

Abstract
Background and Purpose
Specific, high potency receptor antagonists are valuable tools when evaluating animal and human physiology. Within the glucose‐dependent, insulinotropic polypeptide (GIP) system, considerable attention has been given to the presumed GIP receptor antagonist, (Pro3)GIP, and its effect in murine studies. We conducted a pharmacological analysis of this ligand including interspecies differences between the rodent and human GIP system.
Experimental Approach
Transiently transfected COS‐7 cells were assessed for cAMP accumulation upon ligand stimulation and assayed in competition binding using 125I‐human GIP. Using isolated perfused pancreata both from wild type and GIP receptor‐deficient rodents, insulin‐releasing, glucagon‐releasing and somatostatin‐releasing properties in response to species‐specific GIP and (Pro3)GIP analogues were evaluated.
Key Results
Human (Pro3)GIP is a full agonist at human GIP receptors with similar efficacy (E max) for cAMP production as human GIP, while both rat and mouse(Pro3)GIP were partial agonists on their corresponding receptors. Rodent GIPs are more potent and efficacious at their receptors than human GIP. In perfused pancreata in the presence of 7 mM glucose, both rodent (Pro3)GIP analogues induced modest insulin, glucagon and somatostatin secretion, corresponding to the partial agonist activities observed in cAMP production.
Conclusions and Implications
When evaluating new compounds, it is important to consider interspecies differences both at the receptor and ligand level. Thus, in rodent models, human GIP is a comparatively weak partial agonist. Human (Pro3)GIP was not an antagonist at human GIP receptors, so there is still a need for a potent antagonist in order to elucidate the physiology of human GIP.
Abbreviations
- GIP
glucose‐dependent insulinotropic polypeptide (gastric inhibitory peptide)
- GLP‐1
glucagon‐like peptide‐1
- HBS
HEPES‐buffered saline
Tables of links
| TARGETS |
|---|
| GPCRs |
| GIP receptor |
| GLP‐1 receptor |
| GPR119 |
| LIGANDS | |
|---|---|
| GIP | (Pro3)GIP |
| Glucagon | |
| Insulin | |
| Somatostatin | |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Glucose‐dependent insulinotropic polypeptide (also gastric inhibitory peptide; GIP) is a hormone secreted from the K cells of the gut following a meal (Baggio and Drucker, 2007). Like the related hormone glucagon‐like peptide‐1 (GLP‐1), GIP is a potent insulin secretagogue (Holst, 2004). In contrast to the glucagonostatic effect of GLP‐1 (Gutniak et al., 1992; de Heer et al., 2008), GIP has glucagon‐releasing properties (Kreymann et al., 1987; Christensen et al., 2011). The incentive to understand the biology of GIP was intensified by the discovery of an association between rodent GIP receptors and adiposity (Miyawaki et al., 1999; Miyawaki et al., 2002; Nasteska et al., 2014). In humans, although less clear, there is likewise evidence for a role of GIP in fat metabolism with the demonstration of GIP receptor expression in adipose tissue (Ahlqvist et al., 2013), an association between high body mass index and increased GIP levels (Ahlqvist et al., 2013; Calanna et al., 2013), and increased adipose tissue blood flow and triacylglycerol deposition following GIP administration in a state of high insulin and high glucose (Asmar et al., 2010). Furthermore, obese children decrease their basal and postprandial GIP levels, when put on a low‐calorie diet (Deschamps et al., 1980), and healthy young men increase their fasting GIP levels, when put on a high‐fat diet (Brøns et al., 2009).
The availability of the GLP‐1 receptor antagonist exendin‐(9–39) (Raufman et al., 1991; Jørgensen et al., 2013) has been helpful for our understanding of the physiology of GLP‐1, which underlies the pharmaceutical development of GLP‐1 receptor agonists as anti‐diabetic and anti‐obesity agents. Inspired by this, researchers have tried to develop a potent GIP receptor antagonist. Many different strategies have been undertaken in order to inhibit GIP function, including prepro‐GIP gene truncations (Nasteska et al., 2014), administration of low MW receptor antagonists (Nakamura et al., 2012), immunization against GIP (Ebert et al., 1979; Fulurija et al., 2008; Irwin et al., 2009), various truncations and amino acid substitutions of the GIP molecule thought to provide antagonistic properties (Tseng et al., 1996; Gelling et al., 1997; Hinke et al., 2001; Gault et al., 2002; Deacon et al., 2006; Irwin et al., 2006; Kerr et al., 2011), and recently, a potent antagonistic antibody against the GIP receptor was reported (Ravn et al., 2013). However, most attention has been given to the analogue (Pro3)GIP following demonstrations that chronic treatment with this peptide improved diabetic parameters in ob/ob mice (Irwin et al., 2007), improved diabetic parameters and induced weight loss in obese mice previously on a high‐fat diet (McClean, 2007), and reduced weight gain and improved diabetic parameters following the induction of a high‐fat diet (Gault et al., 2007).
Animal models are widely used as basis for predicting biological properties of given receptor‐ligand systems to human (patho)physiology. Often, rodent models are the first choice due to reduced costs and space requirements compared with larger animal models (pigs, dogs and primates). Initial drug screening are similarly often carried out in rodent models. This creates the classical dilemma of whether the chosen rodent model is representative for human physiology or whether the inter‐species differences (stemming from structural, functional, spatial or temporal differences) are too great. Among the seven‐transmembrane receptor systems, such differences have been described for instance for the lipid‐activated G protein‐coupled receptor 119 (GPR119), where a series of compounds, with similar binding properties to human and mouse GPR119, displayed large differences in potency and efficacy (Scott et al., 2013). A similar example can be found within the 5‐HT receptor system, where a single amino acid difference between the human and mouse receptor can account for large differences in binding affinities and potencies (Canal et al., 2013).
Other receptor‐ligand systems, however, display very little interspecies variations, as seen in the GLP‐1 system with a large degree of sequence homology both in terms of the ligand (GLP‐1) and its receptor in humans, mice and rats and where both liraglutide (a long‐acting GLP‐1 receptor agonist) and native GLP‐1 have similar potencies across species‐specific receptors (Knudsen et al., 2012) (Figure 1). Even though the GIP system is closely related to GLP‐1 and the GLP‐1 receptor, the GIP system is noticeably less conserved across species (Figure 1).
Figure 1.
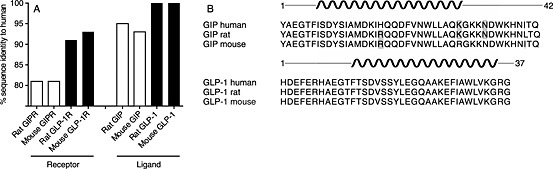
The GIP system is less conserved than the GLP‐1 hormone system. A) Sequence identity between rodent and human GIP receptors (GIPR) and GLP‐1 receptors (GLP‐1R), as well as GIP and GLP‐1 given in percent. The receptors were compared without signalling peptide sequences. The alignment was done in Geneious 6.0.5. B) Sequence alignment between human, rat, and mouse GIP (top) and GLP‐1 (bottom). Grey indicates a mismatch and the black spiral indicates predicted alpha helix structure.
