

Abstract
Organotins are members of the environmental obesogen class of contaminants because they activate peroxisome proliferator-activated receptor γ (PPARγ), the essential regulator of adipogenesis. Exposure to thiazolidinediones, PPARγ ligands used to treat type 2 diabetes, is associated with increased fractures. Diminished bone quality likely results from PPARγ’s role in promoting adipogenesis while suppressing osteogenesis of bone marrow multipotent mesenchymal stromal cells (BM-MSC). We hypothesized that tributyltin (TBT) would be a potent modifier of BM-MSC differentiation and a negative regulator of bone formation. Organotins interact with both PPARγ and retinoid X receptors (RXR) suggesting that they activate multiple nuclear receptor pathways. To investigate the role of RXR in the actions of TBT, the effects of PPARγ (rosiglitazone) and RXR (bexarotene, LG100268) agonists were compared to the effects of TBT in BMS2 cells and primary mouse BM-MSC cultures. In BMS2 cells, TBT induced the expression of Fabp4, Abca1 and Tgm2 in an RXR-dependent manner. All agonists suppressed osteogenesis in primary mouse BM-MSC cultures, based on decreased alkaline phosphatase activity, mineralization and expression of osteoblast-related genes. While rosiglitazone and TBT strongly activated adipogenesis, based on lipid accumulation and expression of adipocyte-related genes, the RXR agonists did not. Extending these analyses to other RXR-heterodimers showed that TBT and the RXR agonists activated the liver X receptor pathway, while rosiglitazone did not. Application of either a PPARγ antagonist (T0070907) or an RXR antagonist (HX531) significantly reduced rosiglitazone-induced suppression of bone nodule formation. Only the RXR antagonist significantly reduced LG100268- and TBT-induced bone suppression. The RXR antagonist also inhibited LG100268- and TBT-induced expression of Abca1, an LXR target gene, in primary BM-MSC cultures. These results provide novel evidence that TBT activates multiple nuclear receptor pathways in BM-MSCs, activation of RXR is sufficient to suppress osteogenesis, and TBT suppresses osteogenesis largely through its direct interaction with RXR.
TOC Image
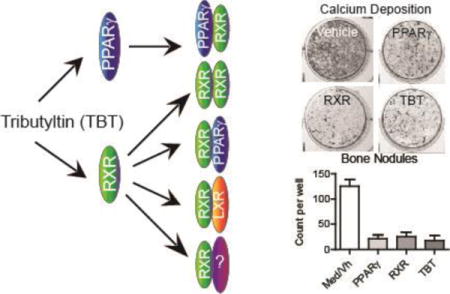
INTRODUCTION
Peroxisome proliferator-activated receptor γ (PPARγ) plays a crucial role in multipotent mesenchymal stromal cell (MSC) differentiation and is a negative regulator of bone formation. PPARγ haploinsufficiency is associated with increased bone mass in vivo,1 which likely results from both an increase in osteogenesis1 and a decrease in osteoclast activity.2 The differentiation of MSCs into adipocytes or osteoblasts is controlled by the balance of the transcriptional activities of PPARγ and runt-related transcription factor 2 (Runx2), an essential mediator of osteogenesis.3 Additionally, the Wnt/β-catenin pathway modulates the expression and function of both PPARγ and Runx2.4 PPARγ both suppresses Runx2 expression and activation5 and interferes with the Wnt pathway.6
Rosiglitazone is a PPARγ ligand and a member of the thiazolidinedione (TZD) class of drugs used to treat type 2 diabetes.7 A significant side effect of TZDs is increased risk of bone fracture.8 Mice treated with rosiglitazone (17–25 mg/kg/day) for 1 to 2 months show reduced bone formation, evidenced by decreased whole body bone mineral density, trabecular bone volume, and osteoblast number. Concomitantly, there is an increase in bone marrow adiposity and in bone resorption. As a result, significant decreases in bone mechanical properties occur. The extent of bone loss may be modified significantly by the age, genetic background, and sex of the mouse.9–12 Accordingly, treatment of human and rodent bone marrow-derived MSC (BM-MSC) models with rosiglitazone results in increased expression and activity of PPARγ and decreased Runx2 expression and activity.9, 13, 14
A growing number of environmental contaminants, including organotins, are being recognized for their ability to activate PPARγ. As such, organotins are members of an emerging class of contaminants called environmental obesogens, contaminants that disrupt the homeostatic controls of adipogenesis and energy homeostasis.15 While organotins have been internationally banned for use as marine antifouling agents, they continue to be used in food crop fungicides, wood preservatives and plastics manufacturing, resulting in significant land-based sources of these pollutants.16 Organotins are readily measurable in house dust.17, 18 Significant human exposure is indicated by the presence of organotins in liver, milk and blood (0.05–400 nM).19–23
Intriguingly, tri-substituted organotins (i.e. tributyltin (TBT) and triphenyltin) are dual agonists of PPARγ and its heterodimerization partners, the retinoid X receptors (RXR).15, 24 TBT and triphenyltin potently activate PPARγ and induce adipocyte differentiation in the BM-MSC line, BMS2 (EC50 10 nM) and in primary mouse and human bone marrow cultures.25, 26 TBT has been shown to suppress osteogenesis in rat calvarial osteoblasts, mouse bone marrow-derived MSC and mouse adipose-derived MSC.27–29 The dual ligand nature of the organotins raises the possibility that RXR could contribute to TBT-induced effects on BM-MSC differentiation, independently of PPARγ. RXRα, β, and γ heterodimerize with and are essential for transcriptional activation of multiple nuclear receptors; further, they also act as homodimers (as reviewed30). Permissive RXR heterodimer partners, such as liver X receptor (LXR) and PPARγ, may be activated by ligand binding to either the partner or to RXR.31 Interestingly, TBT has been shown to activate PPARδ, LXR, and NURR1, all permissive RXR heterodimeric partners.15 In BMS2 cells, both bexarotene (LGD1069), a therapeutic RXR ligand,32 and TBT induced mRNA expression of transglutaminase 2 (Tgm2), an RXR homodimer target, and ATP-binding cassette, sub-family A, member 1 (Abca1), an LXR target.25 It remains to be determined how engagement of RXR by TBT may alter BM-MSC differentiation.
