

Abstract
Inhibition of the cysteine protease cruzain from Trypanosoma cruzi has been studied pre-clinically as a new chemotherapeutic approach to treat Chagas’ Disease. Efficacious effects of vinylsulfone-based cruzain inhibitors in animal models support this therapeutic hypothesis. More recently, substrate-activity screening was used to identify nonpeptidic tetrafluorophenoxymethyl ketone inhibitors of cruzain that showed promising efficacy in animal models. Herein we report efforts to further optimize the in vitro potency and in vivo pharmacokinetic properties of this new class of cruzain inhibitors. Through modifications of the P1, P2 and/or P3 positions, new analogs have been identified with reduced lipophilicity, enhanced potency, and improved oral exposure and bioavailability.
Keywords: Chagas’ Disease, Cruzain, Protease Inhibitors, Pharmacokinetics, Lead Optimization
Graphical Abstract
Chagas’ disease is a parasitic infection caused by the parasite Trpanosoma cruzi and is transmitted to the human host via the triatominae subfamily of reduviidae insects.1–3 While the acute phase of the disease is treatable, the chronic phase of infection is more intractable, requiring extended therapy with high doses of poorly tolerated drugs like nifurtimox and benznidazole.4,5 Moreover, cardiac damage arising from chronic, asymptomatic infection has made Chagas’ disease the leading cause of heart disease in Latin America. Resistance to nifurtimox and benznidazole is also on the rise, highlighting the need for safe and effective new agents that act by distinct molecular mechanisms.
The parasite protease cruzain is the major cysteine protease activity in T. cruzi, with important roles throughout the parasite life cycle.6 The vinylsulfone K777 (1) is an irreversible inhibitor of several mammalian and parasite cysteine proteases, including cruzain (Figure 1). The compound is moderately orally bioavailable (~5–20%) and has proven efficacious in several parasitic disease models. For example, in a dog model of Chagas’ disease, oral treatment with 1 at 50 mg/kg BID for 14 days reduced parasitemia below levels detectable by hemocytometry and partially protected animals from cardiac tissue damage.7
Figure 1.

Structure of the prototypical peptidic cruzain inhibitor 1 (K777), the related activity-based probe 2, and non-peptidic cruzain inhibitors 3 and 4. The P1’-P3 side chains are indicated.
Although 1 has proved invaluable in validating cysteine protease inhibition as a viable therapeutic approach in parasitic diseases, the compound exhibits several suboptimal features from a drug development perspective. In particular, 1 exhibits nonlinear dose-exposure pharmacokinetics, irreversibly inhibits CYP3A4,8 and is a substrate for P-glycoprotein. On the basis of a chemoproteomic analysis employing the N-propargyl analog 2, Renslo and co-workers reported9 that the major target of 1, at least in cell culture, was mammalian cathepsin B of the host cell. This finding may reflect the protonatable, lysosomotrophic nature of 1, a property that has previously derailed clinical development of cathepsin K inhibitors for osteoporosis.10, 11
In parallel with the preclinical development of first-generation protease inhibitor 1, various groups have sought to identify next-generation cruzain inhibitors that are more selective and less peptidic in nature. Thus, in a survey of non-peptidic P2/P3 moieties, Renslo and co-workers identified the vinyl sulfone 3 and solved a high-resolution structure of 3 bound to cruzain.12 Contemporaneously, Ellman and co-workers reported the co-crystal structure of the non-peptidic inhibitor 4,13 which was discovered using substrate-activity screening.14 Comparing these X-ray structures with earlier structures of 1 revealed that both peptidic and non-peptidic inhibitors make contact with the S1’-S3 subsites of cruzain and share important hydrogen bonding interactions, such as with the backbone carbonyl of Asp161. Interestingly, in inhibitor 4 it is the C–H bond at C-5 of the triazole ring that donates this hydrogen bond. The higher affinity of 4 compared to 3 for cruzain may arise in part from an additional hydrogen bond between the P3 quinoline nitrogen atom and Ser61, and from a water-mediated interaction between the basic amine and Glu208. Compound 4 was effective against T. cruzi parasites in two different cell-based assays and when administered to T cruzi infected mice at 20 mg/kg BID (ip) for 27 days, compound 4 afforded a hematological cure in 2/4 treated animals.13
