

Abstract
A novel series of amidinohydrazone-derived furin inhibitors was prepared, the most potent compounds 17 and 21 inhibit furin with Ki values of 0.46 and 0.59 µM, respectively. In contrast to inhibitor 17, which still contains a guanidino residue, compound 21 possesses only weakly basic amidinohydrazone groups.
Keywords: furin, proprotein convertase, protease inhibitor, amidinohydrazone, serine protease
Furin is a type-I transmembrane protein which contains a Ca2+-dependent subtilisin-like serine protease domain. It is ubiquitously found in humans and the best characterized member of the family of proprotein convertases (PCs), which convert numerous precursors of secreted proteins to their active forms. Various studies revealed that furin plays a crucial role in many bacterial and viral diseases, tumorigenesis, neurodegenerative disorders or diabetes.1, 2
Furin possesses a strong preference for substrates with the multibasic cleavage motif Arg-X-Arg/Lys-Arg↓-X. In addition to various types of peptidic substrate-analogues3–5 also first potent nonpeptidic furin inhibitors have been described based on guanylated 2,5-dideoxystreptamines.6 Recently, we have designed a series of highly potent peptidomimetic furin inhibitors, which contain a 4-amidinobenzylamide group as the P1 residue. Using a cell based assay we could demonstrate that these inhibitors are able to reduce the cleavage of the hemagglutinin precursor HA0 in H7N1 fowl plague viruses.7 Correct cleavage of the HA0 precursor is a crucial step during an influenza infection.8
In parallel to the design of these inhibitors we screened various compounds available to us for furin inhibition and could identify a bis(amidinohydrazone)-derivative 1 with a Ki value of 1.82 µM. This compound and several close analogues were originally described for the treatment of trypanosomiasis9 and inflammation processes.10
Interestingly, there exists already an approved amidinohydrazone based drug used for the treatment of hypertension, guanabenz.11 Furthermore, CNI-1493, an anti-inflammatory and anti-parasitic compound that contains four amidinohydrazone groups, reached phase II clinical trials for the treatment of Crohn’s disease.12–14 Very recently, in parallel to our work a related amidinohydrazone derived furin inhibitor 2 was identified by HTS.15
Compared to other furin inhibitors, which often contain strongly basic guanidino or amidino groups, the fact that amidinohydrazones have a significantly decreased basicity might be advantageous; a pKa of 8.1 has been reported for guanabenz16, whereas guanidine has a pKa of approximately 13.

After identification of 1 we prepared several analogues with one or two amidinohydrazone groups by treatment of commercially available carbonyl compounds with aminoguanidine (Table 1). In addition, the known inhibitor 215 was synthesized as reference. For this compound we found a similar potency (Ki = 25 µM) as described in literature. In contrast, the mono-amidinohydrazones 3 and 4 derived from benzaldehyde and benzophenone, as well as the acylated analogue 5 obtained from reaction with benzoyl chloride showed poor inhibition (Ki > 250 µM). Introduction of a second amidinohydrazone group in meta and para position resulted in improved affinity, whereas both acylated aminoguanidines 8 and 11 were less active. Bis-amidinohydrazones 12 derived from 1,3-indandione and 13 obtained from 4,4´-diacyldiphenylether inhibit furin with Ki values > 15 µM and were not further modified.
Table 1.
Amidinohydrazone and acylated aminoguanidine-derived furin inhibitors

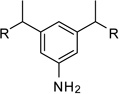
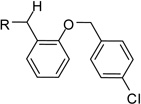
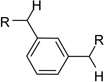
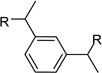
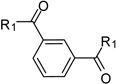
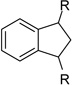
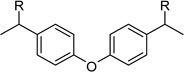
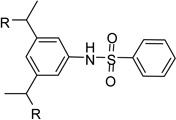
From the X-ray structure of furin in complex with the irreversible inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone it is known that furin has an unusually acidic active site explaining its preference for substrates with basic P6-P1 residues.17 Based on preliminary modelling we assumed that one amidinohydrazone group of 1 should occupy the S1 site, whereas the second one might bind into the S2 pocket. Therefore, we used the simply accessible aniline group of 1 for further modifications with basic residues to address additional acidic binding pockets (Table 2).
Table 2.
Furin inhibitors of the general formula:
 |
||
|---|---|---|
| Compound | R | Ki (µM) |
| 15 | 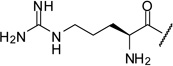 |
1.13 |
| 16 | 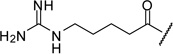 |
1.04 |
| 17 | 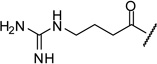 |
0.46 |
| 18 | 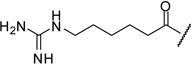 |
1.42 |
| 19 | 1.2 | |
The arginine derivative 15 has slightly enhanced affinity; a similar potency was found for its des-amino analogue 16. Therefore, we also introduced the shorter 4-guanidino-butyryl- and the homologous 6-guanidino-caproyl-residue (17–18).
The most potent inhibitor 17 (preparation see Scheme 1) possesses a Ki value of 0.46 µM, which is approximately 4-fold improved compared to 1. The γ-aminobutyric acid analogue 19, an intermediate from synthesis of compound 17, has slightly reduced activity.
Scheme 1.