The pervasive nature of organotin contamination (i.e. measureable even in house dust and human tissues) and TBT’s potent interaction with nuclear receptors (e.g. affinity for human PPARγ ligand binding domain: Kd 20 nM15; affinity for human RXRα ligand binding domain: Kd 12.5 nM15; activation of mouse PPARγ1 and 2 reporter: EC50 10 nM33; induction of adipocyte differentiation in mouse MSC: EC50 10 nM25) amplify the need to understand their mechanisms of action in bone-forming cells. Here, we investigated the hypothesis that TBT is a potent negative regulator of bone differentiation. Studies in cultured bone marrow stromal cells and in primary mouse bone marrow cultures examined the mechanisms of TBT-induced effects, in comparison to rosiglitazone (a PPARγ agonist), bexarotene (an RXR agonist) and LG100268 (an RXR agonist). The results demonstrate that TBT activates multiple nuclear receptor pathways in an RXR-dependent manner and potently modifies BM-MSC differentiation through its interaction with RXR.
MATERIALS AND METHODS
Materials
Rosiglitazone and T0901317 were from Cayman Chemical (Ann Arbor, MI). Bexarotene was from LC Laboratories (Woburn, MA). The perilipin antibody was from Cell Signaling Technology (Danvers, MA). The ABCA1 antibody was from Novus Biologicals (Littleton, CO). Human insulin, Nile Red, p-nitrophenyl phosphate (pNPP) reagent, LG100268 and tributyltin chloride were from Sigma-Aldrich (St. Louis, MO). HX531 and T0070907 were from Tocris Bioscience (via R&D Systems, Inc., Minneapolis, MN). All other reagents were from Thermo Fisher Scientific (Suwanee, GA) unless noted. Caution: Tributyltin is hazardous and should be handled carefully.
Cell Culture
BMS2 cells are C57BL/6 mouse-derived bone marrow stromal cells34 (provided by P. Kincade, Oklahoma Medical Research Foundation). BMS2 cells were maintained in DMEM with 5% FBS (Sigma-Aldrich), 5 μg/ml plasmocin (Invivogen, San Diego, CA) and 20 mM L-glutamine. Cultures were maintained at 37° C in a humidified, 5% CO2 atmosphere. BMS2 were plated at 160,000 cells/well (6-well plates) in pre-adipocyte medium (DMEM containing 10% FBS, 1mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin) and allowed to become confluent (3–4 days). Prior to dosing, the medium was replaced with pre-adipocyte medium supplemented with insulin (0.5 μg/ml). Cultures received no treatment (Naïve) or were pre-treated with Vh (DMSO, 0.1%) or HX531 (5 μM). After 1 hr, the cultures received no treatment (Naïve) or were treated with Vh, rosiglitazone (100 nM), bexarotene (100 nM), TBT (80 nM) or T0901317 (100 nM) for 24 hrs.
Primary bone marrow cultures were prepared from C57BL/6J mice (male, 8–12 weeks of age) (Jackson Laboratories, Bar Harbor, ME). Studies were reviewed and approved by the Institutional Animal Care and Use Committee at Boston University. All animals were treated humanely and with regard for alleviation of suffering. Mice were housed up to 4 per cage, with a 12 hour light cycle. Water and food (2018 Teklad Global 18% Protein Rodent Diet, Irradiated, Harlan Laboratories, Indianapolis, IN) were provided ad libitum. Animals were euthanized for collection of bone marrow two days after arrival. After euthanasia (cervical dislocation under terminal euthanasia followed by pneumothorax), limbs were aseptically dissected, and soft tissue was removed from the bone. Bone marrow was flushed from the femur, tibia and humerus, strained through a 70 μm cell strainer, diluted in MSC media (α-MEM containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B), and seeded at 6×106/ml in 2 ml per well in a 6-well plate (gene and protein expression assays), 1 ml per well in a 12-well plate (gene expression assays) or 0.5 ml per well in a 24-well plate (phenotype assays) or 0.2 ml per well in a 96-well plate (cell viability assays). Half of the medium was replaced 4 days after plating and the cultures continued for 3 days. Prior to dosing, the medium was changed to include either insulin (500 ng/ml) or osteogenic additives: ascorbate (12.5 μg/ml), β-glycerol phosphate (8 μM), dexamethasone (10 nM), and insulin (500 ng/ml). Naïve wells were maintained in MSC medium, received no treatments and were harvested after 1 week in culture. Medium wells were maintained in differentiation medium but received no treatment and were harvested along with treated cultures. For single exposure experiments, cultures were treated with Vh (DMSO, 0.1% final concentration), rosiglitazone, TBT, LG100268, or bexarotene (10–200 nM). For mixed exposure experiments, cultures were treated first with Vh, or TBT (10–40 nM) and then treated with Vh, rosiglitazone (10–50 nM) or LG100268 (10–50 nM). For antagonist experiments, cultures were treated first with Vh, HX531 (2 μM) or T0070907 (2 μM) and then treated with Vh, rosiglitazone (20 nM (phenotype), 100 nM (gene expression)), LG100268 (20 nM (phenotype), 100 nM (gene expression)), TBT (20 nM (phenotype), 80 nM (gene expression)) or T0901317 (100 nM (gene expression)). Medium was changed, and the cultures were redosed, every other day for a total exposure period of 2–14 days.