In an attempt to develop GIP receptor antagonists to be used in humans, for instance, as a putative therapeutic against obesity (Asmar et al., 2010; Ahlqvist et al., 2013), we set out to investigate functional differences between rodents and human within the GIP system in terms of ligand binding and signalling properties. As agonists, we chose the endogenous agonist GIP(1–42) from human, rat and mouse, and characterized this in parallel with the species‐corresponding previously described antagonist (Pro3)GIP (Gault et al., 2002) on all three GIP receptors (from human, rat and mouse). The goal was to determine whether the beneficial antagonistic effects of (Pro3)GIP in rodents could possibly be transferred to humans.
Methods
Animals
All animal care and experimental procedures complied with institutional guidelines and were approved by the Danish Animal Experiments Inspectorate (2013‐15‐2934‐00833). Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 18 animals were used in the experiments described here.
Male C57Black/6 (B6) mice (25–30 g) or male Wistar rats (220–250 g) were purchased from Taconic (Lille Skensved, Denmark). The animals were housed in plastic‐bottomed, wire‐lidded cages in air‐conditioned (21C) and humidity‐controlled (55%) rooms with a 12:12 h light–dark cycle and free access to standard rat chow and water. Animals were acclimatized for at least 1 week before use.
Transfections and tissue culture
COS‐7 cells (ATCC, Virginia, USA) were cultured at 10% CO2 and 37 °C in DMEM 1885 supplemented with 10% FBS, 2 mM glutamine, 180 units∙mL−1 penicillin and 45 g∙mL−1 streptomycin. Transient transfection of the COS‐7 cells for cAMP accumulation and competition binding was performed using the calcium phosphate precipitation method with the addition of chloroquine (Kissow et al., 2012).
cAMP assay
Transiently transfected COS‐7 cells were seeded out in white 96‐well plates at a density of 3 * 104 cells per well. The day after, the cells were washed twice with HEPES‐buffered saline (HBS) buffer and incubated with HBS and 1 mM IBMX for 30 min at 37 °C. To test agonists, ligands were added and incubated for 30 min at 37 °C. In order to test for antagonistic properties, the antagonist was pre‐incubated for 10 min, and then the agonist was added and incubated for an additional 20 min. The HitHunterTM cAMP XS assay (DiscoveRx, Herlev, Denmark) was carried out according to the manufacturer's instructions.
Competition binding assay
COS‐7 cells were seeded in clear 96‐well plates the day after transient transfection. The number of cells seeded per well, which is determined by the apparent expression efficiency of the receptor, was aimed to result in 5–10% specific binding of the added radioactive ligand (125I‐human GIP). The following day, cells were assayed by competition binding for 4 h at 4 °C using 15–40 pM 125I‐human GIP as well as unlabelled ligand in 50 mM HEPES buffer (pH 7.2) supplemented with 0.5% BSA (binding buffer). After incubation, the cells were washed twice in ice‐cold binding buffer and lysed using 200 mM NaOH with 1% SDS for 30 min. Nonspecific binding was determined as the binding in the presence of 100 nM unlabelled human GIP. The samples were analysed by the Wallac Wizard 1470 Gamma Counter.
Rat or mouse isolated perfused pancreas
Male C57B6 mice (25–30 g) or male Wistar rats (220–250 g) were anaesthetized (mice: ketamine/xylazine 100/10 mg⋅kg−1 i.p.; rats: 0.0158 mg fentanyl citrate + 0.5 mg fluanisone + 0.25 mg midazolam/100 g; Pharmacy Service, Denmark), and the pancreas was dissected and perfused in situ as described previously (Deacon et al., 2006). Briefly, the pancreas was perfused in a single‐pass system through both the coeliac and the superior mesenteric artery via a catheter inserted into the aorta. All other aortic branches were ligated. The venous effluent was collected for 1 min intervals via a catheter in the portal vein, and stored at −20 °C until analysis. The pancreas was perfused with a modified Krebs Ringer bicarbonate buffer [composition: 118 mM NaCl, 3.0 mM KCl, 2.6 mM CaCl2 2H2O, 1.2 mM KH2PO4, 1.2 mM MgSO4 2H2O, 25 mM NaHCO3, 5 mM fumarate, 5 mM pyruvate, 5 mM glutamate, 7 mM glucose, 0.1% BSA, 5% dextran (Pharmacosmos, Holbaek, Denmark)]. Flow rate was kept constant at 1.5 mL⋅min−1 (mouse) or 4 mL⋅min−1 (rat), perfusion buffer was heated and oxygenated (95% O2, 5% CO2), and pressure was continuously measured throughout the experiment. Rodent (rat/mouse) forms of (Pro3)GIP and of GIP were infused as test substances through a sidearm infusion pump at a flow rate of 0.075 mL⋅min−1 (mouse) or 0.2 mL⋅min−1 (rat). Arginine (10 mM) was infused at the end of each experiment as a positive control.
Hormone analysis
Hormone concentrations in the perfusion effluent were measured using in‐house radioimmunoassays. Glucagon was measured using a midregion‐directed glucagon antiserum (code no. 4304) or a COOH‐terminally directed antiserum (code no. 4305), which measures fully processed glucagon as well as N‐terminally extended molecular forms, in addition, a synthetic glucagon for standards, and 125I‐labelled glucagon (a gift from Novo Nordisk A/S) as tracer (Orskov et al., 1991). Insulin was measured using an antibody cross‐reacting strongly with rat insulin I and II (code no. 2006–3). As the standard, we used human insulin, and the tracer was 125I‐labelled human insulin (gift from Novo Nordisk A/S) (Brand et al., 1995). Somatostatin concentrations were determined using a rabbit antiserum (code no. 1758) raised against synthetic cyclic somatostatin, recognizing both somatostatin 14 and somatostatin 28 (Baldissera et al., 1983), somatostatin 14 as standard and 125I‐labelled Tyr11‐somatostatin (NEX389, Perkin‐Elmer, Skovlunde, Denmark) as tracer.
Sequence alignments
The amino acid sequences of the GIP and GLP‐1 systems were acquired from GenBank of NCBI. The alignment was performed in Geneious 6.0.5 using MAFFT v6.814b. The BLOSUM62 matrix was applied with gap open penalty and offset value of 1.53 and 0.123 respectively.
Data analysis
Pharmacological analyses including Schild plot analyses and statistical analyses were carried out with the GraphPad Prism 6.0 software (GraphPad, San Diego, CA, USA) and Microsoft ExcelTM. Sigmoid curves were fitted logistically with a Hillslope of 1.0. The calculations of K i values were based on the Cheng‐Prusoff formula (DeBlasi et al., 1989). Dose ratios (DR) for the Schild analyses (Lazareno and Birdsall, 1993) were based on the potency shift of GIP(1–42) in the presence of a given antagonist concentration, relative to the absence of antagonist.