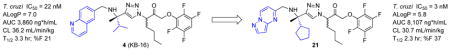
Herein we describe efforts to improve the potency and pharmacokinetic (PK) properties of tetrafluorophenylmethyl ketone inhibitors derived from 4. In vivo PK profiling of 4 in mice revealed moderate half-life and clearance values and reasonable bioavailability. These in vivo data were correlated with in vitro ADME surrogates to guide an optimization strategy focused on reducing lipophilicity while maintaining or improving upon the in vitro biochemical and antiparasitic activity. Structure-aided design was employed to select P1-P3 side chains that would retain key hydrophobic and hydrogen bonding interactions while reducing overall lipophilicity (calculated as ALogP in Vortex, Dotmatics). Guided by this improved analogs such as 21 were developed that exhibit improved anti-trypansomal activity in vitro, combined with superior oral exposure, half-life, and bioavailability as compared to 4.
The in vivo pharmacokinetic properties of 4 were evaluated to establish a baseline for further optimization work. In vitro ADME parameters were also determined in the hope that in vitro surrogates could be correlated with key in vivo parameters. The in vivo PK profile of 4 in mice turned out to be quite reasonable as a starting point for further optimization. Hence, the compound exhibits a reasonably long half-life in mice (T1/2 = 3.3 hr), moderate clearance (CL = 36.2 mL/min/kg), and oral bioavailability of ~20%. A steady-state volume of distribution (Vss) of 4.2 L/kg suggested good tissue penetration, as is desirable for a Chagas’ therapeutic.15 Like 1, compound 4 inhibits CYP3A4 in vitro in the low μM regime (CYP 3A4 IC50 = 3.8 μM). Despite being highly lipophilic (AlogP = 7.0) compound 4 was found to exhibit reasonable stability to cultured liver microsomes, consistent with the long half-life observed in mice (T1/2 ~ 3.3 hr). Permeability in an MDCK cell monolayer assay was modest-to-low and solubility was qualitatively estimated to be low as well, both factors likely contributing to the modest bioavailability observed. Thus, an initial target of the optimization campaign was to reduce lipophilicity, with the expectation that improvements in solubility and permeability would contribute to greater bioavailability and overall exposure on oral dosing.
To improve both potency and in vivo exposure, we sought to identify new P1-P3 moieties that would retain the hydrogen bond to Ser61 while reducing overall lipophilicity (Figure 1). We were aided in this effort by inspection of the previously disclosed X-ray crystal structure of 4 bound to cruzain (pdb 3IUT).13 This structure reveals that the hydrogen bond to Ser61 in S3 is largely solvent exposed, implying that more hydrophilic heterocycles at P3 might retain this interaction while affording the desired reduction in overall lipophilicity. The S2 sub-site of cruzain is the most lipophilic and solvent inaccessible and was therefore expected to contribute more than any other sub-site to a favorable binding free energy. Accordingly, we considered that a larger, more lipophilic group at this position might be beneficial in terms of potency. Among several larger P2 groups explored previously,13 we selected cyclopentyl and isopropyl for the current study. Finally, the S1 sub-site in cruzain is rather more solvent exposed and forms relatively few hydrophobic interactions with the n-Bu side chain of 4 (e.g., a P1 ethyl analog is nearly as potent as 4). We briefly explored small gem-dialkyl substitution at P1 (e.g. cyclopropyl) but found such analogs were devoid of either biochemical or antitrypanosomal activities yet were more potent inhibitors of key CYPs, possibly a consequence of a more exposed triazole ring in such analogs. Our efforts at P1 were thus directed at side chains that retain the larger cone angle of n-butyl (as in 4) but with the introduction of heteroatoms to modulate overall lipophilicity (Figure 2).
