to a greater understanding of the molecular basis of CRC development via the serrated pathway.
Material and Methods
Clinical samples
A total of 73 SSA/P and TSA samples were obtained from patients who underwent endoscopic submucosal dissection at the Yokohama City University Hospital or the Kanto Medical Center, NTT East, between May 2010 and December 2013. The tumors were resected endoscopically using the complete en bloc resection, fixed in 10% formalin and embedded in paraffin. Subsequently, 10 μm‐thick formalin‐fixed, paraffin‐embedded tissue specimens were subjected to laser capture microdissection (Carl Zeiss, Oberkochen, Germany) to dissect the tumor cells. DNA was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA). From each of 45 SSA/P and 14 TSA samples, >1 μg of DNA was obtained, and these DNA samples were used in the subsequent experiments. Samples of 17 high‐methylation CRCs, which were analyzed in our previous study,4 and 108 LSTs, which were characterized by the presence of lateral extensions along the luminal wall with a low vertical axis as well as SSA/P,3 were also included in this study. The clinicopathological characteristics of the patients, including age, sex, tumor size and tumor location, were evaluated at the time of endoscopic or surgical resection. Patients with familial adenomatous polyposis, hereditary nonpolyposis colorectal carcinoma and colitis‐associated carcinoma were excluded. Written informed consent was obtained from each of enrolled patients. This study was approved by the ethics committee of Yokohama City University, Chiba University, The University of Tokyo, Saitama Cancer Center, and Kanto Medical Center, NTT East.
Macroscopic and histological evaluations
The specimens were cut at a thickness of 3 μm and were stained with hematoxylin and eosin. Histopathological examinations were performed independently by two experienced pathologists, who were blinded to the endoscopic findings. The diagnoses were reviewed by both pathologists when a discrepancy occurred. The macroscopic and histological appearances of representative SSA/P and TSA are shown in Figure 1. SSA/P recognized as a flat or low sessile lesion that was frequently covered by a thin layer of mucin.25 Although larger, rounder stellate crypts have been described as a discriminating endoscopic feature of SSA/P, and SSA/P is difficult to distinguish from hyperplastic polyps.26 Histopathologically, SSA/P could be diagnosed based on their architectural features, such as columnar or rectangular dilation of the basal crypt, serration in the lower third of the crypt and a horizontally arranged basal area of the crypt.18, 26 TSA was typically recognized as a rubor exophytic polyp, and was diagnosed by complex villous architecture lined by columnar cells with densely eosinophilic cytoplasm and pencillate nuclei.18 A formation budding vertically from the sides of the villi has been reported to be a specific feature of TSA.
Figure 1.
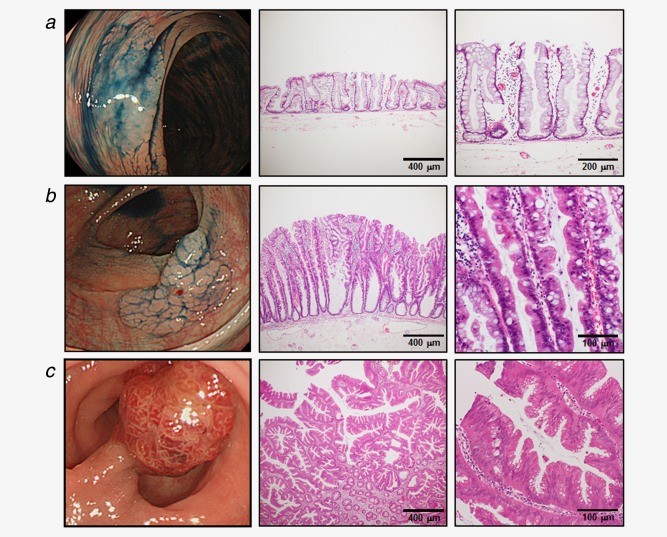
Macroscopic and histopathological appearance of serrated polyps. Endoscopic images after administration of 0.1% indigo carmine solution (left) and HE staining (middle and right). (a) SSA/P without dysplastic change. (b) SSA/P with low‐grade dysplasia. (c) TSA with low‐grade dysplasia. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Assessment of MSI status
MSI status of CRC was evaluated using Bethesda five markers as previously described.4 CRC samples showing instability in two or more markers were defined as MSI‐high, and the 11 high‐methylation CRCs used for the targeted exon sequencing analysis were all found to be MSI‐high.4
Immunohistochemistry
Immunostaining for MLH1 was performed using anti‐MLH1 antibody (Leica Biosystems, Nussloch, Germany), to confirm whether expression of MLH1 protein was retained. Samples showing loss of MLH1 in [mt]30% of the nuclei were considered MLH1‐loss(+). Samples showing limited focal loss of MLH1 in <30% of the nuclei were considered MLH1‐loss(±), and those with MLH protein expression preserved were considered MLH1‐loss(−).