Reagents and conditions: (a) Boc-γ-aminobutyric acid, N-methylmorpholine, isobutylchloroformate, −15°C, 10 min in DMF, followed by addition of 26, 1h at −15°C and overnight at room temperature, (b) 1N HCl in acetic acid, 1h room temperature, (c) 2.6 equiv. aminoguanidine hydrochloride, 5 mol % HCl, in 50 % EtOH reflux for 6h, (d) 3 equiv. 1H-pyrazole-carboxamidine hydrochloride, 6 equiv. diisopropylethylamine in DMF, 16h. Final inhibitors 19 and 17 were purified by preparative reversed phase HPLC to a purity of >95% according to HPLC at 220 nm.
The most efficient non-peptidic furin inhibitor described by Jiao (Ki = 6 nM) contains four guanidine residues,6 therefore, we prepared compound 20 by dimerization of 1 via a malonyl spacer to obtain a first analogue containing four amidinohydrazone groups (Table 3).
Table 3.
Inhibition of furin by amidinohydrazone inhibitors
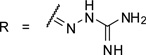 |
||
|---|---|---|
| Compound | Structure | Ki (µM) |
| 20 | 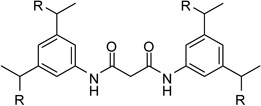 |
1.13 |
| 21 | 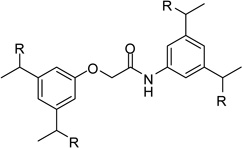 |
0.58 |
| 22 | 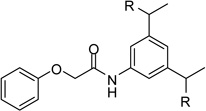 |
5.61 |
| 23 | 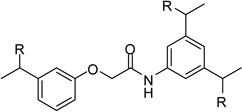 |
1.46 |
| 24 | 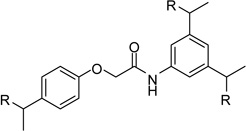 |
1.94 |
| 25 | 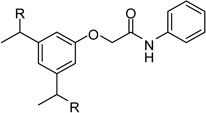 |
1.35 |
However, inhibitor 20 had only marginal improved potency compared to analogue 1. Alternatively, we used chloroacetyl-chloride for the synthesis of inhibitor 21 (preparation see Scheme 2), which also allows access to bi- and trifunctional analogues 22–25. Compound 21 has a slightly improved Ki value of 0.58 µM, which is similar to the potency of the guanidine derivative 17, whereas the other analogues were less potent. The marginal differences in the inhibition constants of the compounds summarized in Table 3 suggest that only amidinohydrazone groups from one phenyl ring are able to make specific interactions with furin.
Scheme 2.