Figure 2.

Summary of the structural chemotypes explored in this work.
New analogs were synthesized using the general synthetic approaches described previously for 4 and similar analogs.13,14 Briefly, amino acid starting materials bearing the desired P1 substituent were converted to α-azido acids and then in four steps to the tetrafluorophenylmethyl azido ketone intermediates A (Scheme 1). Propargyl amines bearing the desired P2 substituent were prepared in non-racemic form using Ellman’s chiral sulfinamide auxiliary.16 The amines were next subjected to reductive amination with heterocyclic aldehydes or alternatively were coupled to heteroaryl carboxylic acids to afford intermediates B bearing the desired P2 and P3 substituents. Finally, copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction between intermediates A and B afforded the final analogs 4-23.
Scheme 1.

General synthetic approach used to prepare compounds 4-23. Conditions: (a) isobutyl chloroformate, N-methylmorpholine, THF, −40 °C; (b) diazomethane, THF, 0 °C; (c) HBr, THF, 0 °C; (d) 2,3,5,6-tetrafluorophenol, KF, DMF, 0 °C; (e) 4Å molecular sieves, toluene, rt; (f) NaBH4, MeOH, 0 °C; (g) when Y = OH; HATU/DIEA/DMF; (h) sodium ascorbate, CuSO4, 1:1 H2O:t-BuOH
The new analogs were tested for cruzain inhibition using a biochemical assay, as described previously.17 To facilitate the rapid evaluation analogs, we determined IC50 values rather than full kinetic parameters. While kinact/Ki values are generally preferred when evaluating irreversible inhibitors, IC50 values can provide useful rank-order SAR, provided that pre-incubation times are consistent. The assay was performed with a final cruzain concentration of 0.1 nM in a pH 5.5 assay buffer comprising 100 mM sodium acetate, 5 mM DTT, 0.01% Triton X-100 and 10 mM EDTA. The substrate Z-FR-AMC was employed at a concentration of 1 uM. Compounds were pre-incubated with cruzain for 5 minutes before addition of substrate. The increase in fluorescence was determined over 5 minutes for each concentration of test compound and IC50 values determined using GraphPad Prism 4.
Antitrypanosomal activity was assessed using a high-content imaging based assay as described elsewhere.18 Briefly, C2C12 cells and T. cruzi parasites were seeded into assay imaging plates containing test compounds, incubated for 72 hours, and fixed in 4% PFA. Cells were stained with DAPI, which labels both host cell nuclei and parasite kinetoplast DNA. Stained and fixed cells were then imaged with an IN Cell Analzyer 2000 (20x/0.75, Plan Apo, CFI/60, 350ex/455em, exposure 100 ms) and analyzed with the IN Cell Developer 1.9 image processing software. Image analysis provides a measure of both host cell viability as well as the number of parasites per host cell.
We were pleased to find that the majority of new analogs exhibited antitrypanosomal activity in the mid-to-low nM range (Table 1). Cellular activity could be roughly correlated with cruzain IC50 values, with some notable exceptions (7, 18, 19, 23). For reasons that remain unclear, cellular and biochemical activities were more strongly correlated for analogs with the lipophilic n-Bu side chain at P1 than for analogs with more hydrophilic side chains at this position. We also found that an amide connection at P2/P3 (when X is C=O) produced analogs of inferior potency in both the antitrypanosomal and biochemical assays. At P2, the cyclopentyl side chain was preferred over isopropyl in terms of potency, but at the cost of higher AlogP values in such analogs. Fortunately a variety of heterocyclic P3 substituents were well tolerated and this allowed for modulation of AlogP values while retaining potency. As alluded to above, introduction of an ether or sulfone function in the P1 side chain produced analogs that were generally very potent in the cellular assay, but unexpectedly weak inhibitors of cruzain in vitro. It is unclear whether these effects are related to differences in permeability, sub-cellular localization, or the engagement of additional targets. Among the most favorable P3/P2 combinations with regard to potency was pyrazolopyrimidine at P3 and cyclopentyl at P2, as in compound 21, which was exceptionally potent in both assays and exhibited a reasonable AlogP of 5.8.