DNA methylation analysis
The bisulfite conversion of 500 ng of genomic DNA was performed using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA), and the resulting product was suspended in 40 μL of distilled water. For the 20 Group‐1 markers and 25 Group‐2 markers established in our previous study,4 pyrosequencing primers were designed, using Pyro Q‐CpG Software (Qiagen), to ensure a product length of <100 bp and to include no or only one CpG site per primer sequence, and used to amplify bisulfite‐treated DNA regions containing several CpG sites.3 For the C in CpG sites within a primer sequence, a nucleotide that does not anneal to C or U was chosen, e.g., adenosine (A). The methylation levels were analyzed quantitatively by pyrosequencing using PyroMark Q96 (Qiagen). The biotinylated PCR product was bound to Streptavidin Sepharose High Performance (Amersham Biosciences, Uppsala, Sweden), washed and denatured using a 0.2 mol/L NaOH solution. After addition of 0.3 µmol/L sequencing primer to the single‐stranded PCR product, pyrosequencing was carried out according to the manufacturer's instructions. Analysis was performed using methylation control samples (0, 25, 50, 75 and 100%), and it was confirmed to be highly quantitative for six Group‐1 markers and 14 Group‐2 markers, whereas markers with correlation coefficient (R 2) < 0.9 were excluded.3, 4, 16 Using these previously established primers, quantitative methylation analyses were performed. The sequence primer information has been described in our previous reports.3, 16 MLH1 methylation was regarded as positive when the methylation rate in a quantitative analysis was >30%.
Mutation analysis
First, we analyzed the genetic mutations of KRAS (34, 35, 37 and 38) and BRAF (1799) using a genotyping assay on the MassARRAY platform as we previously performed.3
We subsequently used the HaloPlex target enrichment system (Agilent Technologies, Santa Clara, CA) to evaluate 126 candidate cancer driver genes in 48 samples (11/17 high‐methylation CRCs, 27/45 SSA/Ps and 10/14 TSAs), according to the manufacturer's instruction. The candidate cancer driver genes were selected using the following criteria: (i) genes containing driver gene mutations according to Vogelstein's report (frequency > 5%),27 (ii) genes identified as frequently (>15%) mutated genes in the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic), (iii) genes known to be associated with carcinogenic pathways (WNT, RTK/RAS, PI3K, TGF‐β, p53, MMR and BMP signaling) and (iv) genes that are frequently mutated in hypermutated CRC.12 A list of the genes and their coverage data is provided in Supporting Information Table S1.
The targeting design was generated using an on‐line design tool for HaloPlex and target enrichment was performed using the HaloPlex standard protocol version 2.0. Briefly, 225 ng of DNA from each sample was aliquoted into eight digestion reactions, each containing two restriction enzymes. DNA from the eight reactions was then pooled, hybridized to the HaloPlex probes and purified using magnetic beads. Fragments were ligated, amplified and barcoded through 19 PCR cycles, and two pools of 48 samples were sequenced using HiSeq1500 (Illumina, San Diego, CA) and a 150‐bp paired‐end protocol. The resulting data were analyzed for quality, coverage, single‐nucleotide variation and insertion/deletion (indel) using a platform provided by SureCall software (Agilent Technologies).
For each of the somatic mutations identified by the HaloPlex target enrichment system, variants with an allele frequency <0.1 and >0.8 were excluded. Known variants reported in the dbSNP (http://www.ncbi.nlm.nih.gov/SNP) were filtered out. Synonymous mutations were also excluded. We then used MutationTaster classification tools28 to predict the functional consequences of amino acid changes or frame‐shift mutations. The mutations that were found to be “disease causing” were defined as significant.
Validation using Sanger sequencing
Mutations in ACVR2A, BMPR2, MSH3 and MSH6 were validated using Sanger sequencing. The mutated sites and the flanking DNA sequences were amplified by PCR, for genomic DNA samples of both the tumor and the corresponding normal tissues. The primer sequences are shown in Supporting Information Table S2. The PCR products were then direct‐sequenced using an ABI automated DNA sequencer and the BigDye Terminator Kit (Life Technologies, Carlsbad, CA).
Statistical analysis
Intra‐observer differences in the histological evaluation were calculated by kappa statistics. The differences of the clinicopathological factors among high‐methylation cancer, SSA/P and TSA were analyzed by Fisher's exact test, except for age and tumor size, which were analyzed by Student's t‐test. The differences of methylation rate in each marker and number of mutations were analyzed by Student's t‐test. The differences of the mutation rate between each tumor were analyzed by Fisher's exact test. Unless otherwise specified, p‐values of <0.05 were considered to denote statistical significance. All the statistical analyses were performed using SPSS, ver. 11.0 (SPSS Inc., Chicago, IL).
Results
Comparison of clinicopathological characteristics
The clinicopathological data are summarized in Table 1. No significant differences in sex or age were observed between patients with high‐methylation cancer and those with SSA/P. However, high‐methylation CRC was identified more frequently in older and female patients, compared with TSA (p = 0.01 and p = 0.01, respectively). Most high‐methylation CRC (88%, p = 0.0003 vs. TSA) and SSA/P (93%, p < 0.0001 vs. TSA) were located in the proximal colon, whereas most TSA (84%) were located in the distal colon. High‐methylation CRC was relatively larger, but no significant difference in size was observed between SSA/P and TSA. Similar to high‐methylation CRC, SSA/P showed significantly higher frequency of BRAF mutation (80%) and a lower frequency of KRAS mutation (4%), compared with TSA (43% for BRAF mutation, p = 0.02, and 50% for KRAS mutation, p = 0.003).
