

Summary
Histone H2AX becomes phosphorylated in chromatin domains flanking sites of DNA double-strand break-age associated with γ-irradiation, meiotic recombination, DNA replication, and antigen receptor rearrangements. Here, we show that loss of a single H2AX allele compromises genomic integrity and enhances the susceptibility to cancer in the absence of p53. In comparison with heterozygotes, tumors arise earlier in the H2AX homozygous null background, and H2AX−/− p53−/− lymphomas harbor an increased frequency of clonal nonreciprocal translocations and amplifications. These include complex rearrangements that juxtapose the c-myc oncogene to antigen receptor loci. Restoration of the H2AX null allele with wild-type H2AX restores genomic stability and radiation resistance, but this effect is abolished by substitution of the conserved serine phosphorylation sites in H2AX with alanine or glutamic acid residues. Our results establish H2AX as genomic caretaker that requires the function of both gene alleles for optimal protection against tumorigenesis.
Introduction
The repair of DNA double-strand breaks (DSBs) is critical for preserving genomic integrity. Chromosomal breaks arise accidentally as products of collapsed replication forks or as programmed events during immune receptor rearrangements in lymphocytes and meiotic recombina tion in germ cells. Antigen receptor diversification in lymphocytes is initiated by the RAG-1/2 endonuclease, which introduces DSBs adjacent to the antigen receptor coding segments (V, D, and J gene segments) (reviewed in Fugmann et al., 2000). The subsequent juxtaposition and ligation of V(D)J ends requires ubiquitously ex pressed DNA repair proteins (Ku80, Ku70, DNA-PKcs, XRCC4, Ligase 4, and Artemis) that function in nonhomologous end joining (NHEJ) (reviewed in Bassing et al., 2002b). The assembly of antigen receptor genes is necessary to drive further development and expansion of lymphocyte precursors. V(D)J recombination is restricted to the G0/G1 phase of the cell cycle (Desiderio et al., 1996), where NHEJ appears to be preferentially active (Takata et al., 1998). Another distinct DNA repair pathway, termed homologous recombination (HR), is critical for resolving DSBs during meiotic recombination (Pâques and Haber, 1999) and in proliferating cells (Sonoda et al., 1998). As such, loss of either NHEJ or HR can lead to unresolved strand breaks, resulting in an arrest in lymphocyte development. Removal of p53-dependent apoptotic function abolishes the block in thymocyte expansion in T cells lacking the HR protein Brca1; however, the surviving T cells harbor extensive chromosomal aberrations and have an increased susceptibility to malignant transformation (Mak et al., 2000). In contrast, inactivation of p53 in NHEJ-deficient mice does not restore lymphocyte development, but these mice rapidly succumb to pro-B cell lymphomas (Guidos et al., 1996; Nacht et al., 1996; Vanasse et al., 1999; Difilippantonio et al., 2000; Frank et al., 2000; Gao et al., 2000; Lim et al., 2000) caused by the aberrant repair of DSBs initiated at the IgH locus during V(D)J recombination (Difilippantonio et al., 2002; Zhu et al., 2002; Gladdy et al., 2003). Even a modest reduction in NHEJ activity results in chromosomal aberrations (Karanjawala et al., 1999), and loss of a single DNA Ligase4 allele sensitizes ink4a/arf−/− mice to sarcomas (Sharpless et al., 2001). These results highlight the fact that physiologically generated DSBs are potent substrates for chromosomal translocations and gene amplifications, which have the potential to drive tumorigenesis when apoptosis is circumvented. Hence, the immediate recognition and appropriate resolution of DSBs is critical for maintaining genomic stability.
Toward this end, thousands of histone H2AX molecules in the vicinity of the break are phosphorylated on serine residues (S136 and S139 in the mouse) located within their unique C-terminal tail (reviewed in Redon et al., 2002). H2AX phosphorylation (termed γ-H2AX) is mediated by the phosphatidylinositol 3-kinase (PI-3K)-like protein kinase family members ATM, ATR, and DNA-PK (Redon et al., 2002). γ-H2AX forms at sites of physio logical DSBs in lymphocytes (Chen et al., 2000; Petersen et al., 2001) and germ cells (Mahadevaiah et al., 2001; Fernandez-Capetillo et al., 2003), and is induced in response to replication stress and external DNA damage (Rogakou et al., 1998; Ward and Chen, 2001; Redon et al., 2002). Many known components of the DNA damage response including Brca1, Nbs1/Mre11/Rad50, 53BP1, MDC1, and Rad51 form foci that colocalize with preexisting γ-H2AX foci (Paull et al., 2000; Chen et al., 2000; Schultz et al., 2000; Rappold et al., 2001; Goldberg et al., 2003; Lou et al., 2003; Stewart et al., 2003). Moreover, H2AX is required for the establishment of damage-induced foci containing many of these factors (Celeste et al., 2002; Bassing et al., 2002a; Celeste et al., 2003; Stewart et al., 2003). Defects in the organization of nuclear foci have been associated with impairment of numerous responses to DSBs (Celeste et al., 2002; Bassing et al., 2002a; Fernandez-Capetillo et al., 2002).
Although H2AX appears to modulate both HR and NHEJ, it is not an essential component of either DSB repair pathway. For example, meiotic recombination is not grossly affected in H2AX−/− mice (Fernandez-Capetillo et al., 2003), although H2AX−/− ES cells, like cells deficient in the HR factor RAD54 (Essers et al., 1997), are sensitive to DNA crosslinking agents and impaired in targeted integration (Celeste et al., 2002; Bassing et al., 2002a). Likewise, elimination of the unique C-terminal H2A serine residue in S. cerevisiae leads to only a mild impairment in NHEJ activity (Downs et al., 2000). Immunoglobulin class-switch recombination, which is also dependent on NHEJ, is defective in H2AX−/− mice (Celeste et al., 2002; Petersen et al., 2001; Reina-San-Martin et al., 2003). Although lymphocyte development and V(D)J recombination are largely intact, there is a 2-fold reduction in the absolute number of lymphocytes in H2AX−/− mice (Bassing et al., 2002a; Celeste et al., 2002). Despite the fact that H2AX is not essential for NHEJ or HR, H2AX deficiency results in genomic insta bility (Celeste et al., 2002; Bassing et al., 2002a). Consis tent with this observation, the H2AX locus maps to chromosome 11q23.3, a region that is commonly deleted in human lymphoid and solid tumors (Monni and Knuutila, 2001). In this study, we investigate the role of H2AX in tumorigenesis using a mouse model system.
Results
Deficiency in H2AX and p53 Cooperate in Tumorigenesis
Initially, a cohort of H2AX+/+, H2AX+/−, and H2AX−/− mice was monitored over a period of 1.5 years. None of the H2AX+/+ (n = 17), H2AX+/− (n = 36), or H2AX−/− (n = 34) mice in this aged population developed tumors (Figure 1A). H2AX−/− peripheral T lymphocytes, however, were found to harbor both random breaks and translocations, some of which involved the antigen receptor loci (Celeste et al., 2002). Given the normal health and lifespan of these animals, we speculated that the observed chromosomal rearrangements in H2AX−/− lymphocytes (Celeste et al., 2002) were insufficient to promote tumori-genesis and that cells harboring unresolved broken chromosomes with the potential to form oncogenic translocations would normally be eliminated in vivo through the activation of p53-dependent apoptosis.