Materials
Rat and mouse (Pro3)GIP (cat. no. 027–29 and 027–49, respectively) and rat and mouse GIP (cat. no. 027–12 and 027–27, respectively) were purchased from Phoenix Pharmaceuticals (Karlsruhe, Germany). Human (Pro3)GIP was synthesized by CASLO ApS (Lyngby, Denmark) and human GIP was purchased from Bachem (H5645: Bubendorf, Switzerland). All peptides had a purity of more than 95% by HPLC analyses and an MS‐controlled molecular weight. cDNAs of the human, rat and mouse GIPR were purchased from Origene (Rockville, MD, USA) (SC110906, RN212314 and MC216211, respectively) and cloned into the pCMV‐Script vector. 125I‐labelled human GIP was purchased from PerkinElmer Life Sciences (Skovlunde, Denmark; NEX402).
Results
Larger structural diversity in the GIP system compared with the GLP‐1 system
It is not clear whether the GLP‐1 and GIP systems in rodents are equally transferable to human physiology. As a crude overview of the potential differences between the systems, the amino acid sequences of both the ligands and the receptors were analysed. At the receptor level, it is apparent that the GIP receptor is less conserved between human and rodents (81%) than the GLP‐1 receptor (91% and 93% for rat and mouse, respectively) (Figure 1A). Also, in regard to ligand sequences, the GLP‐1 system is more conserved than GIP (Figure 1B). Thus, the sequence of GLP‐1 is identical between the analysed species, while human GIP has one non‐conservative amino acid substitution (Arg to His) at position 18 and one additional conservative substitution compared with rat (position 40: Ile to Leu) and two compared with mouse GIP (position 30: Lys to Arg and 34: Asn to Ser).
(Pro3)GIP is an agonist with efficacies dependent on the species providing GIP receptors
Due to the reported antagonistic properties and the effects observed in obese and/or diabetic animal models (Gault et al., 2007; Irwin et al., 2007; McClean, 2007), the Pro3‐analogues were investigated on the human, rat and mouse GIP receptors, in parallel with the respective endogenous GIP agonists. As GIP receptor‐induced cAMP accumulation is well established as an important signalling pathway for GIP function, the effects of species‐specific GIP and (Pro3)GIP analogues were evaluated by measuring cAMP accumulation. Compared with human GIP, human (Pro3)GIP was an efficacious agonist on the human GIP receptor approaching the E max of human GIP (Figure 2A), while both rat (Pro3)GIP (Figure 2B) and mouse (Pro3)GIP (Figure 2C) were partial agonists with E max values of ~50 and ~30%, respectively, on their respective receptors. In addition to the differences in efficacy, there was a dramatic shift in potency between GIP and the corresponding (Pro3)GIP within the three species. On the human GIP receptor, we observed 181‐fold difference between human GIP and human (Pro3)GIP, while much larger differences (1300‐fold and 2636‐fold in rat and mouse, respectively) were observed in the rodent systems. These differences were due to both higher species‐specific GIP potencies (from 26 pM for human GIP on human GIP receptors to 10 and 11 pM for rat and mouse GIP on their GIP receptors) and lower (Pro3)GIP species‐specific potencies (from 4.7 nM on human GIP receptors to 13 and 29 nM on rat and mouse GIP receptors respectively).
Figure 2.
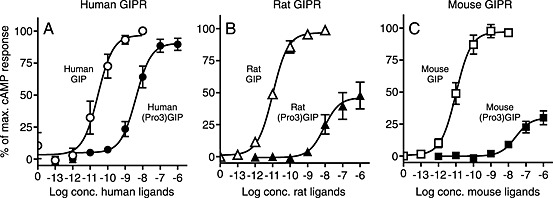
Human (Pro3)GIP is a full agonist, rat and mouse (Pro3)GIP are partial agonists. COS‐7 cells were transiently transfected with either human GIP receptors (GIPR; A), rat GIPR (B), or mouse GIPR (C) and assayed for cAMP accumulation following increasing concentrations of WT GIP and (Pro3)GIP from the corresponding species. The data was normalised to Emax of the endogenous GIP on every receptor and shown as mean±SEM, N≥3. Nonlinear regression was used to calculate the EC50 value and Emax.
(Pro3)GIP is a partial agonist with competitive antagonistic properties in rodent GIP receptors
To determine the potential of the two partial agonists (rat and mouse (Pro3)GIP) as antagonists of GIP‐induced activation, cAMP production was measured as a function of increasing concentration of rat and mouse GIP in the absence or presence of various concentrations of rat and mouse (Pro3)GIP on the corresponding GIP receptors (Figure 3A and B respectively). Reflected by the agonistic properties, (Pro3)GIP increased the cAMP production in the absence or at low GIP concentrations on both receptors. However, a concomitant rightward shift in potency of GIP was observed, which is an indication of a competitive antagonistic nature. At 10, 100, and 1000 nM of rat (Pro3)GIP, the potency of rat GIP was shifted 2‐fold, 4‐fold and 16‐fold compared with the absence of (Pro3)GIP (Figure 3A). Using the calculated EC50 values from these curves, a Schild plot analysis was made (Figure 3C). The Hill coefficient was 0.55 ± 0.20, and the X‐axis intercept, which represents the dissociation constants (K i) for rat (Pro3)GIP, was 13 nM. A similar pattern was observed in the mouse system. Here, the highest concentration of mouse (Pro3)GIP (1 uM) resulted in an 11‐fold right‐shift in the potency of mouse GIP. The Hill coefficient in the Schild Plot in the mouse system was 0.79 ± 0.16 and K i of rat (Pro3)GIP was calculated to be 61 nM (Figure 3D). As the Hill coefficient was <1 in both cases, this indicates confounding factors, presumably determined by the concomitant (partial) agonist and antagonist effects of (Pro3)GIP.
Figure 3.
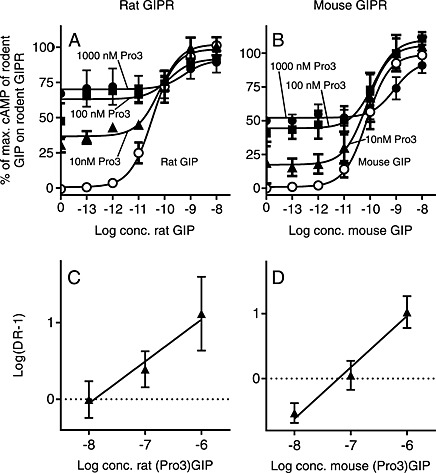
The rodent partial agonist peptides, (Pro3)GIP, are functional antagonists at their corresponding GIP receptors (GIPR). (A) rat GIP and (B) mouse GIP cAMP accumulation concentration–response curves with or without increasing concentrations (10, 100 and 1000nM) of rat (Pro3)GIP or mouse (Pro3)GIP. The results were normalized to E max of the species‐specific GIP in the absence of (Pro3)GIP and shown as mean + SEM, N = 3. Nonlinear regression was used to calculate EC50 values. Schild plot analysis of the dose–response curves of (C) rat GIP and (D) mouse GIP revealed K i‐values of 13 and 61 nM respectively.