Table 1.
Clinicopathological characteristics of cancer and adenoma cases
| High‐methylation CRC | SSA/P | TSA | p‐values | |||
|---|---|---|---|---|---|---|
| CRC vs. SSA/P | CRC vs. TSA | SSA/P vs. TSA | ||||
| No. of cases | 17 | 45 | 14 | |||
| Sex (male/female) | 5/12 | 23/22 | 11/3 | 0.16 | 0.01a | 0.12 |
| Age (years) | 69.0 ± 8.9 | 64.2 ± 11.0 | 61.5 ± 8.0 | 0.11 | 0.01a | 0.39 |
| Tumor location | 0.61 | 0.0003a | <0.0001a | |||
| Proximal (C/A/T) | 15 (4/10/1) | 42 (12/24/6) | 2 (0/1/1) | |||
| Distal (D/S/R) | 2 (1/1/0) | 3 (1/1/1) | 12 (1/8/3) | |||
| Tumor size (mm) | 74.5 ± 31.3 | 27.3 ± 7.8 | 23.4 ± 15.4 | <0.0001a | <0.0001a | 0.21 |
| Genetic aberration, n (%) | ||||||
| KRAS mutation(+) | 3 (18) | 2 (4) | 7 (50) | 0.12 | 0.12 | 0.003a |
| BRAF mutation(+) | 12 (82) | 36 (80) | 6 (43) | 0.50 | 0.16 | 0.02a |
| Dysplastic change, n (%) | n.a. | n.a. | 0.014a | |||
| No dysplastic change | n.a. | 18 (40) | 0 (0) | |||
| Low‐grade dysplasia | n.a. | 21 (47) | 12 (86) | |||
| High‐grade dysplasia | n.a. | 6 (13) | 2 (14) | |||
For comparison of tumor location, tumors were classified into two locations: proximal colon, including cecum (C), ascending (A) and transverse (T) colon and distal colon, including descending (D) and sigmoid (S) colon and rectum (R). The tumor size was recorded as the maximum diameter of the extirpated specimen. p‐values were analyzed by Fisher's exact test or Student's t‐test for age and tumor size.
p < 0.05.
Abbreviation: n.a., not applicable.
Histopathological examination
SSA/P with cytological dysplasia, resembling conventional adenoma,29 was not observed in this study. However, if any of lesion areas displayed enlarged, vesicular nuclei with prominent nucleoli, the lesion was regarded as having undergone a dysplastic change (+). Lesions exhibiting severe atypia were categorized as high‐grade dysplasia, whereas lesions exhibiting low to moderate atypia were categorized as low‐grade dysplasia. SSA/P showed various degrees of dysplasia (18 with no dysplastic change, 21 with low‐grade dysplasia and 6 with high‐grade dysplasia), whereas all the TSA samples showed low‐ to high‐grade dysplasia. Five of the six SSA/P with high‐grade dysplasia exhibited MLH1 methylation, whereas 39 SSA/P with no or low‐grade dysplasia showed significantly lower frequency of MLH1 methylation (83% vs. 31%, p = 0.02). The intra‐observer difference was 0.77, indicating that the diagnosis of serrated polyps was relatively consistent.
Quantitative DNA methylation analysis for epigenotyping
The TSA samples showed high methylation rate in most of the Group‐2 markers but lower methylation rate in Group‐1 markers, which was a characteristic of intermediate‐methylation CRC4 and intermediate‐methylation early colorectal lesions (e.g., LST granular type).3 On the other hand, the SSA/P samples showed generally high methylation rate in both Group‐1 and Group‐2 markers, which was a characteristic of high‐methylation CRC samples (Fig. 2 and Supporting Information Fig. S1).
Figure 2.
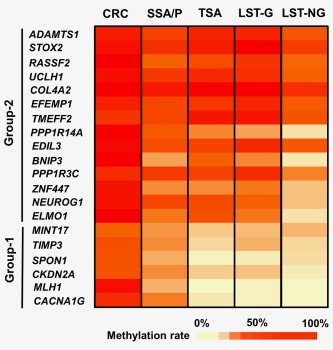
Heatmap of methylation levels of six Group‐1 markers and 14 Group‐2 markers. The color scale is presented as average methylation levels of each marker. Methylation data of CRC and LSTs were quoted from our previous reports.3, 4 As previously described, high‐methylation CRC, LST granular type and LST nongranular type could be classified into high‐, intermediate‐ and low‐methylation epigenotypes, respectively. SSA/P exhibited generally high methylation levels of both Group‐1 and Group‐2 markers, similar to the results of high‐methylation CRC; thus, SSA/P was considered to be high‐methylation epigenotype. TSA exhibited high methylation levels of Group‐2 markers but low methylation levels of Group‐1 markers similar to the results of LST granular type; thus, TSA was considered to be intermediate‐methylation epigenotype. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
When the methylation rates of each of genes were compared, four of the six Group‐1 markers showed significantly lower methylation rates in the TSA samples than in the SSA/P samples, whereas most of the Group‐2 markers were similarly methylated in the TSA and SSA/P samples (Fig. 3). Although SSA/P showed generally high methylation rates in both the Group‐1 and Group‐2 markers, the methylation rates in the high‐methylation CRC samples were even higher, with the exceptions of TIMP3, MINT17, TMEFF2 and PPP1R3C.