Figure 1. H2AX and p53 Deficiency Synergize in Tumorigenesis.

(A) Survival curves for H2AX+/+, H2AX+/−, H2AX−/−, H2AX+/−p53−/−, H2AX+/−p53−/−, and H2AX−/−p53−/− mice. H2AX+/+, H2AX+/−, and H2AX−/− mice (indicated by large diamond) exhibited a tumor free survival for at least 50 weeks. Percent survival is plotted as a function of time in weeks. Number of mice analyzed is indicated.
(B) Tumor spectrum in 15 H2AX+/−p53−/− and 11 H2AX−/−p53−/− mice. Four H2AX−/−p53−/− mice died of unknown causes (data not shown).
To test this hypothesis, we crossed all H2AX geno-types onto a p53−/− background. Loss of one or both alleles of H2AX decreased the average tumor latency of p53−/− mice (Figure 1A). The onset of death was 8 weeks for H2AX−/−p53−/− mice, 10 weeks for H2AX+/+p53−/− mice, and 18 weeks for H2AX+/+p53−/− mice. Moreover, the median survival time of H2AX+/+p53−/− mice (23.4 weeks, n = 21) was significantly longer than either H2AX+/−p53−/− (17 weeks, n = 47) or H2AX−/−p53−/− mice (11.6 weeks, n = 16) (H2AX+/+p53−/− versus H2AX+/− p53−/−, p < 0.05; H2AX+/+p53−/− versus H2AX−/−p53−/−, p < 0.0001). Similar results have been obtained in crosses between p53−/− mice and an independently derived H2AX−/− mouse strain (Bassing et al., 2003 [this issue of Cell]). H2AX−/−p53−/− mice were susceptible predominantly to T and B cell lymphomas (10/11 mice ana lyzed; Figure 1B), as determined by expression of cell surface markers (Thy1.2, CD4 , CD8 , CD3, B220, CD19, IgM, and CD43). Thymic lymphomas expressed Thy1.2, variable amounts of CD4 and/or CD8 and were low for CD3, indicative of an immature T cell phenotype. The two B cell lymphomas were B220-, CD19-, and CD43- positive, and negative for surface IgM (data not shown), and thus were similar to the pro-B cell tumors that consistently develop in mice with dual impairments in p53 and NHEJ (Guidos et al., 1996; Nacht et al., 1996; Vanasse et al., 1999; Difilippantonio et al., 2000; Frank et al., 2000; Gao et al., 2000; Lim et al., 2000). In contrast, the tumor spectrum of H2AX+/−p53−/− mice was broader, and included thymic lymphomas, sarcomas, leukemia, and a brain tumor (Figure 1B). We conclude that partial or complete loss of H2AX synergizes with p53 inactivation to promote tumorigenesis.
Chromosomal Aberrations in H2AX+/−p53−/− and H2AX−/−p53−/− Lymphomas
To clarify the mechanism by which H2AX deficiency modifies cancer susceptibility, we examined metaphases from p53−/−, H2AX+/−p53−/−, and H2AX−/−p53−/− lymphomas by spectral karyotype analysis (SKY). As previously reported (Difilippantonio et al., 2002), p53−/− lymphomas were near tetraploid and rarely harbored clonal chromosomal translocations (average: 0.3 translocations/tumor) (Figures 2A and 2B). In contrast, the majority of H2AX−/−p53−/− lymphomas were near diploid and har bored nonreciprocal clonal rearrangements as well as numerous dicentric chromosomes and centromere fusions, referred to as Robertsonian (Rb) translocations (Figures 2A and 2B, Table 1). In one of the four cases analyzed, telomeric signals remained at the site of fusion (data not shown). H2AX+/−p53−/− lymphomas were generally polyploid but the number of chromosomes gained was less than in p53−/− lymphomas (Figures 2A and 2B, Table 1). In addition, on average fewer clonal translocations were found in H2AX+/−p53−/− lymphomas than in H2AX−/−p53−/− tumors (Figure 2B and Table 1; 1.3 and 2.3, respectively). Thus, the extent of structural and numerical aberrations in H2AX+/−p53−/− lymphomas was intermediate between that observed in H2AX−/−p53−/− and H2AX+/+p53−/− mice.
Figure 2. Structural and Numerical Aberrations in H2AX−/−p53−/−, H2AX+/−p53−/−, and H2AX+/+p53−/− Lymphomas.

(A) SKY analysis of H2AX−/−p53−/− (left), H2AX+/−p53−/− (middle), and H2AX+/+p53−/− (right) thymic lymphomas. Structural aberrations include dicentric (dic) chromosomes, Robertsonian (Rb) fusions, and translocations (T). The total number of chromosomes counted in the metaphase spread is indicated in the lower left-hand corner.
(B) Average number of clonal translocations (left bar graph) and average chromosome number (right bar graph) in H2AX−/−p53−/− (n = 10 tumors), H2AX+/− p53−/− (n = 6), and H2AX+/+p53−/− (n = 3) lymphomas.
Table 1.