Adipogenesis and Osteogenesis Assays
Lipid accumulation was quantified by Nile Red staining.25 Following Nile Red staining, cells were fixed in 2% paraformaldehyde. To quantify alkaline phosphatase activity, cells were incubated with pNPP solution (Sigma). After quenching with NaOH, absorbance (405 nM) was measured. Following the pNPP assay, cells were stained with Alizarin Red (Osteogenesis Quantitation Kit, Millipore, Billerica, MA). Cells were extensively washed and then photographed using the UVP Bioimaging System (UVP, Inc., Upland, CA). The images were analyzed for bone nodule number using Image-Pro Plus (Media Cybernetics, Inc., Rockville, MD). Following image capture, Alizarin Red staining was quantified according to the manufacturer’s instructions. All absorbance and fluorescence measurements were determined using a Synergy2 plate reader (Biotek, Inc.). For lipid accumulation, fluorescence in experimental wells was normalized either by dividing by fluorescence measured in medium wells (Reported as “Fold Change”) or by subtracting fluorescence measured in medium wells (Reported as relative fluorescence units, “RFUs”). Absorbance was normalized by dividing by absorbance measured in medium wells, and data were reported as “Fold Change from Medium.” For bone nodule counts, the medium wells and Vh wells were averaged for each experiment and reported as “Med/Vh.”
mRNA Expression
Total RNA was extracted and genomic DNA was removed using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA). cDNA was prepared from total RNA using the GoScript™ Reverse Transcription System (Promega), with a 1:1 mixture of random and Oligo (dT)15 primers. All qPCR reactions were performed using the GoTaq® qPCR Master Mix System (Promega). Validated primers were purchased from Qiagen Inc. (Valencia, CA)(Table S1). Sequences for the Abca1 and Srebp1c primers, which were synthesized, were previously reported.35 qPCR reactions were performed using a 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA): Hot-Start activation at 95°C for 2 min, 40 cycles of denaturation (95°C for 15 sec) and annealing/extension (55°C for 60 sec). Relative gene expression was determined using the Pfaffl method,36 using the threshold value for 18s ribosomal RNA (Rn18s) for normalization. No significant differences were observed in the expression of Rn18s across the different times or treatments (data not shown). The Cq value from naïve, undifferentiated cultures was used as the reference point, and data were reported as “Fold Change from Naive.” Expression data from medium wells and Vh wells were averaged for each experiment and reported as “Med/Vh.”
Protein Expression
Cells were collected and lysed in Cell Lysis Buffer (Cell Signaling Technology, Beverly, MA) followed by sonication. The lysates were cleared by centrifugation, and the supernatants were used for protein expression analyses. Protein concentrations were determined by the Bradford method. Total proteins (40 μg) were resolved on 7.5% (ABCA1, 220 kDa) or 10% (perilipin, 62 kDa) polyacryalmide gels, transferred to a 0.2 μm nitrocellulose membrane, and incubated with primary antibody (polyclonal rabbit anti-ABCA1 (NB400-105) or monoclonal rabbit anti-perilipin (9349)). Immunoreactive bands were detected using HRP-conjugated goatanti rabbit secondary antibodies (Biorad, Hercules, CA) followed by enhanced chemiluminescence. To control for equal protein loading, blots were re-probed with a β-actin-specific antibody (A5441, Sigma) and analyzed as above.
Statistics
Statistical analyses were performed with Prism 5 (GraphPad Software, Inc., La Jolla, CA). Data are presented as means ± standard error (SE). The n value indicates the number of independent experiments or bone marrow preparations that were evaluated. Gene expression data were log transformed prior to analysis. One-factor ANOVAs (Dunnett’s) and Student’s T tests were used to analyze data.