Figure 3.
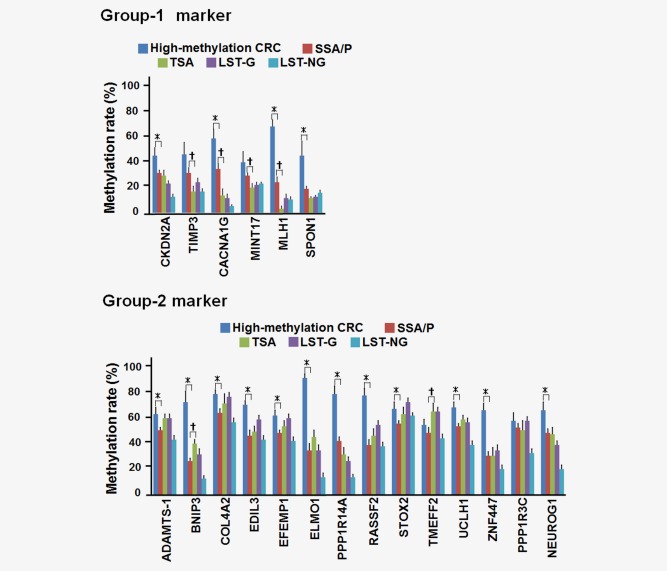
Comparison of the methylation levels of each marker. Methylation data of CRC and LSTs were quoted from our previous reports.3, 4 *p < 0.05 between high‐methylation CRC and SSA/P (Student's t‐test). †p < 0.05 for comparison of SSA/P and TSA (Student's t‐test). Group‐2 markers were highly methylated in both SSA/P and TSA. For Group‐1 markers, SSA/P showed significantly higher methylation rate than TSA in four of the six markers. High‐methylation CRC showed even higher methylation rate than SSA/P in most of the genes. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Because MLH1 hypermethylation reportedly causes MSI in CRC,9, 10 frequency of MLH1‐methylation(+) cases (methylation rate >30%) was compared among the samples. Although most of the high‐methylation CRC samples (14 out of 17, 82%) were MLH1‐methylation(+), 16 of the 45 SSA/P samples (36%, p < 0.0001) and none of the TSA samples (0%, p < 0.0001) were MLH1‐methylation(+).
Although the accumulation of DNA methylation has been previously shown to be mostly completed by the adenoma stage in the case of conventional adenomas,16 the present data indicated that the methylation of genes (e.g., MLH1) could increase during development from adenoma to cancer in the serrated pathway.
The performance of HaloPlex target enrichment analysis
On average, approximately 5.8 million purity‐filtered reads were generated for each sample, and [mt]95% of them were aligned to the target region of 986,662 bp. Samples were sequenced with an average exon coverage of 461‐fold (ranging from 158‐ to 575‐fold). On average per sample, 95% of the targeted bases were covered by at least ten reads. In all the samples except for two SSA/P samples, [mt]60% of the targeted bases were covered by at least 100 reads, and the two SSA/P samples were excluded from subsequent analysis (Supporting Information Fig. S2).
Difference of mutation frequencies
In total, 26,763 somatic mutations were identified in coding regions, including 117 nonsense, 13,863 missense, 12,370 synonymous, 225 frame‐shift indels and 188 in‐frame indels (the average mutation number is shown in Fig. 4). The mutation spectra patterns were similar when compared among the three groups of high‐methylation CRC, SSA/P and TSA (Supporting Information Fig. S3). The predominant type of substitution was a C:G to T:A transition, followed by a T:A to C:G transition.
Figure 4.
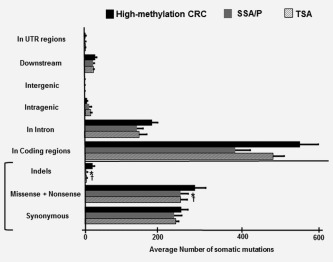
Average number of somatic mutations. The frequency of somatic mutations in high‐methylation CRC was significantly higher than those in SSA/P and TSA, both for nonsynonymous mutations (p < 0.0001 and p = 0.001, respectively) and indels (p < 0.0001 and p < 0.0001, respectively). *p < 0.05 between high‐methylation CRC and SSA/P (Student's t‐test). †p < 0.05 for comparison of high‐methylation CRC and TSA (Student's t‐test).
The frequency of somatic mutations was significantly higher in high‐methylation CRC than in SSA/P or TSA, both in nonsynonymous (missense and nonsense) mutations (282.3 ± 27.5 vs. 245.7 ± 19.3 for SSA/P, p < 0.0001, and vs. 232.7 ± 17.4 for TSA, p = 0.001) and indels (21.0 ± 4.5 vs. 5.1 ± 2.7 for SSA/P, p < 0.0001, and vs. 4.5 ± 2.3 for TSA, p < 0.0001) (Fig. 4 and Supporting Information Fig. S4). However, no significant differences in the mutation frequencies were observed between SSA/P and TSA. Furthermore, no significant difference was observed, when the mutation frequencies were compared between MLH1‐methylated and MLH1‐unmethylated SSA/P samples (247.2 ± 20.1 vs. 244.8 ± 18.6, p = 0.75, and 5.8 ± 2.1 vs. 4.5 ± 2.2, p = 0.12, for nonsynonymous mutations and indels, respectively) (Supporting Information Fig. S4).