Chromosome Gains, Losses, and Rearrangements Observed by SKY
| H2AX+/–p53–/– | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Chr. No. |
Avg. No. |
Ploidy | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | X | Y |
| 3014T | 51–67 | 53.2 | 2n+/– | +1 | +2 | +4 | +5 | +9 | +10 | +11 | +14 | +15 | X | YY | ||||||||||
| +14 | +15 | |||||||||||||||||||||||
| 3151T | 61–64 | 62.8 | 3n+/– | +1 | +4 | +5 | +9 | +11 | –13 | +14 | +15 | –16 | +17 | –18 | del(19) | X | ||||||||
| 3022T | 53–61 | 55 | 3n+/– | +4 | –6 | –7 | –8 | –11 | –18 | XX | ||||||||||||||
| 3149T | 52–73 | 55.8 | 3n+/– | +1 | –3 | +4 | +5 | –6 | –7 | –8 | T(9;15) | +11 | +12 | +14 | +15 | +16 | +17 | T(18;16) | –19 | XX | ||||
| +4 | ||||||||||||||||||||||||
| 3070T | 55–85 | 75 | 4n+/– | +1 | T(2;4) | –3 | +4 | +5 | –13 | –18 | –19 | XX | YY | |||||||||||
| +4 | ||||||||||||||||||||||||
| T(4;1) | ||||||||||||||||||||||||
| T(4;2) | ||||||||||||||||||||||||
| T(4;16) | ||||||||||||||||||||||||
| T(4;X) | ||||||||||||||||||||||||
| 4038T | 54–61 | 56.5 | 3n+/– | –2 | T(3;4) | +4 | Rb(5.5) | –9 | –10 | +11 | –14 | –16 | +17 | –18 | –19 | X | Y | |||||||
| dic(18) | ||||||||||||||||||||||||
|
H2AX–/–p53–/– | ||||||||||||||||||||||||
| 2274B | 39–43 | 40.2 | 2n+/– | +3 | T(12;15) | T(15;12) | der(16) | –19 | X | |||||||||||||||
| 3092B | 37–40 | 38.8 | 2n+/– | T(8;19) | –10 | T(12;15) | T(15;12;15;18) | –19 | XX | |||||||||||||||
| 2280T | 47–51 | 48.3 | 2n+/– | +4 | +9 | +11 | +14 | +15 | XY | |||||||||||||||
| +dic(14) | +dic(15 + T(15;14) | |||||||||||||||||||||||
| T(14;X) | ||||||||||||||||||||||||
| 2929T | 44–47 | 45.5 | 2n+/– | +T(2;6) | +4 | +5 | T(6;16) | +11 | +14 | +15 | XX | |||||||||||||
| +14 | ||||||||||||||||||||||||
| 3150T | 46–48 | 46.4 | 2n+/– | T(2;1) | +4 | +5 | T(7;4) | del(9) | +11 | +12 | +14 | +15 | XY | |||||||||||
| T(2;8) | +15 | |||||||||||||||||||||||
| T(2;14) | ||||||||||||||||||||||||
| 3143T | 43–48 | 46.4 | 2n+/– | +4 | +5 | +7 | +del(9) | +15 | T(19;1) | X | ||||||||||||||
| +5 | +del(9) | T(19;13) | ||||||||||||||||||||||
| 3148T | 37–40 | 38.2 | 2n+/– | –8 | Rb(9.11) | Rb(11.11) | +15 | +16 | T(17;16) | dic(19;8) | X | |||||||||||||
| +11 | Rb(15;15) | Rb(16.16) | ||||||||||||||||||||||
| 3009T | 33–44 | 38.8 | 2n+/– | +15 | Rb(8.16) | XX | ||||||||||||||||||
| Rb(15;15) | T(X;16) | |||||||||||||||||||||||
| T(5;16) | ||||||||||||||||||||||||
| T(17;16) | ||||||||||||||||||||||||
| 2968T | 40–81 | 52.1 | 3n+/– | T(9;4) | T(19;1) | XY | ||||||||||||||||||
| 3154T | 42–54 | 48 | 2n+/– | X | ||||||||||||||||||||
Chromosome gains are highlighted in bold, losses in roman, and rearrangements not affecting the chromosome count are in italics. Listed are those alterations in chromosome number observed in greater than 50% of the metaphases, while the rearrangements were those that were clonal (i.e., found in two or more cells). The exceptions are T(4;1) and T(4;16) in tumor 3070, those translocations involving chromosomes 2 and 19 in tumor 3143, and chromosome 16 in tumor 3009, which demonstrate a prevalence of these chromosomes to be involved in multiple rearrangements. Tumor: tumor name (T, thymic lymphoma; B, pro-B cell lymphoma); Chr. No., range of chromosome number in metaphases from a given tumor; Avg. No., average chromosome number; Ploidy, relative ploidy of tumor with 2n = 40 (normal diploid): chromosomes are indicated across the top row.
In order to assess the consequences of the observed numerical aberrations with respect to genomic imbalances, we used comparative genomic hybridization (CGH), a technique that samples the entire tumor population, to assess gains and losses relative to the most prevalent chromosome ratio (Weaver et al., 1999). CGH analysis revealed very distinct common gains of whole chromosomes or parts of chromosomes 4, 5, 11, and 15 and loss of 19 in thymic lymphomas from p53−/− mice irrespective of the H2AX genotype (Figure 3). The minimal overlapping regions affected were chromosome bands 4C7-D2.2, 5F-G2, 11E1-E2, 15D1-D3, and 19D2-D3. In general, the gains in H2AX+/−p53−/− tumors tended to be entire chromosomes while those in the H2AX−/− p53−/− thymic lymphomas had more regional gains, consistent with an increase in chromosomal translocations detected by SKY analysis (Figure 2 and Table 1). Despite differences in structural chromosomal aberrations and aneuploidy (Figure 2), a similar nonrandom pattern of chromosomal imbalances was found among all H2AX genotypes (Figure 3). Therefore, we hypothesize that the accelerated age of onset of cancer concordant with decreasing amounts of H2AX is due to the increased ability to acquire the minimal set of chromosomal trans-locations necessary for tumor development.
Figure 3. CGH Analysis of H2AX−/−p53−/−, H2AX+/−p53−/−, and H2AX+/+p53−/− Lymphomas.

Summary of CGH analysis using genomic DNA from a H2AX+/+p53−/− thymic lymphoma: designated 2976; four H2AX+/−p53−/− thymic lymphomas: 1, 3014; 2, 3022; 3, 3070; 4, 3149; and eight H2AX−/−p53−/− thymic lymphomas: 1, 2280; 2, 2929; 3, 2968; 4, 3009; 5, 3143; 6, 3148; 7, 3150; and 8, 3154; and two H2AX−/−p53−/− pro-B cell lymphomas: 1, 2274; and 2, 3092. Green bars on the right side of the ideograms indicate chromosome gains and red bars on the left side indicate chromosome losses. Thick bars (e.g., IgH locus in tumor 2274) indicate high-level amplification. All gains and losses for a single tumor are represented in numerical order equidistant from the individual chromosome ideogram. The chromosomal position of the IgH and c-myc loci is indicated in the pro-B cell lymphoma CGH map.