RESULTS
RXR contributes to TBT-induced changes in gene expression
We have shown previously that TBT induced the expression of target genes of multiple nuclear receptors.25 Here, we evaluated the contribution of RXR to TBT-induced changes in gene expression using the bone marrow multi-potent stromal cell line BMS2 and the RXR antagonist HX531.37 Confluent BMS2 cells were pretreated with Vh (DMSO, 0.1%) or HX531 (5 μM) for 1hr and then treated with Vh, TBT (80 nM), or ligands for specific nuclear receptors (100 nM rosiglitazone, PPARγ;7 100 nM bexarotene, RXR;32 100 nM T0901317, LXR38) for 24 hrs. We measured mRNA expression of fatty acid binding protein 4 (Fabp4, PPARγ target)39, Abca1 (LXR target)40, and Tgm2 (RXR target)30. Rosiglitazone and TBT significantly induced the expression of Fabp4 mRNA; however, HX531 only significantly reduced TBT-induced Fabp4 expression (Figure 1A). Bexarotene, TBT and T0901317 significantly induced the expression of Abca1, and HX531 completely suppressed Abca1 expression induced by bexarotene and TBT (Figure 1B). Bexarotene and TBT significantly induced the expression of Tgm2, and this induction was completely suppressed by HX531 (Figure 1C). These data show that TBT induces expression of genes that are targets for PPARγ, LXR and RXR and that activation of RXR contributes significantly to the biological activity of TBT.
Figure 1. Antagonism of RXR suppresses expression of multiple TBT-induced genes.
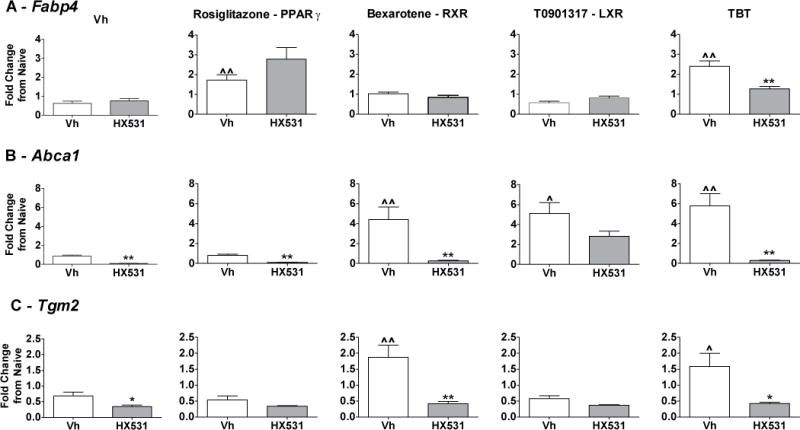
Confluent BMS2 cells were treated first with Vh (DMSO, 0.1%) or HX531 (5 μM) for 1hr and then treated with Vh, rosiglitazone (100 nM), bexarotene (100 nM), TBT (80 nM) or T0901317 (100 nM) in the presence of insulin (0.5 μg/ml) for 24 hrs. Gene expression was quantified by RT-qPCR. (A) PPARγ target. (B) LXR target gene. (C) RXR target gene. Data are presented as means ± SE (n=4–5). Statistically different from Vh+Vh-treated (ˆp<0.05, ˆˆp<0.01, ANOVA, Dunnett’s). Statistically different from Vh+agonist-treated (*p<0.05, **p<0.01, ANOVA, Dunnett’s).
TBT suppresses osteogenesis in primary BM-MSCs
We evaluated the physiological effects of TBT using primary, BM-MSCs. Given TBT’s ability to interact directly with both PPARγ and its heterodimerization partner RXR,15, 24 we compared its actions to agonists specific for either PPARγ (rosiglitazone) or RXR (bexarotene) to help dissect the mechanisms by which TBT exerts its effects on BM-MSC differentiation. Based upon previous dose response analyses for adipocyte differentiation in a bone marrow MSC line (BMS2), TBT concentrations of 10 nM (EC50) and 100 nM (maximal non-toxic dose) were used.25 In that study, TBT was shown to have similar potency but lower efficacy in inducing lipid accumulation relative to rosiglitazone (EC50 60 nM) and similar potency but greater efficacy in inducing lipid accumulation relative to bexarotene (EC50 5 nM). Given the similar potencies of the experimental compounds, responses to TBT were compared to responses to 10 and 100 nM rosiglitazone and bexarotene. Most importantly, these TBT concentrations are well within organotin levels measured in human liver, milk, and blood (0.05–400 nM).19–23
We determined that the concentrations of the ligands used were not toxic to BM-MSC. Cellularity, apoptosis, and necrosis were assessed by MTT labeling, caspase-3 activity, and dead cell protease release, respectively (Figure S1). None of the ligands at the tested concentrations caused a loss of cellularity or overt toxicity.
Next, we examined the effect of the PPARγ and RXR ligands on BM-MSC osteogenic differentiation. Primary bone marrow cultures were prepared from male C57BL/6J mice and induced to undergo osteogenesis. At the initiation of differentiation, cultures were treated with Vh (DMSO, 0.1%), rosiglitazone, bexarotene, or TBT (10–100 nM), incubated for 12 days and assessed for osteogenesis (alkaline phosphatase activity, mineralization and bone nodule number). All three ligands (rosiglitazone, bexarotene and TBT) reduced osteogenesis in BM-MSCs (Figure 2). Dose response analyses revealed that a dose as low as 10 nM was sufficient for either rosiglitazone or TBT to suppress alkaline phosphatase activity (Figure 2A) and bone nodule number, while a concentration of 100 nM was required to suppress calcium deposition (Figure 2B–C). Bexarotene was less potent at suppressing osteogenesis, with only the 100 nM dose significantly reducing alkaline phosphatase activity and bone nodule number (Figure 2A–C). When applied in a mixed exposure, TBT had no effect on rosiglitazone- or LGD100268 (an RXR ligand41)-induced changes in alkaline phosphatase activity (Figure S2A). TBT enhanced the reduction in bone nodule formation induced by LG100268 (Figure S2B).