Reagents and conditions: (a) 1.2 equiv. 26, 1.2 equiv. K2CO3 in dry dichloromethane, reflux 5h, (b) 2 equiv. 29, 2 equiv. Cs2CO3 in acetonitrile, reflux 5h, (c) i: 5.2 equiv. aminoguanidine hydrochloride, 5 mol % HCl, in 50 % EtOH reflux for 6h, ii: purification by preparative reversed phase HPLC.
Selected compounds were tested also towards several trypsin like serine proteases, such as thrombin, trypsin, plasmin and FXa. For compound 21 we found an inhibition constant of 0.86 µM towards thrombin, whereas all other Ki values were > 10 µM.
Although we could obtain only slightly improved inhibitors compared to our original screening hit, we assume that such amidinohydrazones might be a suitable start point for the design of non-peptidic furin inhibitors with relatively high selectivity towards trypsin-like serine proteases. Despite several attempts we failed to obtain a crystal structure of furin in complex with these types of inhibitors; therefore, we presently have no information regarding their binding mode. However, their reduced basicity may lead to improved pharmacokinetic properties compared to guanidine and amidine-derived furin inhibitors.
Chemistry
In general, all carbonyl compounds were converted to final inhibitors by treatment with aminoguanidine hydrochloride in ethanol and catalytic amounts of hydrochloric acid.9 The synthesis of amidinohydrazone inhibitors is exemplarily described for inhibitors 17 and 19 (Scheme 1) and 21 (Scheme 2).
Protected amino acids were coupled to intermediate 269 using the mixed anhydride procedure, followed by removal of the protecting group and conversion to the amidinohydrazone (Scheme 1). Reaction of 19 with pyrazol carboxamidine18 provided inhibitor 17.
Inhibitor 21 was prepared according to Scheme 2. Reaction of 26 with chloroacetyl-chloride provided intermediate 28, which was converted with the phenol derivative 299 to compound 30. Final conversion to the amidinohydrazones was performed as described in Scheme 1.
Acylated compounds 5, 8 and 11 were synthesized from reaction of corresponding acid chlorides with aminoguanidine × HCO3− in pyridine.19
Acknowledgments
The authors are grateful to Prof. Wolfram Bode and Prof. Jörg Stürzebecher for their initial contributions to this project.
References
- 1.Fugere M, Day R. Trends Pharmacol Sci. 2005;26:294. doi: 10.1016/j.tips.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas G. Nat Rev Mol Cell Biol. 2002;3:753. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angliker H. J Med Chem. 1995;38:4014. doi: 10.1021/jm00020a016. [DOI] [PubMed] [Google Scholar]
- 4.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. Nature. 1992;360:358. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 5.Basak A. J Mol Med. 2005;83:844. doi: 10.1007/s00109-005-0710-0. [DOI] [PubMed] [Google Scholar]
- 6.Jiao GS, Cregar L, Wang J, Millis SZ, Tang C, O'Malley S, Johnson AT, Sareth S, Larson J, Thomas G. Proc Natl Acad Sci U S A. 2006;103:19707. doi: 10.1073/pnas.0606555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker GL, Sielaff F, Than ME, Lindberg I, Routhier S, Day R, Lu Y, Garten W, Steinmetzer T. J Med Chem. 2010;53:1067. doi: 10.1021/jm9012455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garten W, Klenk HD. In: Avian Influenza. Klenk HD, Matrosovich M, Stech J, editors. Vol. 27. Karger: Basel; 2008. p. 156. [Google Scholar]
- 9.Ulrich P, Cerami A. J Med Chem. 1984;27:35. doi: 10.1021/jm00367a007. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi M, Ulrich P, Bloom O, Meistrell M, 3rd, Zimmerman GA, Schmidtmayerova H, Bukrinsky M, Donnelley T, Bucala R, Sherry B, et al. Mol Med. 1995;1:254. [PMC free article] [PubMed] [Google Scholar]
- 11.Baum T, Eckfeld DK, Metz N, Dinish JL, Rowles G, Van Pelt R, Shropshire AT, Fernandez SP, Gluckman MI, Bruce WF. Experientia. 1969;25:1066. doi: 10.1007/BF01901433. [DOI] [PubMed] [Google Scholar]
- 12.Specht S, Sarite SR, Hauber I, Hauber J, Goerbig UF, Meier C, Bevec D, Hoerauf A, Kaiser A. Parasitol Res. 2008;102:1177. doi: 10.1007/s00436-008-0891-x. [DOI] [PubMed] [Google Scholar]
- 13.Lowenberg M, Verhaar A, van den Blink B, ten Kate F, van Deventer S, Peppelenbosch M, Hommes D. J Immunol. 2005;175:2293. doi: 10.4049/jimmunol.175.4.2293. [DOI] [PubMed] [Google Scholar]
- 14.Ulloa L. Nat Rev Drug Discov. 2005;4:673. doi: 10.1038/nrd1797. [DOI] [PubMed] [Google Scholar]
- 15.Komiyama T, Coppola JM, Larsen MJ, van Dort ME, Ross BD, Day R, Rehemtulla A, Fuller RS. J Biol Chem. 2009;284:15729. doi: 10.1074/jbc.M901540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soll RM, Lu T, Tomczuk B, Illig CR, Fedde C, Eisennagel S, Bone R, Murphy L, Spurlino J, Salemme FR. Bioorg Med Chem Lett. 2000;10:1. doi: 10.1016/s0960-894x(99)00632-0. [DOI] [PubMed] [Google Scholar]
- 17.Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME. Nat Struct Biol. 2003;10:520. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- 18.Bernatowicz MS, Wu Y, Matsueda GR. The Journal of Organic Chemistry. 1992;57:2497. [Google Scholar]
- 19.Bignon E. Patent No. WO 02/34743 A1. 2002