H2AX Monitors Chromosomal Breaks during Lymphocyte Development
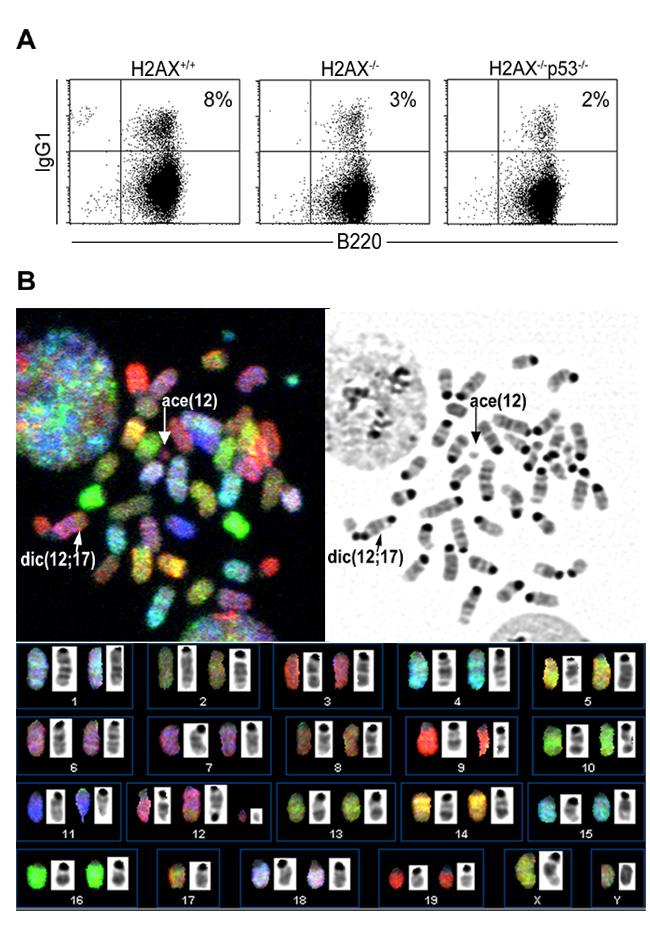
In NHEJ-/p53-deficient lymphomas, oncogenic translocations arise as byproducts of aberrant V(D)J recombination (Difilippantonio et al., 2002; Gladdy et al., 2003; Zhu et al., 2002). To determine whether DNA rearrange ments in H2AX−/−p53−/− mice juxtaposed oncogenes with antigen receptor loci, we analyzed metaphase spreads using a combination of whole chromosome painting probes together with probes for c-myc, IgHCα, and TCRα genes, which reside on chromosomes 15, 12, and 14, respectively. Both pro-B cell lymphomas (designated 3092 and 2274) contained a recurrent T(12; 15) translocation with the distal portion of chromosome 15 fused to the derivative chromosome 12, and an IgHCα signal near the breakpoint (Figure 4). In addition, these tumors exhibited complex translocations that contained coamplified IgHCα and c-myc sequences (Figure 4, Table 1; see also Supplemental Figures S1A and S1B online at http://www.cell.com/cgi/content/full/114/3/■■■/DC1). For both tumors, the breakpoint occurred more than 440 kb 3′ of c-myc (Supplemental Figure S1C). These cytogenetic features of H2AX−/−p53−/− pro-B cell lymphomas are the hallmarks of RAG-dependent lympho mas that invariably develop in NHEJ-/p53-deficient mice (Difilippantonio et al., 2002; Zhu et al., 2002; Gladdy et al., 2003). However, while NHEJ-/p53-deficient pro-B cell lymphomas contain short homologies directly at the translocation junctions (Difilippantonio et al., 2002; Zhu et al., 2002), the cloned chromosome 12 and 15 fusion from tumor 3092 did not have sequence homology immediately at the junction (Supplemental Figure S1D), suggesting that this joining may have been mediated by classical NHEJ. These results are similar to the analysis of translocation breakpoints in pro-B cell lymphomas from an independently derived H2AX−/−p53−/− mouse strain (Bassing et al., 2003). Thus, H2AX and p53 cooper ate to limit the oncogenic potential of DSBs during anti gen receptor rearrangements, a finding that is consis tent with the localization of γ-H2AX to sites of V(D)J recombination (Chen et al., 2000).
Figure 4. Fusions of the c-myc Oncogene to Antigen Receptor Loci in H2AX−/−p53−/− B and T Cell Lymphomas.

Top: SKY analysis of H2AX−/−p53−/− pro-B cell (3092 and 2274) and thymic (2280) lymphomas. Translocations in pro-B and T cell lymphomas are similar to those observed in NHEJ-/p53-deficient lymphomas and ATM−/− thymomas respectively. Bottom: FISH analysis using chromosome 12, 15, and 14 painting probes combined with locus- (IgHCα, c-myc, and TCRα) specific probes. IgH and c-myc are juxtaposed and amplified (indicated by three arrows) in the pro-B cell lymphomas, and TCRα/c-myc are fused at the T(15; 14) translocation breakpoint in the thymic lymphoma.
Amplification of IgH/c-myc in NHEJ-/p53-deficient tumors is driven via breakage-fusion-bridge (BFB) cycles initiated by aberrant processing of RAG-induced DSBs (Difilippantonio et al., 2002; Zhu et al., 2002; Gladdy et al., 2003). Based on CGH, the largest region involved in the duplication/amplification of chromosome 12 occurred in tumor 2274 (Figure 3). This was approximately 45 Mb in size occurring roughly between nucleotide position 62,000,000 (band D2) and 107,000,000 (band F2 containing the IgH locus). In tumor 3092, the region of chromosome 12 gained (~25Mb) was between 82,000,000 (band E) and 107,000,000 (band F2). In 3092 the gain observed on chromosome 15 was larger than in 2274, and was approximately 20 Mb from the middle of band D1 (46,000,000) to D3 (66,000,000). For tumor 2274 the area on chromosome 15 near c-myc that was amplified (as seen by FISH and Southern blot; Supplemental Figure S1) was below the limit of resolution of mouse chromosome CGH (4 Mb). The variations in the boundaries as well as the number of copies of the amplified sequences are likely to depend on where the breaks occur and the number of cycles of BFB (Coquelle et al., 1997). Although activation of fragile sites may determine the boundaries of amplification (Coquelle et al., 1997), the heterogeneity in the distance between amplified units (Supplemental Figure S1) argues against a single site of breakage being utilized for the resolution of the bridge during BFB cycles.
Primary H2AX−/− T lymphocytes carry both random chromosomal rearrangements and more rarely TCRα-associated translocations (Celeste et al., 2002). Consistent with this, the majority of H2AX−/−p53−/− thymomas analyzed (n = 6) harbored clonal translocations without the involvement of antigen receptor loci (Table 1; data not shown). However, one of the H2AX−/−p53−/− thymic lymphomas (2280) harbored clonal rearrangements involving chromosome 14 in the vicinity of the TCRα locus (Table 1). Moreover, on the T(15;14) chromosome, the TCRα gene colocalized precisely with c-myc at the breakpoint, but neither gene appeared amplified as in the pro-B cell lymphomas (Figure 4). FISH mapping, using two BAC probes 96F18 and 212H11 (Difilippantonio et al., 2002), revealed that the breakpoint on chro mosome 15 was within 200 kb telomeric of c-myc. Thus, tumorigenesis in H2AX−/−p53−/− mice can be caused ei ther by failure to correctly repair RAG-mediated DSBs or via other physiological breaks that might arise spontaneously during DNA replication.
Loss of One H2AX Allele Compromises Genomic Integrity
If H2AX behaved as a canonical tumor suppressor, loss of the second H2AX allele would be expected in H2AX+/− p53−/− tumors. However, the wild-type allele remained intact in all of the tumors tested (n = 4), as determined by Western blotting analysis of γ-H2AX (Supplemental Figure S2). An alternative explanation for why one copy of H2AX can synergize with p53 inactivation to promote tumorigenesis is that the efficiency of DSB signaling through H2AX phosphorylation is decreased in H2AX+/− mice. Support for this hypothesis comes from Western blot analysis in which primary H2AX+/− thymocytes clearly expressed less than the normal content of unphosphorylated and phosphorylated H2AX (Figure 5A).
Figure 5. Loss of One Copy of H2AX Leads to Genomic Instability.

(A) Western blot analysis of H2AX (left) and γ-H2AX (right) protein in H2AX+/+, H2AX+/−, and H2AX−/− thymocytes before (C) and 30 min after treatment with 10 Gy γ-irradiation (IR). Nbs1 protein levels are shown below as loading controls. The position of phosphorylated Nbs1 relative to the unphosphorylated form is noted with arrows (upper and lower, respectively).