Figure 2. PPARγ and RXR ligands suppress osteogenic differentiation in BM-MSCs.
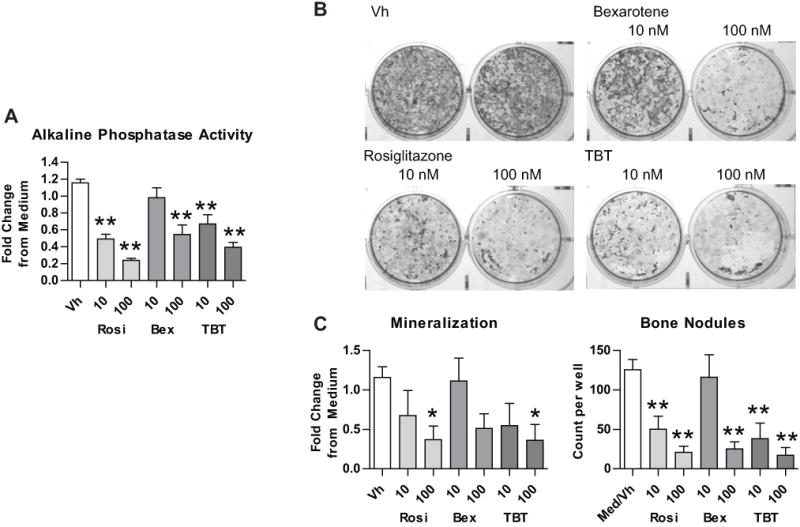
Primary bone marrow cells were isolated from 8–10 week old male C57BL/6 mice, plated, and allowed to adhere for 7 days. The medium was replaced with MSC medium containing the osteogenic additives, β-glycerol phosphate, ascorbate, insulin and dexamethasone. “Medium” wells were left untreated. Cultures were treated with Vh (DMSO, 0.1%), rosiglitazone (Rosi), bexarotene (Bex) or TBT (10–100 nM), cultured for 12 days and analyzed for osteogenesis. (A) Alkaline phosphatase activity. (B) Alizarin staining. (C) Quantification of mineralization and bone nodule number from Alizarin staining. Data are presented as means ± SE (n=3–6). Statistically different from Vh-treated (*p<0.05, **p<0.01, ANOVA, Dunnett’s).
To examine changes in gene expression, established BM-MSC cultures were transitioned to osteogenic medium and were treated as above for 2–10 days. Total RNA was isolated, and gene expression relative to that of naïve, undifferentiated cultures was determined by RT-qPCR (Figure 3). Runx2 is the master regulator of osteogenesis and controls the expression of osterix (Osx), which also is an essential osteogenic transcription factor. Osteocalcin (bone gamma-carboxyglutamate protein, Bglap) is expressed by late osteoblasts and is involved in mineralization. Dentin matrix phosphoprotein 1 (Dmp1) is expressed by mineralizing osteocytes and regulates biomineralization and mineral metabolism.42 The expression of Osx, Bglap, and Dmp1 significantly increased over the course of osteogenesis. Rosiglitazone reduced the expression of Runx2 and suppressed the upregulation of Osx, Bglap, and Dmp1 expression. While bexarotene and TBT did not significantly affect Runx2 expression, they suppressed the upregulation of Osx, Bglap, and Dmp1 expression. Taken together, these results suggest that TBT potently suppresses osteogenesis and that RXR ligands can suppress osteogenesis.
Figure 3. PPARγ and RXR ligands suppress osteogenic gene expression in BM-MSCs.
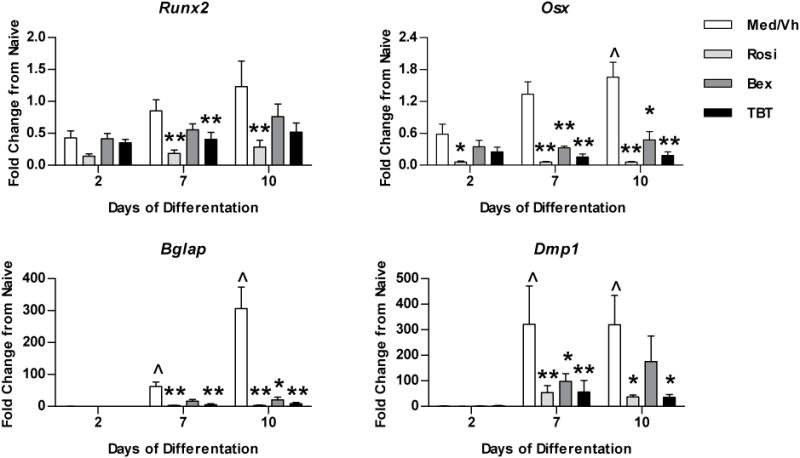
Primary bone marrow cultures were established and osteogenesis was initiated as in Figure 2. Cells were treated with Vh (DMSO, 0.1%), rosiglitazone (Rosi), bexarotene (Bex) or TBT (100 nM), cultured for 2–10 days and analyzed for gene expression by RT-qPCR. Data are presented as means ± SE (n=4–8). Statistically different from day 2, Vh-treated (ˆp<0.05, ˆˆp<0.01, ANOVA, Dunnett’s). Statistically different from Vh-treated on the same day (*p<0.05, **p<0.01, ANOVA, Dunnett’s).