Lower activities of all three (Pro3)GIP in the rodent GIP systems compared with the human system
To determine whether the species‐specific differences of (Pro3)GIPs were due to ligand or receptor differences, we investigated the effects of the three (Pro3)GIP ligands across all three GIP receptors. The approaching full agonism as observed for human (Pro3)GIP at the human GIP receptor was not seen with the two rodent (Pro3)GIP ligands, which both demonstrated an E max of ~70% as compared with human GIP (Figure 4A). In contrast, the (Pro3)GIP analogues were equally potent at the human GIP receptor (Table 1). At the rat GIP receptor, there was a lower efficacy of all three (Pro3)GIP analogues, with human (Pro3)GIP again showing the highest efficacy (67 vs. ~40%), but with similar potencies for the three (Pro3)GIP analogues (Figure 4B, Table 1). The same tendency was seen at the mouse GIP receptor with an even lower efficacy of human (Pro3)GIP (41%) and rodent (Pro3)GIP (~30%) compared with mouse GIP (Figure 4C).
Figure 4.
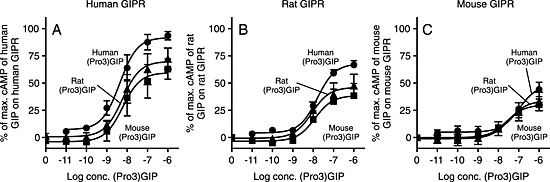
Human (Pro3)GIP is a partial agonist on rodent receptors. COS‐7 cells were transiently transfected with either human GIPR (A), rat GIPR (B), or mouse GIPR (C) and assayed for cAMP accumulation following increasing concentrations of human, rat, and mouse (Pro3)GIP. The data was normalised to Emax of the species specific GIP on every receptor and shown as mean±SEM, N≥3. Nonlinear regression was used to calculate the EC50 value and Emax.
Table 1.
cAMP accumulation induced by human, rat, and mouse GIP and (Pro3)GIP
| Human GIPR | Rat GIPR | Mouse GIPR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| logEC50± SEM | EC50 (nM) | E max± SEM | (N) | logEC50± SEM | EC50 (nM) | E max± SEM | (N) | logEC50± SEM | EC50 (nM) | Emax± SEM | (N) | |
| Human GIP | −11 ± 0.26 | 0.026 | 100 ± 0 | 7 | −10 ± 0.34 | 0.071 | 100 ± 0 | 7 | −9.6 ± 0.26 | 0.25 | 100 ± 0 | 7 |
| Rat GIP | −11 ± 0.11 | 0.0076 | 113 ± 9 | 4 | −11 ± 0.06 | 0.010 | 117 ± 12 | 4 | −11 ± 0.16 | 0.011 | 144 ± 12 | 4 |
| Mouse GIP | −11 ± 0.11 | 0.0052 | 99 ± 3 | 4 | −11 ± 0.13 | 0.0072 | 120 ± 10 | 4 | −11 ± 0.16 | 0.011 | 156 ± 13 | 4 |
| Human (Pro3)GIP | −8.3 ± 0.25 | 4.7 | 90 ± 4 | 4 | −7.8 ± 0.01 | 15 | 80 ± 4 | 3 | −7.0 ± 0.054 | 92 | 71 ± 8 | 3 |
| Rat (Pro3)GIP | −7.9 ± 0.32 | 12 | 73 ± 11 | 5 | −7.9 ± 0.21 | 13 | 64 ± 15 | 5 | −7.4 ± 0.24 | 38 | 57 ± 14 | 5 |
| Mouse (Pro3)GIP | −8.0 ± 0.31 | 11 | 67 ± 8 | 5 | −7.8 ± 0.16 | 15 | 61 ± 12 | 5 | −7.5 ± 0.30 | 29 | 59 ± 8 | 4 |
cAMP accumulation is shown following ligand stimulation of COS‐7 cells transiently transfected with either human, rat, or mouse GIPR cDNA. Data are presented as mean logEC50±SEM and Emax±SEM. N, number of experiments. Data in bold indicate that the receptor and ligands are from the same species.
Higher activities of GIP in the rodent GIP system as compared with the human system
Inspired by the findings that the glutamic acid to proline substitution at the third position in GIP resulted in a lower activity in the rodent systems, we decided to examine whether such species‐dependent differences were present for native GIP. Interestingly, the opposite picture arose here, as the human GIP displayed a lower potency and efficacy on all three receptors compared with the rodent GIP ligands (Figure 5A–C respectively). The rodent GIPs were equally potent and efficacious on all tested receptors, without any considerable change in potency between the receptors (EC50 ~ 10 pM) (Table 1). In contrast, human GIP had a lower efficacy on all three receptors with the largest difference in the rodent receptors with an E max of 75% on the rat GIP receptor and 60% on mouse GIP receptors compared with the corresponding rodent GIP. Likewise, human GIP had a lower potency than the rodent GIPs on all three receptors with the largest difference being 23‐fold on the mouse GIP receptor followed by 10‐fold and 5‐fold on the rat and human GIP receptors respectively.
Figure 5.
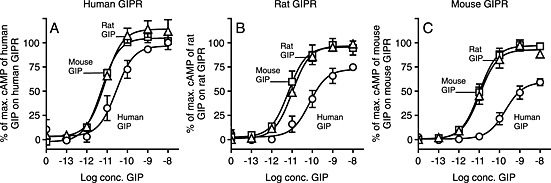
Human GIP is less efficacious and has a lower potency than rodent GIP. COS‐7 cells were transiently transfected with either human GIP receptors (GIPR; A), rat GIPR (B), or mouse GIPR (C) and assayed for cAMP accumulation following increasing concentrations of human, rat and mouse GIP. The data was normalized to Emax of the species specific GIP on every receptor and shown as mean±SEM, N≥3. Nonlinear regression was used to calculate the EC50 value and Emax.
No inter‐species differences in the binding affinities of GIP analogues or (Pro3)GIP analogues
In order to determine whether the differences at both the receptor and the ligand level in terms of cAMP accumulation were due to differences in binding affinity, competition binding experiments were conducted using 125I‐labelled human GIP as the radioligand. Human GIP was found to bind to all three receptors with equally high affinities (K d of 0.90 to 1.1 nM) (Table 2). Likewise, both rodent GIP analogues were similar to human GIP on the tested receptors with a K i in the nM range from 0.27 nM of rat GIP on the mouse GIP receptor to 2.0 nM for mouse GIP on the human GIP receptor. Thus, the functional discrepancies between human and rodent GIP are not due to actual differences in affinity. The (Pro3)GIP analogues displayed in average 10‐fold lower affinities than the native GIP analogues on all three receptors with the lowest affinity observed for the human ligand on mouse GIP receptor (K i of 39 nM) whereas the rest displayed affinities (K i) between 4.8 and 14 nM. Thus, similar to the native GIP ligands, the functional differences between the human (Pro3)GIP and the rodent counterparts are not caused by differences in binding affinities.
Table 2.