(B) Lymph node T cells from H2AX+/+, H2AX+/−, and H2AX−/− littermates were cultured for 2 days, and metaphase spreads were examined for chromosomal aberrations. A total of 100 metaphases from two mice of each genotype were scored.
(C) Passage 1 MEFs were either untreated or irradiated with graded doses of γ-irradiation, and metaphases were prepared 16 hr later. Average number of chromosome aberrations (breaks, fragments, and exchanges) is plotted. For each dose, at least 30 metaphases were examined for each genotype.
(D) Examples of metaphases from H2AX+/+p53−/−, H2AX+/−p53−/−, and H2AX−/−p53−/− passage 1 MEFs after treatment with 1 Gy (top) and 5 Gy (lower panels) γ-irradiation. Arrowheads, chromosome type aberrations; arrows, chromatid type aberrations.
(E) Growth kinetics of H2AX+/+, H2AX+/−, H2AX−/−, H2AX+/+p53−/−, H2AX+/−p53−/−, and H2AX−/−p53−/− MEFs at passage 1. The cell number is an average of duplicates from two independent cells lines of each genotype. The slopes (number of cells per day), calculated from early time points in curves, were: H2AX+/+, 8.67 × 105; H2AX+/−, 4.88 × 105; H2AX−/−, 3.10 × 104; H2AX+/+p53−/−, 9.98 × 105; H2AX+/−p53−/−, 5.11 × 105; and H2AX−/−p53−/−, 2.22 × 105.
(F) Radiation sensitivity of H2AX+/+p53−/−, H2AX+/−p53−/−, and H2AX−/−p53−/− passage 1 MEFs, plotted as a fraction of surviving cells relative to unirradiated samples of the same genotype. Data were obtained from an average of duplicates from two independent cell lines.
To determine whether H2AX haploinsufficiency affects chromosomal stability, we examined metaphase spreads derived from T cells isolated from two indepen dent sets of H2AX+/+, H2AX+/−, and H2AX−/− littermates. H2AX+/− T cells (n = 100) exhibited excess numbers of chromosome breaks, fragments, and fusions relative to H2AX+/− cells (n = 100) and approximately half as many aberrations as in H2AX−/− T cells (n = 100) (Figure 5B). Similarly, the average number of chromosomal aberrations in primary fibroblast cultures from H2AX+/−p53−/− mice (1.2) was intermediate between that found in H2AX+/+p53−/− (0.37) and H2AX−/−p53−/− (2.1) MEFs (Figure 5C). Treatment with 0.5-1.5 Gy of γ-irradiation further increased the number of chromosomal aberrations (Figure 5C). Asymmetric chromatid-type exchanges, which are the result of the fusions between two or more broken chromatids, were prominent in H2AX−/−p53−/− and H2AX+/−p53−/− MEFs but were rarely found in irradiated p53−/− MEFs (Figure 5D). Thus, H2AX haploinsufficiency results in genomic instability.
Defects in DSB repair are associated with decreased proliferation in vitro and radiation sensitivity (Barlow et al., 1996; Nussenzweig et al., 1996). Consistent with an increased genomic instability, primary H2AX+/− MEFs exhibited a longer population doubling time than H2AX+/+ controls (Figure 5E), and H2AX+/− ES cells exhibited an intermediate sensitivity to agents that cause DSBs (Supplemental Figure S3). Loss of p53 only partially alleviated the growth defects in H2AX−/− MEFs (Figure 5E). This is in contrast to the complete rescue of proliferation defects in Ku80−/−p53−/− and Xrcc4−/−p53−/− MEFs (Difilippantonio et al., 2000; Frank et al., 2000) but similar to the partial rescue observed in ATM−/−p53−/− MEFs (Xu et al., 1998). Like their p53-wild-type counterparts, primary H2AX+/−p53−/− MEFs exhibited a longer doubling time and were more sensitive than H2AX+/+p53−/− MEFs to ionizing radiation (Figures 5E and 5F). In total, these in vitro results demonstrate that the level of DNA damage, rates of cellular proliferation, and radiation sensitivity are proportional to the gene dosage of H2AX. This is consistent with the earlier onset and in creased frequency of chromosome translocations observed in tumors from H2AX−/−p53−/− versus H2AX+/− p53−/− mice.
Phosphorylation of H2AX Is Critical for Maintaining Genomic Stability
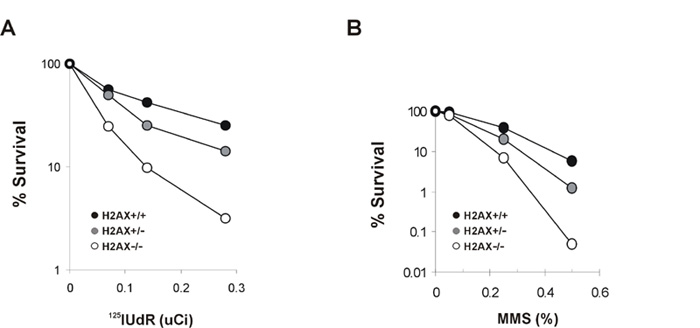
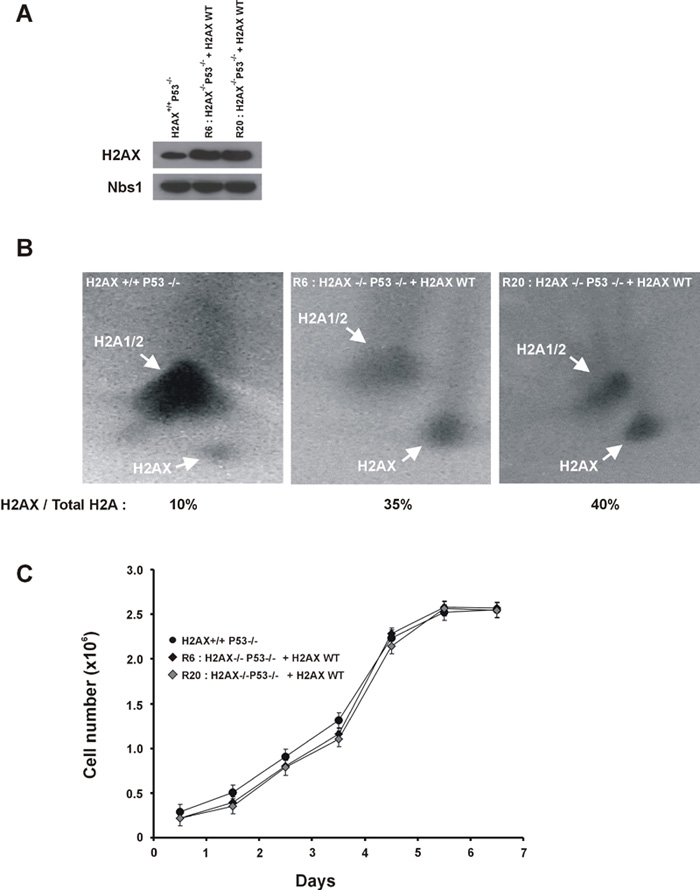
To identify the functional domains of H2AX required for maintaining genomic stability, we simultaneously mutated the SQ motifs (S136/S139) in the C terminus to either alanine (S136/139A) to eliminate phosphorylation or to glutamic acid (S136/139E) to mimic constitutive phosphorylation (Downs et al., 2000). Immortalized H2AX−/−p53−/− MEFs were reconstituted with either wild-type, S136/S139A, or S136/139E H2AX constructs (Celeste et al., 2003). Restoration of H2AX with the wild-type transgene significantly decreased the amount of spontaneous and ionizing radiation-induced DNA dam age observed in metaphases spreads (Figure 6A). Consistent with reconstitution of H2AX function, both the irradiation-induced survival (Figure 6B) and the ability to form 53BP1, Brca1 (Figure 6C), and Nbs1 irradiation- induced foci (IRIF) (Celeste et al., 2003) were rescued by the addition of wild-type H2AX. Levels of H2AX in the wild-type reconstituted H2AX−/−p53−/− lines were at least 3.5-fold higher than in H2AX+/+p53−/− lines (35%–40% of the total H2A content versus 10%, respectively), but this overexpression did not affect the population doubling time or irradiation-induced survival (Supplemental Figure S4; Figure 6B). In marked contrast, substitution of H2AX serine residues by either alanine or glu tamic acid failed to rescue the chromosomal defects observed in the parental H2AX−/−p53−/− cells (Figure 6A). Furthermore, both S136/S139A and S136/139E mutant lines were still sensitive to γ-irradiation (Figure 6B), and failed to form IRIF (Figure 6C) (Celeste et al., 2003). Thus, the placement of negatively charged residues in the COOH-terminal tail does not mimic constitutive phosphorylation. Rather, radiation resistance, foci formation, and genomic integrity require the specific transfer of phosphate groups to serine 136/139 in H2AX.