Aberrations in individual genes
The results of the targeted exon sequencing are summarized in Supporting Information Table S3. When mutations in individual genes were compared, 32 of the 126 genes showed a significantly higher frequency of mutations in high‐methylation CRC compared with SSA/P (Fig. 5 and Supporting Information Fig. S5). These frequently mutated genes included candidate cancer driver genes (e.g., BRAF, APC, ACVR2A, MSH3 and MSH6), as previously detected in hypermutated CRC by the TCGA group.12 Interestingly, a frame‐shift mutation of ACVR2A was identified in all the high‐methylation CRC cases, and the mutations could be summarized into two hot spots (c.1303delA and c.278delA). These mutations were validated by Sanger sequencing using genomic DNA samples of cancer tissues and the corresponding normal tissues, and the mutations were confirmed to have occurred in all the cancer samples but in none of the normal samples (Supporting Information Figs. S6 and S7).
Figure 5.
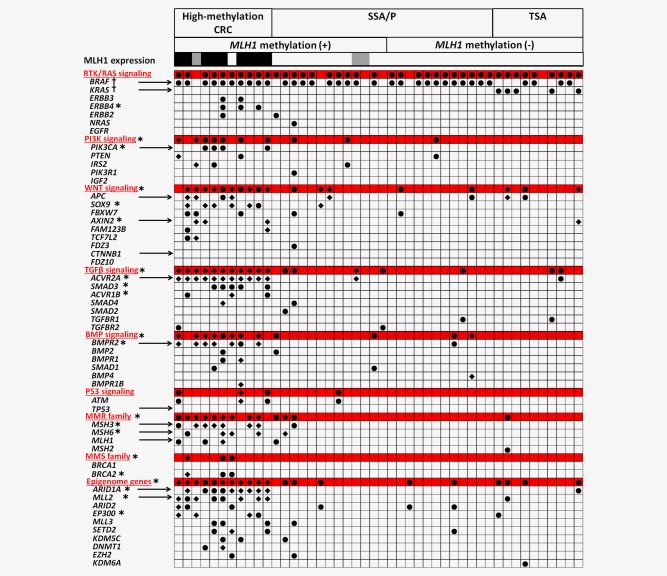
Genetic alterations in individual genes and carcinogenic signaling pathways. Black circle, nonsynonymous mutations. Black rhombus, frame‐shift indels. For MLH1 expression, MLH1‐loss(+), ‐loss(±) and ‐loss(−) were shown by black, gray and blank, respectively. The alterations that are shown were predicted to be “disease causing” by MutationTaster.28 *p < 0.05 for comparison of high‐methylation CRC and SSA/P. †p < 0.05 for comparison of SSA/P and TSA. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
A high mutation frequency of MMR family genes (82%) was specifically detected in high‐methylation CRC, whereas these genes were not frequently mutated in MLH1‐methylated or MLH1‐unmethylated SSA/P (Fig. 5 and Supporting Information Fig. S4). None of the individual genes showed a significantly higher frequency of mutation in MLH1‐methylated SSA/P than in MLH1‐unmethylated SSA/P. Considering that no difference in the frequency of nonsynonymous mutations and indels was observed between MLH1‐methylated and MLH1‐unmethylated SSA/P, MLH1 methylation in SSA/P might be insufficient for the development of the hypermutation phenotype seen in high‐methylation CRC.
When SSA/P and TSA were compared, no significant differences in the frequencies of gene mutations were observed except for BRAF and KRAS. Overall, 74% of the SSA/P were BRAF‐mutation(+), similar to the findings of high‐methylation CRC (91%, p = 0.6), meanwhile, a lower BRAF mutation frequency was detected in TSA (40%, p = 0.02). On the other hand, although all the high‐methylation CRC and SSA/P samples were KRAS‐mutation(−), a significantly higher frequency of KRAS mutation was detected in TSA (50%, p = 0.01 and p = 0.0008, respectively).
Aberrations in several signaling pathways
To evaluate how some well‐defined carcinogenic pathways are associated with the development of high‐methylation cancer, we compared alterations in RTK/RAS, PI3K, WNT, TGF‐ β, BMP and TP53 signaling between high‐methylation cancer and SSA/P (Fig. 5).
The RTK/RAS signaling pathway was frequently altered in both high‐methylation CRC (91%) and SSA/P (78%). However, mutations in the ERBB receptors were identified only in high‐methylation CRC.
Consistent with the TCGA database, genes involved in PI3K, WNT and TGF‐β signaling were altered more frequently in high‐methylation CRC than SSA/P (64% vs. 11%, p = 0.003; 91% vs. 19%, p < 0.0001; and 100% vs. 19%, p < 0.0001, respectively). The frequency of BMP signaling alteration was also significantly higher in high‐methylation CRC (82% vs. 19%, p = 0.0008). We also found that epigenetic modifier genes (e.g., ARID1A and EP300) were more frequently mutated in high‐methylation CRC. The frequency of TP53 signaling alterations was relatively low (27%) in high‐methylation CRC and was not significantly higher than SSA/P.