Similar displacement of 125I‐labeled hGIP by GIP analogues or (Pro3)GIP analogues.
| Human GIPR | Rat GIPR | Mouse GIPR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| logIC50 ± SEM | K i/K d (nM) | (N) | logIC50 ± SEM | K i/K d (nM) | (N) | logIC50 ± SEM | K i/K d (nM) | (N) | |
| Human GIP | −9.0 ± 0.093 | 0.90 | 7 | −8.9 ± 0.095 | 1.1 | 3 | −9.1 ± 0.17 | 0.72 | 3 |
| Rat GIP | −8.9 ± 0.12 | 1.2 | 3 | −9.2 ± 0.094 | 0.67 | 3 | −9.6 ± 0.099 | 0.27 | 3 |
| Mouse GIP | −8.7 ± 0.30 | 2.0 | 3 | −8.9 ± 0.37 | 1.2 | 3 | −9.1 ± 0.42 | 0.72 | 3 |
| Human (Pro3)GIP | −8.0 ± 0.36 | 10 | 4 | −7.8 ± 0.42 | 14 | 3 | −7.4 ± 0.59 | 39 | 3 |
| Rat (Pro3)GIP | −7.9 ± 0.20 | 12 | 3 | −7.9 ± 0.13 | 12 | 3 | −8.1 ± 0.077 | 8.8 | 3 |
| Mouse (Pro3)GIP | −8.1 ± 0.06 | 7.7 | 3 | −8.1 ± 0.093 | 8.8 | 3 | −8.3 ± 0.26 | 4.8 | 3 |
The binding of 125I‐labeled hGIP to the transiently transfected COS‐7 cells with either human, rat, or mouse GIPR cDNA, was tested in the presence of increasing amounts of human, rat, or mouse GIP and (Pro3)GIP. Data are presented as mean logIC50±SEM. N, number of experiments. Data in bold indicate that the receptor and ligands are from the same species.
(Pro3)GIP stimulates insulin, glucagon and somatostatin release
As most of the studies describing (Pro3)GIP were carried out in rodent models (Gault et al., 2007; Irwin et al., 2007; McClean, 2007), we determined how the receptor binding and activation would translate into hormone secretion, using perfused pancreata from rodents. In brief, the tested analogues were perfused through the pancreata of the rodents, and the venous effluent was collected at 1 min interval and analysed for insulin, glucagon and somatostatin content. In both perfusion models, we used 100‐fold higher concentrations of (Pro3)GIP (100 nM), compared with that of GIP (1 nM), because of the 1000‐fold difference in potencies (Table 1) and the 10‐fold difference in affinity (Table 2). In the mouse pancreas, (Pro3)GIP as well as GIP‐induced secretion of insulin, glucagon and somatostatin (Figure 6A–C) as expected from the agonistic nature of (Pro3)GIP (Figure 2C). However, compared with GIP, there were differences in the magnitude of secretion. Although given at 100‐fold higher concentration, (Pro3)GIP did not reach the same levels of insulin secretion as GIP (Figure 6A), while there was no difference in somatostatin and glucagon release (Figure 6B and C). Similar results were obtained in the rat perfused pancreas model with a significantly lower insulin and somatostatin secretion by (Pro3)GIP compared with GIP (Figure 6D and F, respectively) but with an equal glucagon release by both ligands (Figure 6E).
Figure 6.
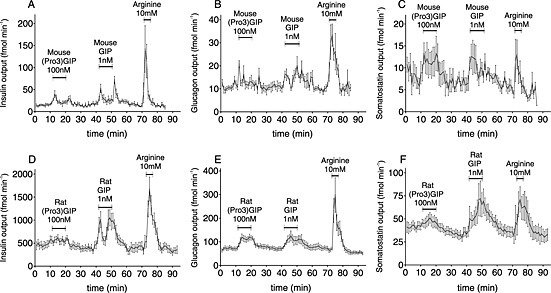
(Pro3)GIP is a agonist on rodent pancreata. (A/D) insulin, (B/E) glucagon, and (C/F) somatostatin secretion from perfused mouse (A, B, C) and rat (D, E, F) pancreata following stimulation with either species specific 100 nM (Pro3)GIP, 1 nM GIP, or 10 mM arginine (N=6‐9). The glucose concentration was 7 mM and data are mean±SEM.
In order to confirm that the observed hormone secretion from perfused pancreata was mediated through the GIP receptors, the same experimental set‐up was repeated using GIP receptor KO mice. As seen in Figure 7, GIP receptor‐deficient mice did not respond to mouse GIP or mouse (Pro3)GIP with either insulin, glucagon or somatostatin secretion (Figure 7A–C respectively). As with the pancreata from wild‐type mice and rats, the positive control, arginine, induced secretion of the respective hormones, validating the experimental set‐up.
Figure 7.
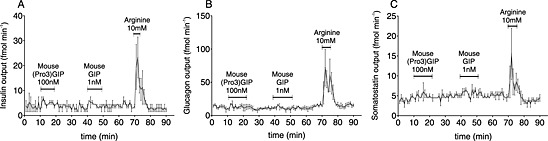
The effects seen in perfused pancreata are mediated through GIP receptors (GIPR). A) insulin, B) glucagon, and C) somatostatin secretion following stimulation of perfused pancreata from GIPR KO mice by either 100 nM mouse (Pro3)GIP, 1 nM mouse GIP, or 10 mM arginine (N=3). The glucose concentration was 7 mM and data are mean±SEM.
Discussion
Our study demonstrated that human (Pro3)GIP was not an antagonist at human GIP receptors, but rather approaches full agonism. At rodent GIP receptors, (Pro3)GIP was a partial agonist with competitive antagonistic properties when evaluated in terms of cAMP accumulation. In terms of its effect on pancreatic hormone secretion, (Pro3)GIP followed the partial agonistic pattern seen in cAMP with regard to insulin, glucagon and somatostatin secretion. However, (Pro3)GIP induced a glucagon release equal to that induced by native GIP in rats.
(Pro3)GIP is not an effective GIP receptor antagonist for future studies of the GIP system
The first GIP receptor antagonists that showed promise were the truncated and amidated forms, GIP(6–30)‐NH2 and GIP(7–30)‐NH2 with affinities (IC50 values) between 3 and 200 nM obtained in heterologous binding experiments (Tseng et al., 1996; Gelling et al., 1997; Hinke et al., 2001). However, elaborate in vivo characterization has not been carried out. The naturally occurring GIP metabolite GIP(3–42) binds with similar affinity (IC50 of 22 nM); however, no antagonistic effect could be demonstrated in vivo in pigs (Deacon et al., 2006). Low MW compounds have also been presented, e.g. the antagonist SKL‐14959 with an affinity (K i value) of 55 nM and the ability to increase blood glucose levels and inhibit insulin secretion during an oral glucose tolerance test in mice (Nakamura et al., 2012). Further development of this compound has not been published. Recently, a promising GIP receptor antibody, Gipg013, with a K i of 6.8 nM was reported (Ravn et al., 2013). Based on the numerous rodent studies that displayed promising improvements in the diabetic and/or obese state, (Pro3)GIP showed potential to fill the role as a promising antagonist in the context of GIP physiology (Gault et al., 2007; Irwin et al., 2007; McClean, 2007; De Toro‐Martin et al., 2014). In ob/ob mice, chronic treatment with (Pro3)GIP improved glucose tolerance and insulin sensitivity (Irwin et al., 2007), while treatment in mice previously on a high‐fat diet resulted in weight loss, improved insulin sensitivity and glucose tolerance (McClean, 2007).