Figure 6. Phosphorylation of H2AX at Serine 136 and 139 Is Essential for Maintaining Genomic Integrity, Radiation Resistance, and Irradiation-Induced Foci Formation.

(A) Average number of chromosome aberrations in untreated and irradiated H2AX−/−p53−/−cells and H2AX−/−p53−/− cells reconstituted with wild-type H2AX (+H2AX wt), serine to alanine substituted H2AX (+H2AX S136/139A), and serine to glutamic acid substituted H2AX (+H2AX S136/139E). For each dose and genotype, at least 50 metaphases were examined.
(B) Radiation sensitivity of immortalized reconstituted lines plotted as a fraction of surviving cells relative to unirradiated samples of the same genotype. The calculated survival represents the average from two independent cell lines (H2AX−/−p53−/−, DKO13 and DKO15; H2AX+/+p53−/−, PW4 and PW16; +H2AX wt, R6 and R20; +H2AX S136A/S139A, A7 and A8; and +H2AX S136E/S139E, E2 and E5) of each genotype.
(C) 53BP1 (green) and Brca1 (red) staining pattern in reconstituted lines 2 hr after treatment with 10 Gy irradiation. Images were merged (right) to determine colocalization. The wild-type, but not the serine to glutamic acid or serine to alanine reconstituted lines, restores 53BP1 and Brca1 foci formation. Similar results were obtained at 6 hr postirradiation.
Discussion
We have shown that H2AX phosphorylation is critical for protecting the genome from spontaneous-, irradiation- and V(D)J recombination-induced DSBs. In concordance with this, the observed phenotypes tightly correlate with H2AX phosphorylation at sites of DSBs, irrespective of their origin (Chen et al., 2000; Mahadevaiah et al., 2001; Petersen et al., 2001; Rogakou et al., 1998; Ward and Chen, 2001). γ-H2AX is associated with nuclear foci containing factors that are essential for DNA repair, replication, and cell cycle regulation. Although focus formation is dependent on H2AX phosphorylation, H2AX−/− mice exhibit a relatively mild phenotype. H2AX−/− mice are viable and show modest sensitivity to γ-irradiation and impairment in class-switch recombination but display no detectable abnormalities in the for mation of V(D)J recombination signal or coding joints (Celeste et al., 2002; Bassing et al., 2002a). However, since only productive V(D)J joints are selected for during lymphocyte development, subtle defects in the capacity to repair DSBs during V(D)J recombination (or DNA replication) would be difficult to detect. The 50% reduction in the absolute number of T and B lymphocytes in H2AX−/− mice (Celeste et al., 2002) bolsters the idea that there is a reduced efficiency in the repair of RAG- or replication-induced lesions, and that those H2AX−/− cells harboring genome-destabilizing chromosome breaks are eliminated in vivo. Our demonstration that H2AX deficiency synergizes with p53 inactivation to promote tumorigenesis strongly supports this hypothesis.
The T(12;15) rearrangement and coamplification of c-myc/IgH in pro-B cell lymphomas from H2AX−/−p53−/− and NHEJ-/p53-deficient mice (Difilippantonio et al., 2002; Zhu et al., 2002; Gladdy et al., 2003) are strikingly similar. By contrast, it was surprising that only 1 of 14 H2AX-/p53-deficient thymic lymphomas contained a translocation involving the TCRα and c-myc) loci. We speculate that the majority of translocations observed in H2AX−/−/−/− T cells arose via misrepair of replicative DSBs during the rapid expansion of thymocyte precursors rather than during V(D)J recombination. Consistent with this, we have shown an accumulation of DSBs and chromosomal abnormalities in cultured H2AX−/− and H2AX+/− T cells. Interestingly, chromosome breaks were not detected in H2AX−/− B cells stimulated to undergo class-switch recombination, despite the fact that class switching is reduced in the absence of H2AX (Reina-San Martin et al., 2003). We find that p53 deficiency does not rescue the H2AX−/− class-switching defect (Supplemental Figure S5); however, an increased fre quency of H2AX−/−p53−/− B cells (13%) exhibited breaks or translocations, some in the vicinity of the IgH locus (Supplemental Figure S5). Based on these results, we would predict that H2AX/p53 deficiency should result in susceptibility to B cell lymphomas of a more advanced developmental stage than observed in the context of NHEJ/p53 deficiency. Consistent with this, Bassing et al. (2003) have observed H2AX-/p53-deficient B cell lymphomas harboring breakpoints in the vicinity of the switch regions.