Comparison of MLH1 expression by immunohistochemistry
MLH1 protein expression was analyzed by immunohistochemistry (Fig. 5 and Supporting Information Fig. 8). Although MLH1 expression was lost in most of high‐methylation CRC samples (9/11, 82%), MLH1 expression was preserved in all the TSA samples. MLH1 expression was also preserved in most of SSA/P samples, but two MLH1‐methylated SSA/P samples (2/25, 8%) showed limited focal loss of MLH1. Although these focal lesions might be regarded as precursor of high‐methylation CRC, MLH1 methylation was generally considered to be insufficient in SSA/P and further methylation increase might be required to silence MLH1 expression.
Discussion
To clarify the involvement of epigenetic and genetic alterations in the serrated pathway of CRC, we performed a quantitative methylation analysis to determine the epigenotype of SSA/P and TSA samples and compared them with other colorectal tumors, including LST and high‐methylation CRC. We also performed a targeted exon sequencing analysis of 126 candidate CRC driver genes to evaluate their involvement in the progression of serrated adenoma to cancer.
SSA/P and TSA constituted approximately 10% of all polyps.25 Here, we confirmed that most SSA/P had BRAF mutation, and occurred predominantly in the proximal colon, similar to high‐methylation CRC. On the other hand, most TSA were located in the distal colon, and had lower frequency of BRAF mutation and a significant higher frequency of KRAS mutation, with mutually exclusive manner. A quantitative methylation analysis revealed that SSA/P showed high‐methylation epigenotype, and was thus considered to be a precursor lesion of high‐methylation CRC, whereas TSA clearly showed intermediate‐methylation epigenotype. Consistent with our report, Gaiser et al.24 found similar methylation profiles in SSA/P and MSI‐high CRC, whereas TSA also revealed aberrant methylation pattern, but clustered more heterogeneously and closer to microsatellite‐stable CRCs. The serrated pathway is also believed to follow two routes: one characterized by BRAF mutation and one characterized by KRAS mutation30; TSA might perhaps be a precursor of KRAS/BRAF‐mutated intermediate‐methylation CRC at the distal/proximal colon, respectively. Recently, Bettington et al.31 reported that TSA frequently present CIMP, whereas majority of them retain enzyme function of MMR and indicate microsatellite‐stable phenotype. In contrast to our report, BRAF‐mutated TSA was reported to show frequent TP53 mutation and CTNNB1 activation. Although no TP53 mutation or rare WNT signaling alteration was detected in BRAF‐mutated TSA in this study, the major purpose of this study was investigation of genetic profiles in SSA/P and high‐methylation CRC, and only four BRAF‐mutated TSA were included. Additional studies are needed to reveal whether TSA could be precursors of proximal BRAF‐mutated microsatellite‐stable CRC.
Although the rates of the presence of CIMP in SSA/P varied depending on used marker panel, CIMP has been reportedly observed in 44–80% of SSA/P.22, 23, 32 Although we detected high levels of methylation accumulation in SSA/P, our data also suggested that DNA methylation accumulation was not completed by the adenoma stage, and the methylation of genes, including MLH1, may increase during development from adenoma to cancer in the serrated pathway. Compared with our results, lower frequency of MLH1 methylation in SSA/P have been reported by other groups.20, 33 This result might be explained, in part, by differences in the tumor size and histological appearance. Dhir et al.34 recently reported that methylation events accumulate with the progression of serrated lesions; the stepwise increment of MLH1, TLR2 and CDX2 methylation was observed from HP to SSA/P, with the highest scores observed in SSA/P with cytological dysplasia. Muto et al.35 reported a significant increment in the epigenetic silencing of AXIN2 with progression from SSA/P to MSI cancer. In this study, MLH1 methylation in SSA/P was significantly correlated with high‐grade dysplasia. Although we and other group36 showed that DNA methylation accumulation was mostly completed by the adenoma stage in the adenoma–carcinoma sequence from conventional tubular adenoma to cancer, the further accumulation of DNA methylation might occur during the progression of adenoma to the development of cancer in the serrated pathway.
When the number of somatic mutations was compared between MSI‐high and microsatellite‐stable CRC, MSI‐high CRC reported to show a significantly higher number of mutations.12, 13, 37 Consistent with these previous reports, we detected frequent mutations in high‐methylation CRC. Although MLH1 methylation was reported to contribute to MSI‐high CRC development,9, 10 MLH1‐methylated SSA/P did not show higher frequency of mutation than MLH1‐unmethylated SSA/P in any of the individual genes, or all the somatic mutations combined. In contrast, high‐methylation CRC had significant higher frequencies of mutation in many individual genes and also for all the somatic mutations combined. As reported in hypermutation CRC,12 MMR family genes (e.g., MSH3 [64%] and MSH6 [45%]) were frequently mutated in high‐methylation CRC. MSI has been reported to be acquired during the progression from precursor to malignant lesion.38 Although an advanced pathology was observed in MLH1‐methylated SSA/P compared with MLH1‐unmethylated SSA/P, the epigenetic silencing of MLH1 may be insufficient in SSA/P to acquire a hypermutation phenotype. In fact, when methylation level of MLH1 was compared between MLH1‐methylated SSA/P and high‐methylation CRC, the methylation level in MLH1‐methylated SSA/P was significantly lower than that in high‐methylation CRC (44.4 ± 12.6% vs. 78.1 ± 16.7%, p < 0.0001). As for immunostaining of MLH1 protein, although most of high‐methylation CRC (82%) showed loss of MLH1 expression, only two MLH1‐methylated SSA/P samples showed focal loss of MLH1. Whereas these focal lesions may be regarded as precursor of high‐methylation CRC, MLH1 methylation level was suggested to be still low in SSA/P, and further methylation increase might be necessary for development of hypermutation and malignant tumor. The additional abrogation of MMR family genes (e.g., MSH3 and MSH6 mutations) might also be necessary for the development of hypermutation and malignant tumor in this pathway.