Consistent with previous rodent studies (Gault et al., 2002; Gault et al., 2003; Gault Victor et al., 2007), we find that rodent (Pro3)GIP ligands act as competitive antagonists on the rodent receptors (Figure 3). But when it comes to the human GIP system, human (Pro3)GIP acted as a efficacious agonist (Figure 2A), which is in line with very recent studies that reported substantial agonist activity of human (Pro3)GIP with efficacies up to 83% of human GIP in cAMP release from transiently transfected HEK293 cells (Ravn et al., 2013) or CHL cells (Pathak et al., 2015), and in the reporter gene expression for cAMP‐response element (Al‐Sabah et al., 2014). However, this contrasts to a previous study demonstrating (Pro3)GIP to have 9% of GIP's efficacy on transiently transfected Chinese hamster lung cells (CHL) expressing the human GIP receptor (Gault Victor et al., 2007). These efficacy discrepancies may rely on differences in cell types (CHL, HEK283 and COS‐7 cells); however, consistent for all studies is the finding that (Pro3)GIP was not a neutral antagonist, but has agonist properties in cAMP‐dependent pathways.
Structural GIP divergence between species has markedly affects the pharmacology
Our study emphasizes important interspecies variations within the GIP system. The rodent GIP receptorligands were more potent and efficacious than human GIP on all the tested receptors (Figure 5), with up to 22‐fold and 25‐fold increase in potency of rat GIP and mouse GIP on the mouse GIP receptors (Figure 8A and B). These changes were not matched by a similar increase in binding affinities (Table 2). Only few amino acids are altered among the three GIP receptor ligands (Figure 1A). The most dramatic change is located at position 18 where human GIP has a histidine instead of an arginine, which is found in rat and mouse GIP. Among the few non‐identities observed (in positions 18, 30, 34 and 40), this is the only case where the rodent sequences are identical and, because of the non‐conservative nature (Arg/His), a significant structural divergence is possible. A previous study supports the functional importance of this position, as an alanine substitution in human GIP increased insulin secretion from BRIN‐BD11 cells compared with native GIP (Venneti et al., 2011). Thus, position 18 seems to have a role in the activation mechanism of the GIP receptor, as also supported by a recent study predicting a direct interaction between His18 of human GIP and Ala32 of the N‐terminus of the GIP receptor (Tikhele et al., 2010). Thus, these findings add some detail to the general view that hydrophobic interactions between the C‐terminal part of GIP and the N‐terminus receptor parts account for binding, while the N‐terminus of GIP interacts with other extracellular receptor domains inducing activation (Malde et al., 2007; Parthier et al., 2007). This so‐called two‐step receptor activation not only describes GIP receptor activation but presents a global mechanism for ligand‐binding and receptor activation among class B1 receptors (e.g. glucagon, GLP‐1, secretin, vasoactive intestinal polypeptide and parathyroid hormone receptor) (Gourlet et al., 1996; Hjorth and Schwartz, 1996; Gardella and Jüppner, 2001; Castro et al., 2005; Vilardaga et al., 2011). It also extends beyond class B1, as also some class A seven‐transmembrane receptors (e.g. chemokine, C3a and C5a receptors) are activated by their endogenous peptide ligands in a two (or multi)‐step model involving receptor recognition and activation by different regions in the ligands (Allen et al., 2007; Klos et al., 2013; Thiele and Rosenkilde, 2014).
Figure 8.
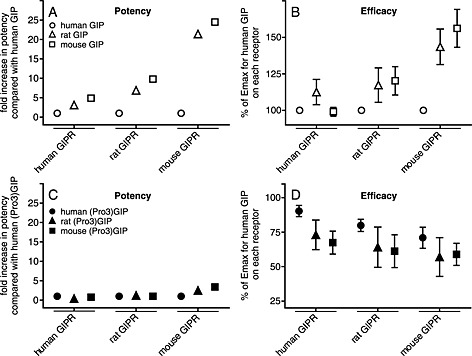
The changes in potency and efficacy of the tested GIP receptors (GIPR) and GIP analogues. The calculated EC50 (A/C) and Emax (B/D) values determined from the cAMP accumulation assays and normalised to human GIP on each species receptor (A, B, and D) or human (Pro3)GIP on each species receptor.
Taken together, the rodent GIP systems seem more active compared with the human system, in terms of GIP (1–42)‐mediated activation and more sensitive to GIP ligand modifications, as judged by the lower activity of (Pro3)GIP. This is relevant and should be considered in studies using rodent models. As our study indicates, when using human GIP in rodent models, researchers are in fact injecting a less potent partial agonist (compared with rodent GIP) that may lead to underestimation of the true GIP activity in such experimental systems.
The therapeutic potential of targeting the GIP receptor
In the wake of the GIP receptor KO studies demonstrating resistance to diet‐induced obesity (Miyawaki et al., 2002), many have tried to antagonize the GIP receptors as a potential target for treating obesity. Accumulating evidence links GIP biology to adiposity and due to the lack of an effective GIP receptor antagonist, the therapeutic potential of antagonizing this receptor in obese patients remains to be assessed. In this context, our study brings attention to a significant interspecies difference within the GIP system, and thus, pharmacological evaluation of novel compounds in rodent models should interpreted cautiously with respect to their relevance for humans.
Conflicts of interest
A. H. S. U., L. S. H., B. S., M. C., B. H. and M. M. R. declare that they have no conflict of interest. J. J. H. has served as a consultant or advisor to Novartis Pharmaceuticals, Novo Nordisk, Merck Sharp & Dohme and Roche and has received fees for lectures from Novo Nordisk, Merck Sharp & Dohme and GlaxoSmithKline. F. K. K. has received fees for consultancy, lectures or being part of an advisory board from AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Eli Lilly, Gilead, Merck Sharp & Dohme, Novo Nordisk, Sanofi and Zealand Pharma, and has received research support from Sanofi and Novo Nordisk.
Acknowledgements
This work was supported by the Novo Nordisk Foundation Center for Basic Metabolic Research and the University of Copenhagen.
Sparre‐Ulrich, A. H. , Hansen, L. S. , Svendsen, B. , Christensen, M. , Knop, F. K. , Hartmann, B. , Holst, J. J. , and Rosenkilde, M. M. (2016) Species‐specific action of (Pro3)GIP – a full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. British Journal of Pharmacology, 173: 27–38. doi: 10.1111/bph.13323.