TBT engages the PPARγ pathway in primary BM-MSCs
TBT is a PPARγ ligand,15 and PPARγ ligands are known to suppress osteogenesis and activate adipogenesis.9 Thus, we investigated TBT-induced adipogenesis and PPARγ-mediated gene transcription. Both cultured and primary BM-MSCs from mouse undergo PPARγ ligand-driven adipocyte differentiation without the addition of exogenous hormones.13 Furthermore, another environmental PPARγ ligand, mono-(2-ethylhexyl) phthalate, has been shown to stimulate adipocyte differentiation in NIH 3T3 L1 cells in the presence of serum and insulin alone.43 Thus, we began by investigating the effects of the ligands on adipogenesis by testing their effect on BM-MSC differentiation in medium that was supplemented only with serum and insulin. Established cultures were treated with Vh (DMSO, 0.1%), rosiglitazone, bexarotene or TBT (10–100 nM). Lipid accumulation was assessed by Nile Red staining after 12–14 days in culture. Consistent with our and other’s studies in mouse bone marrow cell lines and primary cultures,10, 25, 26 rosiglitazone and TBT significantly increased lipid accumulation in primary BM-MSCs with minimal hormonal support (Figure 4A). However, in contrast to the bone marrow stromal cell line, bexarotene did not induce lipid accumulation in primary BM-MSCs.
Figure 4. Rosiglitazone and TBT stimulate adipocyte differentiation in BM-MSCs even in the presence of osteogenic signals.
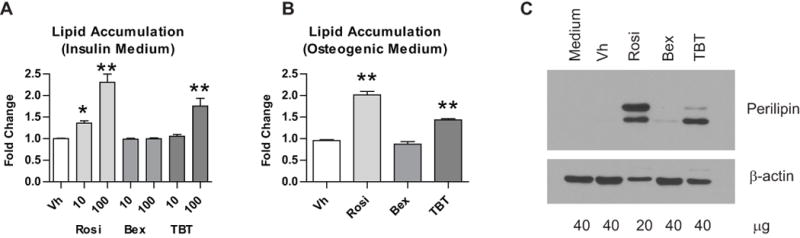
(A) Primary bone marrow cells were isolated from 8–10 week old male C57BL/6 mice, plated, and allowed to adhere for 7 days. The medium was replaced with MSC medium containing insulin (500 ng/ml). “Medium” wells were left untreated. Cultures were treated with Vh (DMSO, 0.1%), rosiglitazone (Rosi), bexarotene (Bex) or TBT (10–100 nM), cultured for 12–14 days and analyzed for lipid accumulation (Nile Red staining). Data are presented as means ± SE (n=4–9). Statistically different from Vh-treated (*p<0.05, **p<0.01, ANOVA, Dunnett’s). (B) Primary bone marrow cultures were established and osteogenesis was initiated as in Figure 2. Cultures were treated Vh, rosiglitazone, bexarotene or TBT (100 nM), cultured for 12 days and analyzed for lipid accumulation. Data are means ± SE (n=3). Statistically different from Vh-treated (** p<0.01, ANOVA, Dunnett’s). (C) Primary bone marrow cultures were established and osteogenesis was initiated as in Figure 2. Cultures were treated with Vh, rosiglitazone, bexarotene or TBT (100 nM) and cultured for 5 days. Perilipin expression was determined in whole cell lysates by immunoblot. Rosiglitazone-treated samples were loaded with 2-fold less protein. Data are representative of 4 independent bone marrow preparations.
Next, we assessed the ability of the ligands to divert BM-MSC differentiation toward adipogenesis in cultures grown in osteogenic conditions. Even in the presence of osteogenic additives, rosiglitazone and TBT both induced lipid accumulation, indicative of adipocyte differentiation, in BM-MSCs, but bexarotene did not (Figure 4B). Terminal adipocyte differentiation was confirmed by assessing perilipin protein expression by immunoblot (Figure 4C). Rosiglitazone and TBT induced the expression of perilipin, while bexarotene did not. Perilipin is a phosphoprotein, thus the multiple bands observed are likely phosphorylated and non-phosphorylated forms of the protein.44
We have previously shown that bexarotene and TBT each have a small but significant effect on adipocyte differentiation stimulated by rosiglitazone in BMS2 cells.25 When applied as a mixed exposure (Figure S2C), TBT had little effect on rosiglitazone-induced lipid accumulation in primary mouse BM-MSCs. TBT and LG100268 each had little impact on the lipid accumulation stimulated by the other compound; additive action was not apparent.