Although H2AX, as a histone, probably does not directly mediate enzymatic DNA repair, it clearly plays a critical role in suppressing oncogenic translocations. H2AX phosphorylation at Ser136/139 does not provide the initial signal that attracts proteins to damaged DNA (Celeste et al., 2003). However, once repair proteins have migrated to DSBs, the phosphorylated residues on H2AX may serve as docking sites for the assembly of proteins in chromatin regions distal to the lesion, resulting in the formation of nuclear foci. A high concentration of proteins in a locally damaged chromatin domain may facilitate the repair of a critical subset of (persistent) lesions, which might otherwise be substrates for aber rant recombination reactions. For example, H2AX phosphorylation and its associated foci may directly modulate the higher order chromatin structure in a manner that keeps the broken DNA ends tethered together. In our irradiation experiments, we found that reducing the amount of H2AX increases the propensity for asymmet ric chromosomal interchanges. This could be explained by a failure to keep the ends in close proximity long enough for them to be re-ligated. The availability of these ends to recombine with broken ends from other chromosomes would result in translocations. According to this model, γ-H2AX might be more critical for the response to spontaneous DSBs than during programmed V(D)J recombination, where tethering by the RAG-1/-2 proteins facilitate efficient and rapid rejoining of broken DNA intermediates in a postcleavage synaptic complex (Agrawal and Schatz, 1997; Hiom and Gellert, 1998). While it is unclear precisely how chromatin remodeling facilitates normal DNA repair, there is mounting evidence that γ-H2AX promotes changes in the structural configuration of chromatin (Downs et al., 2000; Fernandez-Capetillo et al., 2003; Reina-San Martin et al., 2003). Thus, chromatin reorganization by γ-H2AX could prevent the premature separation of broken ends, a function that would safeguard against potentially tumorigenic chromosome rearrangements.
Tumor suppressor genes have been classified into two groups: gatekeepers and caretakers (Kinzler and Vogelstein, 1997). Gatekeepers are involved in limiting cell growth and survival, whereas caretakers are DNA repair genes whose inactivation leads to genetic alterations including mutations, rearrangements, and gene amplifications. Although haploinsufficiency is predicted to be a common feature of cancer susceptibility genes (Cook and McCaw, 2000; Quon and Berns, 2001), the majority of known haploinsufficient tumor suppressors are gatekeepers (Bai et al., 2003; Cook and McCaw, 2000; Fero et al., 1998; Goss et al., 2002; Alt et al., 2003; Gruber et al., 2002; Inoue et al., 2001; Magee et al., 2003; Quon and Berns, 2001; Venkatachalam et al., 1998). H2AX modulates DSB repair, and tumorigenesis is synergistically enhanced in H2AX+/−p53−/− mice, even though H2AX levels are 50% of wild-type. The resultant decrease in γ-H2AX formation is insufficient to maintain genomic stability and leads to increased levels of chro mosomal aberrations, reduced growth rates, and radia tion sensitivity. Thus, histone H2AX is a caretaker that does not conform to Knudson's classic two-hit model of tumor suppression (Knudson, 1985).
We have found that combined deficiency in H2AX and p53 in mice results in both lymphoid and solid tumors. Interestingly, the histone H2AX gene, located 11 Mb telomeric to ATM at 11q23.3, is in a region commonly deleted or translocated in several human hematological malignancies and solid tumors (Monni and Knuutila, 2001). Heterozygous deletion of chromosome bands 11q22-q23 is detected at a particularly high frequency in B cell chronic lymphocytic leukemia (B-CLL), mantle cell lymphoma (MCL), and T cell prolymphocytic leuke mia (T-PLL), and is associated with rapid disease pro gression and poor survival in B-CLL (Monni and Knuutila, 2001; Stankovic et al., 2002). Since somatic disruption of ATM only accounts for a distinct subset of tumors involving alterations in 11q (Monni and Knuutila, 2001), other tumor suppressor genes in this chromosomal region are likely to play a pathogenic role. Further studies are therefore needed to directly address the contribution of H2AX haploinsufficiency in human malignancies associated with 11q deletions.
Experimental Procedures
Generation of Mice, MEFs, and T Cells
p53−/− mice (C57BL/6J) were purchased from Taconic Laboratories. H2AX+/− mice (Celeste et al., 2002) were intercrossed to generate H2AX+/−, H2AX+/+, and H2AX−/− mice. H2AX−/−p53−/− mice and primary e10.5 MEFs were generated from intercrosses between H2AX+/−p53+/− × H2AX+/−p53+/−, H2AX−/−p53+/− × H2AX+/−p53+/−, or H2AX−/−p53+/− × H2AX+/−p53−/− mice, where H2AX−/− breeders were female. For growth assays, passage 1 MEFs (105 cells) were plated in duplicate 6-well dishes and counted daily without changing the media. The plating efficiencies, measured after 24 hr, were similar among the different genotypes (H2AX+/+, 219%; H2AX+/−, 191%; H2AX−/−, 119%; H2AX+/+p53−/−, 209%; H2AX+/−p53−/−, 213%; and H2AX−/−p53−/−, 109%). For radiation survival, cells were counted 5 days after treatment with graded doses of γ-irradiation. Counts were normalized to the number of cells in unirradiated controls of the same genotype. Primary lymph node T cells (2 × 106 cells/ml) were stimulated with soluble anti-CD3 and -CD28 antibodies (5 μg/ml each; Pharmingen) and harvested after 48 hr. Immortalized H2AX−/−p53−/− MEFs were generated by a 3T9 protocol. Reconstituted stable MEF lines expressing H2AX wt, H2AX S136/139, or H2AX S136/139E were generated by cotransfection with a puromycin-containing plasmid into immortalized H2AX−/−p53−/− cells as described (Celeste et al., 2003). Immunofluorescent staining of MEFs after methanol fixation using rabbit polyclonal 53BP1 and mouse monoclonal Brca1 antibodies was performed as described (Celeste et al., 2002, 2003).
Histopathological and Flow Cytometric Analysis of Tumors
Mice were examined daily for evidence of morbidity. Tissues were fixed, sectioned, stained, and examined by light microscopy. Single-cell suspensions from lymphomas were stained for various lineage specific cell surface markers (B220, Thy1.2, CD3, CD4, CD8, CD19, CD43, and IgM) and analyzed by flow cytometry as described (Nussenzweig et al., 1996).
Southern Blotting, Spectral Karyotyping, FISH, and CGH Analysis
Southern blotting was performed as described (Difilippantonio et al., 2000; Zhu et al., 2002) using Jh4, c-myc, and 3′IgH RR (3′ of Cα) probes. Mitotic chromosome spreads from primary passage 1 MEFs (H2AX+/+, H2AX+/− H2AX−/−, H2AX+/+p53−/−, H2AX+/−p53−/−, and H2AX−/−p53−/−), immortalized MEFs (H2AX−/−p53−/− and H2AX−/− p53−/− MEFs reconstituted with serine mutant forms of H2AX), primary T and B cells, and lymphomas were prepared as described (Celeste et al., 2002). Spectral karyotype (SKY) was performed as described (Liyanage et al., 1996). For FISH analysis, metaphases were hybridized with BAC probes for the following regions: IgHCα, c-myc, TCRα, and the breakpoint region on chromosome 15 previously identified in Ku−/−p53−/− pro-B cell lymphomas (96F18 and 212H11) (Difilippantonio et al., 2002). Flow-sorted single chromosomes were used for painting probes as described (Difilippantonio et al., 2002). A telomere repeat-specific peptide nucleic acid (PNA) probe labeled with Cy3 (Applied Biosystems, Framingham, MA) was hybridized and detected as described (Difilippantonio et al., 2002). The mouse CGH hybridization protocol followed that of Weaver et al. (1999).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to F. Alt for discussions of unpublished data. We thank L. Stapleton for SKY kits and chromosome painting probes; M. Cowan and H. Chen for assistance in culturing lymphocytes and Southern blotting; S. Jay for mouse caretaking; J. Chen and S. Ganesen for sharing 53BP1 and Brca1 antibodies; C. Bassing for the 3′IgH RR probe; and A. Singer, L. Staudt, M. Nussenzweig, and E. Max for critical comments on the manuscript.