Several epigenetic modifier genes were frequently mutated in high‐methylation CRC (e.g., ARID1A, MLL2 and EP300). ARID1A encodes a member of the SWI/SNF family, the members of which have helicase and ATPase activities and are thought to regulate the transcription of certain genes by altering the chromatin structure around those genes, and these members have recently been reported in MSI‐high cancers: not only CRC, but also gastric and ovarian cancers.39, 40 In addition to the genes that we evaluated, Tahara et al.41 reported that CIMP(+) high‐methylation CRCs were frequently mutated in the chromatin regulation genes CHD7 and CHD8. These epigenetic aberrations might also contribute to the development of cancer from adenoma.
The signal pathways altered by hypermutation included RTK/RAS, PI3K, WNT and TGF‐β signaling, in close agreement with the findings in a previous report.12 For RTK/RAS signaling, though BRAF mutation was detected at similarly high frequencies in both SSA/P and high‐methylation CRC, mutations in the ERBB receptors were identified only in high‐methylation CRC. For TGF‐β signaling, all the cancer samples showed a frame‐shift mutation of ACVR2A, which could be summarized into two hot spots; one was previously identified in a TCGA report,12 whereas the other was a novel mutation. The mutation of TP53 or an alteration of TP53 signaling was not frequently involved in high‐methylation CRC, as we and others have previously reported.2, 4, 15
Alterations of BMP signaling were also significantly frequent among high‐methylation CRCs (9 out of 11, 82%), including mutations of BMPR2, BMP2 and SMAD4. It was reported that BMP2 expression was lost in microadenoma of familial adenomatous polyposis, whereas BMP2 was expressed in mature colonic epithelial cells, promoting differentiation and inhibiting proliferation.42 Inactivation of BMPR2, BMPR1A and SMAD4 was frequently observed in sporadic CRC, correlating to loss of Smad1/5/8 phosphorylation.43 Juvenile polyposis syndrome, an inherited syndrome with high risk of CRC, is caused by germline mutation in BMP signaling.44, 45 We previously found that activation of BMP signaling was essential in oncogene‐induced senescence, and inhibition of BMP signaling resulted in an escape from oncogene‐induced senescence.46 In addition to PI3K, WNT and TGF‐β signaling, the alteration of BMP signaling might also contribute to tumorigenesis in high‐methylation CRC with BRAF mutation through the abrogation of senescence mechanism.
In summary, SSA/P, but not TSA, showed high‐methylation epigenotype and was strongly correlated with BRAF mutation, and thus was considered to be a precursor of high‐methylation CRC. Although the abrogation of MMR family genes was suggested to be important for tumorigenesis in the serrated pathway, MLH1 methylation in SSA/P might be insufficient to acquire a hypermutation phenotype; further increase of MLH1 methylation and possibly additional abrogation of MMR family genes, such as mutations of MSH3 and MSH6, might be necessary. Significantly frequent mutations in PI3K, WNT, TGF‐β and BMP signaling, but not in TP53 signaling, were thought to occur during the development of high‐methylation CRC from SSA/P.
Supporting information
Supporting Information
Acknowledgements
The authors thank Hiromi Tanaka, Yuko Hosaka, Rikiya Okuyama, Kaori Nakano, Machiko Hiraga and Ayaka Miura for technical assistance. All authors have no potential conflict of interests to disclose.