Table 1.
Calculated lipophilicity, biochemical, and antitrypanosomal activity of cruzain inhibitors described in this work.
| compound | P3 | X | P2 | P1 | ALogPa | cruzain IC50 (μM)b | T. cruzi IC50 (μM)c |
|---|---|---|---|---|---|---|---|
| 4 | Quin | CH2 | i-Pr | n-Bu | 7.0 | 0.37 | 0.022 |
| 5 | Quin | C=O | i-Pr | n-Bu | 6.7 | 1.35 | 0.70 |
| 6 | Quin | CH2 | i-Pr | -CH2CH2SO2Me | 4.8 | 0.56 | 0.38 |
| 7 | Quin | CH2 | i-Pr | -CH2CH2OMe | 5.1 | 0.34 | 0.002 |
| 8 | Quin | CH2 | c-Pent | n-Bu | 7.5 | 0.027 | 0.015 |
| 9 | Py | CH2 | i-Pr | n-Bu | 5.6 | 1.10 | 0.143 |
| 10 | Py | C=O | i-Pr | n-Bu | 5.3 | 23 | 1.6 |
| 11 | BnThia | CH2 | i-Pr | n-Bu | 6.8 | 0.037 | 0.002 |
| 12 | BnThia | C=O | i-Pr | n-Bu | 6.6 | 3.59 | 0.65 |
| 13 | BnThia | CH2 | i-Pr | -CH2CH2SO2Me | 4.8 | 0.125 | 0.012 |
| 14 | BnThia | CH2 | i-Pr | -CH2CH2OMe | 4.9 | 0.063 | 0.044 |
| 15 | BnThia | CH2 | c-Pent | n-Bu | 7.4 | 0.005 | 0.003 |
| 16 | BnThia | CH2 | c-Pent | -CH2CH2SO2Me | 5.3 | 0.025 | 0.010 |
| 17 | BnThia | CH2 | c-Pent | -CH2CH2OMe | 5.6 | 0.124 | 0.007 |
| 18 | BnFur | CH2 | i-Pr | -CH2CH2SO2Me | 5.0 | 0.88 | 0.003 |
| 19 | BnFur | CH2 | c-Pent | -CH2CH2SO2Me | 5.5 | 0.79 | 0.005 |
| 20 | PzPm | CH2 | i-Pr | n-Bu | 5.3 | 0.075 | 0.008 |
| 21 | PzPm | CH2 | c-Pent | n-Bu | 5.8 | 0.003 | 0.003 |
| 22 | PzPm | CH2 | i-Pr | -CH2CH2SO2Me | 3.2 | 0.467 | 2.34 |
| 23 | PzPm | CH2 | c-Pent | -CH2CH2OMe | 4.0 | 2.82 | 0.020 |
Calculated in Vortex, from Dotmatix, LLC.
Inhibition of recombinant cruzain in a biochemical assay, as described in text.
Antitrypanosomal activity of compounds against intracellular T. cruzi in C2C12 host cells using high-content imaging as described in the text and in ref 18.
Selected analogs with reduced AlogP values and/or that exhibited in vitro antitrypanosomal activity superior to 4 were further evaluated in a panel of in vitro ADME assays (Table 2). The more potent analogs unfortunately also inhibited CYP3A4 with IC50 values lower than for 4, and regardless of the particular heterocyclic group at P3. It seems plausible that CYP inhibition may be related to the triazole ring shared by all of these analogs.
Table 2.