To examine changes in adipocyte-related gene expression during osteogenesis, established BM-MSC cultures were transitioned to osteogenic medium and were treated as above for up to 10 days. Total RNA was isolated, and gene expression relative to that of naïve, undifferentiated cultures was determined by RT-qPCR (Figure 5). PPARγ (Pparg) is the master regulator of adipocyte differentiation. RXRs (Rxra) are the ubiquitously expressed heterodimeric partner of nuclear receptors. CCAAT/enhancer binding protein alpha (Cebpa) is an early adipogenic transcription factor. Fabp4 is a PPARγ target gene. Perilipin (Plin1) regulates lypolysis and is expressed by differentiated adipocytes.44, 45 Low level adipogenesis occurred concurrently with osteogenesis in Vh-treated cultures, as indicated by the significant increase in expression of the adipogenic transcription factors, Pparg and Cebpa, and the adipocyte markers, Fabp4 and Plin1, on days 7 and 10 of differentiation (Figure 5). The ligands exerted little effect on the expression of the adipogenic transcription factors, Pparg and Cebpa, with increases in expression stimulated by rosiglitazone and TBT only at day 2 of differentiation, and had no effect on the expression of Rxra. Rosiglitazone, bexarotene and TBT all significantly increased the expression of the PPARγ gene target Fabp4, although expression induced by rosiglitazone and TBT was more robust and sustained. Only rosiglitazone and TBT strongly induced the expression of Plin1, a marker of terminal adipocyte differentiation. Bexarotene appeared to have a much lower efficacy than rosiglitazone and TBT at simulating adipogenic gene transcription (Figure 5), consistent with the lack of lipid accumulation and adipocyte-specific protein expression (Figure 4). These results indicate that TBT potently suppresses osteogenesis while concomitantly activating adipogenesis, but that an RXR ligand may suppress osteogenesis without strongly inducing adipogenesis.
Figure 5. Rosiglitazone and TBT engage the PPARγ pathway during osteogenesis.
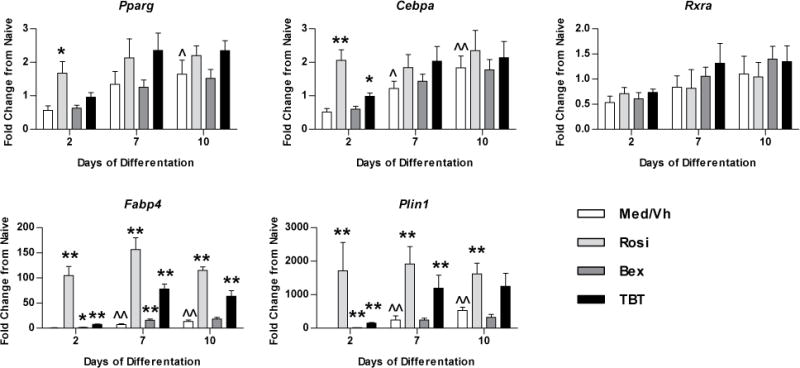
Primary bone marrow cultures were established and osteogenesis was initiated as in Figure 2. Cultures were treated with Vh (DMSO, 0.1%), rosiglitazone (Rosi), bexarotene (Bex) or TBT (100 nM), cultured for 2–10 days and analyzed for gene expression by RT-qPCR. Data are presented as means ± SE (n=3–4). Statistically different from day 2, Vh-treated (ˆp<0.05, ˆˆp<0.01, ANOVA, Dunnett’s). Statistically different from Vh-treated on the same day (*p<0.05, **p<0.01, ANOVA, Dunnett’s).
Direct interaction of TBT with RXR contributes significantly to TBT’s effects on BM-MSC differentiation
Given the results that both rosiglitazone, a PPARγ ligand, and bexarotene, an RXR ligand, could modify BM-MSC differentiation, we hypothesized the TBT can directly engage each of these receptors to mediate its effects on MSC differentiation. Therefore, we investigated the relative contribution of PPARγ and RXR activation to the bone suppressive effects of rosiglitazone, LG100268 and TBT using a PPARγ antagonist (T0070907, IC 1 nM46) and an RXR antagonist (HX531, IC50 18 nM37). GW9662, a more commonly used PPARγ antagonist, is not appropriate to use in this context because it does not block the anti-osteogenic effects of PPARγ.47 The maximum tolerated concentration (2 μM) of each antagonist was used. Established BM-MSC cultures were transitioned to osteogenic medium and were treated first with Vh (DMSO, 0.1%), T0070907 (2 μM) or HX531 (2 μM) and then treated with Vh, rosiglitazone (100 nM), LG100268 (100 nM) or TBT (80 nM) for 7 days. Alkaline phosphatase activity and bone nodule number were determined (Figure 6A–B, Figure S3). While rosiglitazone, LG100268 and TBT all significantly decreased alkaline phosphatase activity, neither antagonist had a significant effect (Figure 6A). In contrast, treatment with the PPARγ antagonist alone significantly increased bone nodule number and significantly counteracted the effect of rosiglitazone; however, it did not have a significant effect on either the LG100268- or TBT-induced reduction in bone nodule number (Figure 6B). Interestingly, the RXR antagonist also showed a trend toward increasing bone nodule number alone and significantly reduced loss of bone nodule formation by all three agonist types (Figure 6B). Surprisingly, lipid accumulation was relatively resistant to suppression by either the PPAR or RXR antagonist (Figure S4A). Indeed, the RXR antagonist alone and in the presence of LG100268 enhanced lipid accumulation, which is in contrast to previous reports.48 We confirmed that there was no contamination with a PPARγ ligand using a mouse PPARγ2-based reporter assay (Figure S4B). Lipid accumulation stimulated by rosiglitazone and TBT was marginally reduced in the presence of the PPARγ antagonist, T0070907, even though the antagonist blocked early Fabp4 expression (Figure S4C). Taken together, the data show that effects on bone nodule formation were most sensitive to inhibition by the antagonists and that direct interaction with RXR contributes to TBT-induced loss of bone formation.