References
- Ahlqvist E, Osmark P, Kuulasmaa T, Pilgaard K, Omar B, Brøns C et al. (2013). Link between GIP and osteopontin in adipose tissue and insulin resistance. Diabetes 62: 2088–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Crown SE, Handel TM (2007). Chemokine:receptor structure, interactions, and antagonism. Annu Rev Immunol 25: 787–820. [DOI] [PubMed] [Google Scholar]
- Al‐Sabah S, Al‐Fulaij M, Ahmed HA (2014). Selectivity of peptide ligands for the human incretin receptors expressed in HEK‐293 cells. Eur J Pharmacol 741: 311–315. [DOI] [PubMed] [Google Scholar]
- Asmar M, Simonsen L, Madsbad S, Stallknecht B, Holst JJ, Bülow J (2010). Glucose‐dependent insulinotropic polypeptide may enhance fatty acid re‐esterification in subcutaneous abdominal adipose tissue in lean humans. Diabetes 59: 2160–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ (2007). Biology of incretins: GLP‐1 and GIP. Gastroenterology 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- Baldissera FGA, Munoz‐Perez MA, Holst JJ (1983). Somatostatin 1–28 circulates in human plasma. Regul Pept 6: 63–69. [DOI] [PubMed] [Google Scholar]
- Brand CL, Jorgensen PN, Knigge U, Warberg J, Svendsen I, Kristensen JS et al. (1995). Role of glucagon in maintenance of euglycemia in fed and fasted rats. Am J Physiol 269: E469–E477. [DOI] [PubMed] [Google Scholar]
- Brøns C, Jensen CB, Storgaard H, Hiscock NJ, White A, Appel JS et al. (2009). Impact of short‐term high‐fat feeding on glucose and insulin metabolism in young healthy men. J Physiol 587: 2387–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calanna S, Christensen M, Holst JJ, Laferrère B, Gluud LL, Vilsbøll T et al. (2013). Secretion of glucose‐dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta‐analysis of clinical studies. Diabetes Care 36: 3346–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Cordova‐Sintjago T, Liu Y, Kim MS, Morgan D, Booth RG (2013). Molecular pharmacology and ligand docking studies reveal a single amino acid difference between mouse and human serotonin 5‐HT2A receptors that impacts behavioral translation of novel 4‐phenyl‐2‐dimethylaminotetralin ligands. J Pharmacol Exp Therapeut 347: 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga J‐P (2005). Turn‐on switch in parathyroid hormone receptor by a two‐step parathyroid hormone binding mechanism. Proc Natl Acad Sci U S A 102: 16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M, Vedtofte L, Holst JJ, Vilsboell T, Knop FK (2011). Glucose‐Dependent insulinotropic polypeptide: a bifunctional glucose‐dependent regulator of glucagon and insulin secretion in humans. Diabetes 60: 3103–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Toro‐Martin J, Fernandez‐Millan E, Lizarraga‐Mollinedo E, Lopez‐Oliva E, Serradas P, Escriva F et al. (2014). Predominant role of GIP in the development of a metabolic syndrome‐like phenotype in female Wistar rats submitted to forced catch‐up growth. Endocrinology 155: 3769–3780. [DOI] [PubMed] [Google Scholar]
- Deacon CF, Plamboeck A, Rosenkilde MM, deHeer J, Holst JJ (2006). GIP‐(3–42) does not antagonize insulinotropic effects of GIP at physiological concentrations. Am J Physiol Endocrinol Metabol 291: E468–E475. [DOI] [PubMed] [Google Scholar]
- Deblasi A, O'reilly K, Motulsky HJ (1989). Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol Sci 10: 227–229. [DOI] [PubMed] [Google Scholar]
- Deschamps I, Heptner W, Desjeux JF, Baltakse V, Machinot S, Lestradet H (1980). Effects of diet on insulin and gastric inhibitory polypeptide levels in obese children. Pediatr Res 14: 300–303. [DOI] [PubMed] [Google Scholar]
- Ebert R, Illmer K, Creutzfeldt W (1979). Release of gastric inhibitory polypeptide (GIP) by intraduodenal acidification in rats and humans and abolishment of the incretin effect of acid by GIP‐antiserum in rats. Gastroenterology 76: 515–523. [PubMed] [Google Scholar]
- Fulurija A, Lutz TA, Sladko K, Osto M, Wielinga PY, Bachmann MF et al. (2008). Vaccination against GIP for the treatment of obesity. PLoS One 3: e3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardella TJ, Jüppner H (2001). Molecular properties of the PTH/PTHrP receptor. Trends Endocrinol Metabol 12: 210–217. [DOI] [PubMed] [Google Scholar]
- Gault Victor A, Hunter K, Irwin N, Greer B, Green Brian D, Harriott P et al. (2007). Characterisation and glucoregulatory actions of a novel acylated form of the (Pro3)GIP receptor antagonist in type 2 diabetes. Biol Chem 388: 173. [DOI] [PubMed] [Google Scholar]
- Gault VA, O'harte FPM, Harriott P, Flatt PR (2002). Characterization of the cellular and metabolic effects of a novel enzyme‐resistant antagonist of glucose‐dependent insulinotropic polypeptide. Biochem Biophys Res Commun 290: 1420–1426. [DOI] [PubMed] [Google Scholar]
- Gault VA, O'harte FPM, Harriott P, Mooney MH, Green BD, Flatt PR (2003). Effects of the novel (Pro3)GIP antagonist and exendin(9–39)amide on GIP‐ and GLP‐1‐induced cyclic AMP generation, insulin secretion and postprandial insulin release in obese diabetic (ob/ob) mice: evidence that GIP is the major physiological incretin. Diabetologia 46: 222–230. [DOI] [PubMed] [Google Scholar]
- Gault VA, Mcclean PL, Cassidy RS, Irwin N, Flatt PR (2007). Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high‐fat and cafeteria diets. Diabetologia 50: 1752–1762. [DOI] [PubMed] [Google Scholar]
- Gelling RW, Coy DH, Pederson RA, Wheeler MB, Hinke S, Kwan T et al. (1997). GIP(6‐30amide) contains the high affinity binding region of GIP and is a potent inhibitor of GIP1‐42 action in vitro. Regul Pept 69: 151–154. [DOI] [PubMed] [Google Scholar]
- Gourlet P, Vilardaga J‐P, De Neef P, Waelbroeck M, Vandermeers A, Robberecht P (1996). The C‐terminus ends of secretin and VIP interact with the N‐terminal domains of their receptors. Peptides 17: 825–829. [DOI] [PubMed] [Google Scholar]
- Gutniak M, Orskov C, Holst JJ, Ahrén B, Efendic S (1992). Antidiabetogenic effect of glucagon‐like peptide‐1 (7–36)amide in normal subjects and patients with diabetes mellitus. New Engl J MedMassachusetts Medical Society 326: 1316–1322. [DOI] [PubMed] [Google Scholar]
- de Heer J, Rasmussen C, Coy DH, Holst JJ (2008). Glucagon‐like peptide‐1, but not glucose‐dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 51: 2263–2270. [DOI] [PubMed] [Google Scholar]
- Hinke SA, Manhart S, Pamir N, Demuth HU, Gelling W, Pederson RA et al. (2001). Identification of a bioactive domain in the amino‐terminus of glucose‐dependent insulinotropic polypeptide (GIP). Biochim Biophys Acta Protein Struct Mol Enzymol 1547: 143–155. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Schwartz TW (1996). Glucagon and GLP‐1 receptors: lessons from chimeric ligands and receptors. Acta Physiol Scand 157: 343–345. [DOI] [PubMed] [Google Scholar]