Antitrypanosomal Activity and in vitro ADME properties of selected cruzain inhibitors.
| compound | MDCK (Papp cm/s * 10−6) | CYP 3A4 IC50 (μM) | T. cruzi IC50 |
|---|---|---|---|
| 4 | 3.02 | 3.8 | 0.022 |
| 6 | 3.49 | 1.2 | 0.38 |
| 8 | 0.48 | 0.26 | 0.015 |
| 11 | 2.26 | 0.3 | 0.037 |
| 16 | 6.8 | 0.20 | 0.01 |
| 20 | 15.1 | 0.314 | 0.01 |
| 21 | 2.9 | 0.263 | 0.003 |
Permeability (A to B) in MDCK cell monolayers.
Inhibition of CYP3A4 in vitro.
Antitrypanosomal activity of compounds against intracellular T. cruzi in C2C12 host cells using high-content imaging as described in the text and in ref 18.
Three new analogs exhibiting improved potency compared to 4 and reasonable in vitro ADME profiles were chosen for evaluation in PK studies in mice. Compound 21 was an easy choice, given its exceptional potency in both antitrypanosomal and biochemical assays, combined with a lower AlogP value of 5.8. Analogs 8 and 11 were selected as very close analogs of 4, differing at P2 (cyclopentyl for isopropyl) or P3 (benzthiazole for quinoline), respectively.
Dosing for the PK studies was at 40 mg/kg for the PO arm and 10 mg/kg for the IV arm. Compared to 4, the three new analogs 8, 11, and 21 exhibited similar or superior AUC, T1/2, CL, and F% values. Exposure following PO dosing was two-fold higher for 8 and 21 than for compound 4. The higher lipophilicity of analogs 8 and 11 could be correlated with higher volume of distribution (Vss) in vivo. Notably, compound 21 retained Vss comparable to 4, despite its more favorable (lower) AlogP value. Interestingly, AlogP also seemed to correlate with oral bioavailability (%F). Hence, compounds 4, 8, and 11, all with AlogP values close to 7, exhibited similar %F values in the range of 19.1–23. Compound 21 with an AlogP of 5.8 by contrast, showed a significantly improved %F of 37. Overall, the ~10-fold improved in vitro potency of compound 21, combined with its superior in vivo PK properties suggest this analog as a promising candidate for further preclinical evaluation.
In summary, we have described the optimization of both in vitro and in vivo properties of non-peptidic tetrafluorophenoxy ketones with potent activity against T. cruzi parasites in cell culture. The introduction of the pyrazolopyrimidine ring system at P3, when combined with a cyclopentyl group at P2 afforded analog 21 with significantly enhanced potency and reduced AlogP values as compared to progenitor compound 4. These modifications also translated into improved in vivo exposure and bioavailability following oral administration. Although none of the modifications explored in this effort could divorce CYP3A4 inhibition from the desired anti-parasitic effects, compounds like 21 exhibit antitrypanosomal activity at concentrations ~100-fold lower than are required to significantly inhibit CYP3A4. This suggests that a useful therapeutic window may already be in hand and that further pre-clinical evaluation of such analogs is warranted.
Table 3.
In vivo pharmacokinetic properties of selected cruzain inhibitors.
| Compound | Route | Dose (mg/kg) | Cmax (ng/mL) | AUC (ng*h/mL) | T1/2 | CL (mL/min/kg) | Vss (L/kg) | %F |
|---|---|---|---|---|---|---|---|---|
| 4 | IV | 10 | 4133 | 4673 | 2.95 | 36.2 | 4.2 | |
| PO | 40 | 833 | 3860 | 3.34 | 21 | |||
| 8 | IV | 10 | 8706 | 4.2 | 19 | 5.4 | ||
| PO | 40 | 8096 | 3 | 23 | ||||
| 11 | IV | 10 | - | 9613 | 5.67 | 16.6 | 6.5 | |
| PO | 40 | 1228 | 6566 | 3.41 | 19.1 | |||
| 21 | IV | 10 | 5370 | 3.04 | 30.7 | 4.19 | ||
| PO | 40 | 8107 | 2.32 | 37 |
Acknowledgments
ARR acknowledges funding from the Sandler Foundation. SP was an AMGEN Summer Undergraduate Research Fellow. JAE acknowledges funding from the NIH (R01-GM054051).