References
- Agrawal A, Schatz DG. RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell. 1997;89:43–53. doi: 10.1016/s0092-8674(00)80181-6. [DOI] [PubMed] [Google Scholar]
- Alt JR, Greiner TC, Cleveland JL, Eischen CM. Mdm2 haploinsufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J. 2003;22:1442–1450. doi: 10.1093/emboj/cdg133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai F, Pei XH, Godfrey VL, Xiong Y. Haploinsufficiency of p18(INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol. Cell. Biol. 2003;23:1269–1277. doi: 10.1128/MCB.23.4.1269-1277.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA. 2002a;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002b;109:S45–S55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114(this issue) doi: 10.1016/s0092-8674(03)00566-x. ■■■. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T, Nussenzweig A. Response to RAG-mediated VDJ cleavage by NBS1 and g-H2AX. Science. 2000;290:1962–1965. doi: 10.1126/science.290.5498.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WD, McCaw BJ. Accommodating haploinsufficient tumor suppressor genes in Knudson's model. Oncogene. 2000;19:3434–3438. doi: 10.1038/sj.onc.1203653. [DOI] [PubMed] [Google Scholar]
- Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- Desiderio S, Lin WC, Li Z. The cell cycle and V(D)J recombination. Curr. Top. Microbiol. Immunol. 1996;217:45–59. doi: 10.1007/978-3-642-50140-1_4. [DOI] [PubMed] [Google Scholar]
- Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J. Exp. Med. 2002;196:469–480. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- Essers J, Hendriks RW, Swagemakers SM, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JH, Kanaar R. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 1997;89:195–204. doi: 10.1016/s0092-8674(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu. Rev. Immunol. 2000;18:495–527. doi: 10.1146/annurev.immunol.18.1.495. [DOI] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- Gladdy RA, Taylor MD, Williams CJ, Grandal I, Karaskova J, Squire JA, Rutka JT, Guidos CJ, Danska JS. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell. 2003;3:37–50. doi: 10.1016/s1535-6108(02)00236-2. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP, Groden J. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–2053. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- Gruber SB, Ellis NA, Scott KK, Almog R, Kolachana P, Bonner JD, Kirchhoff T, Tomsho LP, Nafa K, Pierce H, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297:2013. doi: 10.1126/science.1074399. Erratum: Science 297(5594), 2002. [DOI] [PubMed] [Google Scholar]
- Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996;10:2038–2054. doi: 10.1101/gad.10.16.2038. [DOI] [PubMed] [Google Scholar]
- Hiom K, Gellert M. Assembly of a 12/23 paired signal complex: a critical control point in V(D)J recombination. Mol. Cell. 1998;1:1011–1019. doi: 10.1016/s1097-2765(00)80101-x. [DOI] [PubMed] [Google Scholar]
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001;15:2934–2939. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol. 1999;9:1501–1504. doi: 10.1016/s0960-9822(00)80123-2. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- Knudson AG., Jr. Hereditary cancer, oncogenes, and anti-oncogenes. Cancer Res. 1985;45:1437–1443. [PubMed] [Google Scholar]
- Lim DS, Vogel H, Willerford DM, Sands AT, Platt KA, Hasty P. Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol. Cell. Biol. 2000;20:3772–3780. doi: 10.1128/mcb.20.11.3772-3780.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage M, Coleman A, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, et al. Multicolour spectral karyotyping of mouse chromosomes. Nat. Genet. 1996;14:312–315. doi: 10.1038/ng1196-312. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- Magee JA, Abdulkadir SA, Milbrandt J. Haploinsufficiency at the Nkx3.1 locus. A paradigm for stochastic, dosage-sensitive gene regulation during tumor initiation. Cancer Cell. 2003;3:273–283. doi: 10.1016/s1535-6108(03)00047-3. [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS. Recombinational DNA double-strand breaks in mice precede synapsis. Nat. Genet. 2001;27:271–276. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- Mak TW, Hakem A, McPherson JP, Shehabeldin A, Zablocki E, Migon E, Duncan GS, Bouchard D, Wakeham A, Cheung A, et al. Brcal required for T cell lineage development but not TCR loci rearrangement. Nat. Immunol. 2000;1:77–82. doi: 10.1038/76950. [DOI] [PubMed] [Google Scholar]
- Monni O, Knuutila S. 11q deletions in hematological malignancies. Leuk. Lymphoma. 2001;40:259–266. doi: 10.3109/10428190109057924. [DOI] [PubMed] [Google Scholar]
- Nacht M, Strasser A, Chan YR, Harris AW, Schlissel M, Bronson RT, Jacks T. Mutations in the p53 and SCID genes cooperate in tumorigenesis. Genes Dev. 1996;10:2055–2066. doi: 10.1101/gad.10.16.2055. [DOI] [PubMed] [Google Scholar]
- Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–555. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/g-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quon KC, Berns A. Haplo-insufficiency? Let me count the ways. Genes Dev. 2001;15:2917–2921. doi: 10.1101/gad.949001. [DOI] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 2001;153:613–620. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner WM. Histone H2A variants H2AX and H2AZ. Curr. Opin. Genet. Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, Ferguson DO, O'Hagan RC, Castrillon DH, Lee C, Farazi PA, Alson S, Fleming J, Morton CC, Frank KM, et al. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol. Cell. 2001;8:1187–1196. doi: 10.1016/s1097-2765(01)00425-7. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic T, Stewart GS, Byrd P, Fegan C, Moss PA, Taylor AM. ATM mutations in sporadic lymphoid tumours. Leuk. Lymphoma. 2002;43:1563–1571. doi: 10.1080/1042819021000002884. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanasse GJ, Halbrook J, Thomas S, Burgess A, Hoekstra MF, Disteche CM, Willerford DM. Genetic pathway to recurrent chromosome translocations in murine lymphoma involves V(D)J recombinase. J. Clin. Invest. 1999;103:1669–1675. doi: 10.1172/JCI6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J. 1998;17:4657–4667. doi: 10.1093/emboj/17.16.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- Weaver ZA, McCormack SJ, Liyanage M, du Manoir S, Coleman A, Schrock E, Dickson RB, Ried T. A recurring pattern of chromosomal aberrations in mammary gland tumors of MMTV-cmyc transgenic mice. Genes Chromosomes Cancer. 1999;25:251–260. [PubMed] [Google Scholar]
- Xu Y, Yang EM, Brugarolas J, Jacks T, Baltimore D. Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm–/– mice. Mol. Cell. Biol. 1998;18:4385–4390. doi: 10.1128/mcb.18.7.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.