- Holst JJ (2004). On the Physiology of GIP and GLP‐1. Horm Metab Res 36: 747–754. [DOI] [PubMed] [Google Scholar]
- Irwin N, Green BD, Parker JC, Gault VA, O'harte FPM, Flatt PR (2006). Biological activity and antidiabetic potential of synthetic fragment peptides of glucose‐dependent insulinotropic polypeptide, GIP(1–16) and (Pro3)GIP(1–16). Regul Pept 135: 45–53. [DOI] [PubMed] [Google Scholar]
- Irwin N, Mcclean PL, O'harte FPM, Gault VA, Harriott P, Flatt PR (2007). Early administration of the glucose‐dependent insulinotropic polypeptide receptor antagonist (Pro3)GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia 50: 1532–1540. [DOI] [PubMed] [Google Scholar]
- Irwin N, Mcclean PL, Patterson S, Hunter K, Flatt PR (2009). Active immunisation against gastric inhibitory polypeptide (GIP) improves blood glucose control in an animal model of obesity‐diabetes. Biol Chem 390: 75. [DOI] [PubMed] [Google Scholar]
- Jørgensen NB, Dirksen C, Bojsen‐Møller KN, Jacobsen SH, Worm D, Hansen DL et al. (2013). Exaggerated glucagon‐like peptide 1 response is important for improved β‐cell function and glucose tolerance after roux‐en‐Y gastric bypass in patients with type 2 diabetes. Diabetes 62: 3044–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BD, Flatt AJS, Flatt PR, Gault VA (2011). Characterization and biological actions of N‐terminal truncated forms of glucose‐dependent insulinotropic polypeptide. Biochem Biophys Res Commun 404: 870–876. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissow H, Hartmann B, Holst JJ, Viby NE, Hansen LRS, Rosenkilde MM et al. (2012). Glucagon‐like peptide‐1 (GLP‐1) receptor agonism or DPP‐4 inhibition does not accelerate neoplasia in carcinogen treated mice. Regul Pept 179: 91–100. [DOI] [PubMed] [Google Scholar]
- Klos A, Wende E, Wareham KJ, Monk PN (2013). International Union of Basic and Clinical Pharmacology. LXXXVII. Complement Peptide C5a, C4a, and C3a Receptors. Pharmacol Rev 65: 500–543. [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Hastrup S, Underwood CR, Wulff BS, Fleckner J (2012). Functional importance of GLP‐1 receptor species and expression levels in cell lines. Regul Pept 175: 21–29. [DOI] [PubMed] [Google Scholar]
- Kreymann B, Ghatei MA, Williams G, Bloom SR (1987). Glucagon‐like peptide‐1 7–36: A physiological incretin in man. Lancet 330: 1300–1304. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ (1993). Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng‐Prusoff equations. Br J Pharmacol 109: 1110–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malde AK, Srivastava SS, Coutinho EC (2007). Understanding interactions of gastric inhibitory polypeptide (GIP) with its G‐protein coupled receptor through NMR and molecular modeling. J Pept Sci 13: 287–300. [DOI] [PubMed] [Google Scholar]
- Mcclean PLI (2007). GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high‐fat diet. Am J Physiol Endocrinol Metabol 293: E1746–E1755. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y et al. (1999). Glucose intolerance caused by a defect in the entero‐insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci 96: 14843–14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H et al. (2002). Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8: 738–742. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Tanimoto H, Mizuno Y, Tsubamoto Y, Noda H (2012). Biological and functional characteristics of a novel low‐molecular weight antagonist of glucose‐dependent insulinotropic polypeptide receptor, SKL‐14959, in vitro and in vivo . Diabetes Obes Metabol 14: 511–517. [DOI] [PubMed] [Google Scholar]
- Nasteska D, Harada N, Suzuki K, Yamane S, Hamasaki A, Joo E et al. (2014). Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high‐fat diet conditions. Diabetes 63: 2332–2343. [DOI] [PubMed] [Google Scholar]
- Orskov C, Jeppesen J, Madsbad S, Holst JJ (1991). Proglucagon products in plasma of noninsulin‐dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest 87: 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D et al. (2007). Crystal structure of the incretin‐bound extracellular domain of a G protein‐coupled receptor. Proc Natl Acad Sci 104: 13942–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak V, Gault VA, Flatt PR, Irwin N (2015). Antagonism of gastric inhibitory polypeptide (GIP) by palmitoylation of GIP analogues with N‐ and C‐terminal modifications improves obesity and metabolic control in high fat fed mice. Mol Cell Endocrinol 401: 120–129. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raufman JP, Singh L, Eng J (1991). Exendin‐3, a novel peptide from Heloderma horridum venom, interacts with vasoactive intestinal peptide receptors and a newly described receptor on dispersed acini from guinea pig pancreas. Description of exendin‐3(9–39) amide, a specific exendin receptor antagonist. J Biol Chem 266: 2897–2902. [PubMed] [Google Scholar]
- Ravn P, Madhurantakam C, Kunze S, Matthews E, Priest C, O'brien S et al. (2013). Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose‐dependent insulinotropic polypeptide receptor. J Biol Chem 288: 19760–19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JS, Brocklehurst KJ, Brown HS, Clarke DS, Coe H, Groombridge SD et al. (2013). Conformational restriction in a series of GPR119 agonists: differences in pharmacology between mouse and human. Bioorg Med Chem Lett 23: 3175–3179. [DOI] [PubMed] [Google Scholar]
- Thiele S, Rosenkilde M (2014). Interaction of chemokines with their receptors – from initial chemokine binding to receptor activating steps. Curr Med Chem 21: 3594–3614. [DOI] [PubMed] [Google Scholar]
- Tikhele SH, Pissurlenkar RRS, Srivastava S, Saran A, Coutinho EC (2010). Mapping interactions of gastric inhibitory polypeptide with GIPR N‐terminus using NMR and molecular dynamics simulations. J Pept Sci 16: 383–391. [DOI] [PubMed] [Google Scholar]
- Tseng CC, Kieffer TJ, Jarboe LA, Usdin TB, Wolfe MM (1996). Postprandial stimulation of insulin release by glucose‐dependent insulinotropic polypeptide (GIP). Effect of a specific glucose‐dependent insulinotropic polypeptide receptor antagonist in the rat. J Clin Invest 98: 2440–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti KC, Malthouse JP, O'harte FPM, Hewage CM (2011). Conformational, receptor interaction and alanine scan studies of glucose‐dependent insulinotropic polypeptide. Biochim Biophys Acta Protein Proteonomics 1814: 882–888. [DOI] [PubMed] [Google Scholar]
- Vilardaga J‐P, Romero G, Friedman P, Gardella T (2011). Molecular basis of parathyroid hormone receptor signaling and trafficking: a family B GPCR paradigm. Cell Mol Life Sci 68: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]