References and notes
- 1.Chagas C. Mem Inst Oswaldo Cruz. 1909;1:159. [Google Scholar]
- 2.Rassi A, Jr, Rassi A, Marin-Neto JA. Lancet. 2010;375:1388. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 3.Coura JR, Vinas PA. Nature. 2010;465:S6–S7. doi: 10.1038/nature09221. [DOI] [PubMed] [Google Scholar]
- 4.Castro JA, Diaz de Toranzo EG. Biomed Environ Sci. 1988;1:19. [PubMed] [Google Scholar]
- 5.Viotti R, Vigliano C, Lococo B, Alvarez MG, Petti M, Bertocchi G, Armenti A. Expert Rev Anti-infect Ther. 2009;7:157. doi: 10.1586/14787210.7.2.157. [DOI] [PubMed] [Google Scholar]
- 6.Harth G, Andrews N, Mills AA, Engel JC, Smith R, McKerrow JH. Mol Biochem Parasitol. 1993;58:17. doi: 10.1016/0166-6851(93)90086-d. [DOI] [PubMed] [Google Scholar]
- 7.Barr SC, Warner KL, Kornreic BG, Piscitelli J, Wolfe A, Benet L, McKerrow JH. Antimicrob Agents Chemother. 2005;49:5160. doi: 10.1128/AAC.49.12.5160-5161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson W, Christians U, Benet L. Drug Metab Dispos. 2000;28:1343. [PubMed] [Google Scholar]
- 9.Choy JW, Bryant C, Calvet CM, Doyle PS, Gunatilleke SS, Leung SSF, Ang KKH, Chen S, Gut J, Oses-Prieto Ja Johnston JB, Arkin MR, Burlingame AL, Taunton J, Jacobson MP, McKerrow JH, Podust LM, Renslo AR. Beilstein J Org Chem. 2013;9:15–25. doi: 10.3762/bjoc.9.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black W, Percival M. ChemBioChem. 2006;7:1525. doi: 10.1002/cbic.200600149. [DOI] [PubMed] [Google Scholar]
- 11.Falgueyret JP, Desmarais S, Oballa R, Black WC, Cromlish W, Khougaz K, Lamontagne S, Massé F, Riendeau D, Toulmond S, Percival MD. J Med Chem. 2005;48:7535. doi: 10.1021/jm0504961. [DOI] [PubMed] [Google Scholar]
- 12.Bryant C, Kerr ID, Debnath M, Ang KKH, Ratnam J, Ferreira RS, Jaishankar P, Zhao D, Arkin MR, McKerrow JH, Brinen LS, Renslo AR. Bioorg Med Chem Lett. 2009;19:6218. doi: 10.1016/j.bmcl.2009.08.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brak K, Kerr ID, Barrett KT, Fuchi N, Debnath M, Ang K, Engel JC, McKerrow JH, Doyle PS, Brinen LS, Ellman JA. J Med Chem. 2010;53:1763. doi: 10.1021/jm901633v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brak K, Doyle PS, McKerrow JH, Ellman JA. J Am Chem Soc. 2008;130:6404. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbina JA. Acta Tropica. 2010;115:6404. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 16.Patterson AW, Ellman JA. J Org Chem. 2006;71:7110. doi: 10.1021/jo061160h. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira RS, Bryant C, Ang KKH, McKerrow JH, Shoichet BK, Renslo AR. J Med Chem. 2009;52:5005. doi: 10.1021/jm9009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel JC, Ang KK, Chen S, Arkin MR, McKerrow JH, Doyle PS. Antimicrob Agents Chemother. 2010;54:3326. doi: 10.1128/AAC.01777-09. [DOI] [PMC free article] [PubMed] [Google Scholar]