References
- 1. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008;135:1079–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hinoue T, Weisenberger DJ, Lange CP, et al. Genome‐scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012;22:271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sakai E, Ohata K, Chiba H, et al. Methylation epigenotypes and genetic features in colorectal laterally spreading tumors. Int J Cancer 2014;135:1586–95. [DOI] [PubMed] [Google Scholar]
- 4. Yagi K, Akagi K, Hayashi H, et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res 2010;16:21–33. [DOI] [PubMed] [Google Scholar]
- 5. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal‐tumor development. N Engl J Med 1988;319:525–32. [DOI] [PubMed] [Google Scholar]
- 6. Toyota M, Ahuja N, Ohe‐Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer 2005;5:223–31. [DOI] [PubMed] [Google Scholar]
- 8. Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006;38:787–93. [DOI] [PubMed] [Google Scholar]
- 9. Hawkins N, Norrie M, Cheong K, et al. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology 2002;122:1376–87. [DOI] [PubMed] [Google Scholar]
- 10. Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair‐defective human tumor cell lines. Cancer Res 1997;57:808–11. [PubMed] [Google Scholar]
- 11. Samowitz WS. The CpG island methylator phenotype in colorectal cancer. J Mol Diagn 2007;9:281–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007;446:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314:268–74. [DOI] [PubMed] [Google Scholar]
- 15. Shen L, Toyota M, Kondo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A 2007;104:18654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yagi K, Takahashi H, Akagi K, et al. Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am J Pathol 2012;180:616–25. [DOI] [PubMed] [Google Scholar]
- 17. Torlakovic E, Snover DC. Serrated adenomatous polyposis in humans. Gastroenterology 1996;110:748–55. [DOI] [PubMed] [Google Scholar]
- 18. Snover DC, Ahnen DJ, Burt RW. Serrated polyps of the colon and rectum and serrated polyposis. WHO classification of the tumours of the digestive system. Lyon: IARC, 2010. 160–5. [Google Scholar]
- 19. Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol 2011;42:1–10. [DOI] [PubMed] [Google Scholar]
- 20. Burnett‐Hartman AN, Newcomb PA, Potter JD, et al. Genomic aberrations occurring in subsets of serrated colorectal lesions but not conventional adenomas. Cancer Res 2013;73:2863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kambara T, Simms LA, Whitehall VL, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004;53:1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim YH, Kakar S, Cun L, et al. Distinct CpG island methylation profiles and BRAF mutation status in serrated and adenomatous colorectal polyps. Int J Cancer 2008;123:2587–93. [DOI] [PubMed] [Google Scholar]
- 23. O'Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 2006;30:1491–501. [DOI] [PubMed] [Google Scholar]
- 24. Gaiser T, Meinhardt S, Hirsch D, et al. Molecular patterns in the evolution of serrated lesion of the colorectum. Int J Cancer 2013;132:1800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spring KJ, Zhao ZZ, Karamatic R, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology 2006;131:1400–7. [DOI] [PubMed] [Google Scholar]
- 26. Bettington M, Walker N, Rosty C, et al. Critical appraisal of the diagnosis of the sessile serrated adenoma. Am J Surg Pathol 2014;38:158–66. [DOI] [PubMed] [Google Scholar]
- 27. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods 2014;11:361–2. [DOI] [PubMed] [Google Scholar]
- 29. Ban S, Mitomi H, Horiguchi H, et al. Adenocarcinoma arising in small sessile serrated adenoma/polyp (SSA/P) of the colon: clinicopathological study of eight lesions. Pathol Int 2014;64:123–32. [DOI] [PubMed] [Google Scholar]
- 30. O'Brien MJ, Zhao Q, Yang S. Colorectal serrated pathway cancers and precursors. Histopathology 2015;66:49–65. [DOI] [PubMed] [Google Scholar]
- 31. Bettington ML, Walker NI, Rosty C, et al. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod Pathol 2015;28:414–27. [DOI] [PubMed] [Google Scholar]
- 32. Rosenberg DW, Yang S, Pleau DC, et al. Mutations in BRAF and KRAS differentially distinguish serrated versus non‐serrated hyperplastic aberrant crypt foci in humans. Cancer Res 2007;67:3551–4. [DOI] [PubMed] [Google Scholar]
- 33. Maeda T, Suzuki K, Togashi K, et al. Sessile serrated adenoma shares similar genetic and epigenetic features with microsatellite unstable colon cancer in a location‐dependent manner. Exp Ther Med 2011;2:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dhir M, Yachida S, Van Neste L, et al. Sessile serrated adenomas and classical adenomas: an epigenetic perspective on premalignant neoplastic lesions of the gastrointestinal tract. Int J Cancer 2011;129:1889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muto Y, Maeda T, Suzuki K, et al. DNA methylation alterations of AXIN2 in serrated adenomas and colon carcinomas with microsatellite instability. BMC Cancer 2014;14:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo Y, Wong CJ, Kaz AM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology 2014;147:418.e8–29.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One 2010;5:e15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamamoto E, Suzuki H, Yamano HO, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol 2012;181:1847–61. [DOI] [PubMed] [Google Scholar]
- 39. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011;11:481–92. [DOI] [PubMed] [Google Scholar]
- 40. Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 2013;3:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tahara T, Yamamoto E, Madireddi P, et al. Colorectal carcinomas with CpG island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology 2014;146:530.e5–8.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hardwick JC, Van Den Brink GR, Bleuming SA, et al. Bone morphogenetic protein 2 is expressed by, and acts upon, mature epithelial cells in the colon. Gastroenterology 2004;126:111–21. [DOI] [PubMed] [Google Scholar]
- 43. Kodach LL, Wiercinska E, de Miranda NF, et al. The bone morphogenetic protein pathway is inactivated in the majority of sporadic colorectal cancers. Gastroenterology 2008;134:1332–41. [DOI] [PubMed] [Google Scholar]
- 44. Haramis AP, Begthel H, van den Born M, et al. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 2004;303:1684–6. [DOI] [PubMed] [Google Scholar]
- 45. Howe JR, Bair JL, Sayed MG, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001;28:184–7. [DOI] [PubMed] [Google Scholar]
- 46. Kaneda A, Fujita T, Anai M, et al. Activation of Bmp2‐Smad1 signal and its regulation by coordinated alteration of H3K27 trimethylation in Ras‐induced senescence. PLoS Genet 2011;7:e1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information