Figure 6. TBT’s effects on osteogenesis are dependent upon direct interaction with RXR.
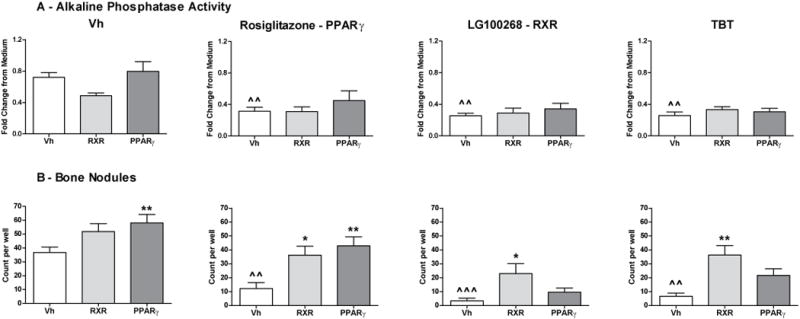
Primary bone marrow cultures were established and osteogenesis was initiated as in Figure 2. Cultures first were treated with Vh (DMSO, 0.1%), HX531 (RXR, 2 μM) or T0070907 (PPARγ, 2 μM) and then treated with Vh, rosiglitazone, LG100268 or TBT (20 nM), cultured for 10 days and analyzed for alkaline phosphatase activity (A) and bone nodule number (Alizarin staining, B). Data are presented as means ± SE (n=4–7). Statistically different from Vh+Vh-treated (ˆp<0.05, ˆˆp<0.01, ˆˆˆp<0.001, ANOVA, Dunnett’s). Statistically different from Vh+agonist-treated (*p<0.05, **p<0.01, ANOVA, Dunnett’s).
TBT engages an alternative permissive RXR heterodimer nuclear receptor pathway in BM-MSCs
Because TBT can bind and activate transcription via RXR,15 we hypothesized that TBT may activate permissive nuclear receptors in BM-MSCs during differentiation. Interestingly, a small but growing body of evidence suggests that other nuclear receptors such as LXR may regulate bone homeostasis.49 The LXRs (Lxra, Lxrb) are physiological mediators of lipid, cholesterol, and glucose homeostasis and metabolism and regulate expression ABCA1, a cholesterol transporter, and steroid regulatory element binding transcription factor 1 (Srebf1), a lipogenic transcription factor.50 In BM-MSC cultures induced to undergo osteogenesis, expression of Lxra, Abca1 and Srebf1 increased significantly over time in Vh-treated cells, while Lxrb expression did not change (Figure 7A). All of the ligands induced the expression of Lxra, although expression induced by rosiglitazone and TBT was more robust and sustained than expression induced by bexarotene. None of the ligands significantly modified expression of Lxrb. All three ligands induced expression of Srebf1, although only bexarotene and TBT induced a sustained increase in expression. Only TBT and bexarotene induced Abca1 expression (Figure 7A). Increased expression of ABCA1 protein was confirmed by immunoblot (Figure 7B). An RXR antagonist (HX531), but not a PPARγ antagonist (T0070907), significantly suppressed the expression of Abca1 stimulated by LG100268 and TBT (Figure 7C). Unlike LXR, retinoic acid receptor (RAR) forms a non-permissive heterodimer with RXR and thus cannot be activated by ligand binding to RXR alone.51 We examined the ability of rosiglitazone, bexarotene and TBT to stimulate gene expression in the RAR pathway, for comparison. All three of the ligands enhanced Rarb expression and decreased Alpl expression; otherwise TBT did not uniquely upregulate RAR target genes (Figure S5). The results suggest that activation of RXR, but not PPARγ, contributed to ABCA1 upregulation. These data show that TBT is capable of activating at least three nuclear receptor pathways (PPARγ, RXR and LXR), opening the possibility for multifaceted effects on BM-MSC differentiation and function.
Figure 7. TBT engages a permissive nuclear receptor, LXR, in BM-MSCs, in an RXR-dependent manner.
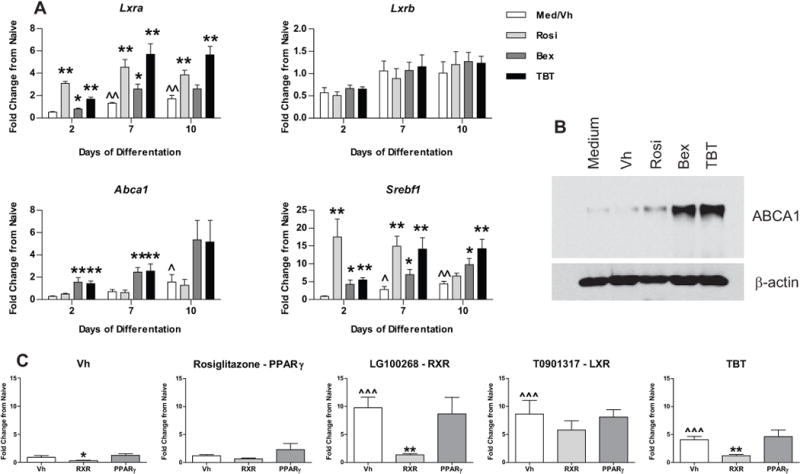
Primary bone marrow cultures were established and osteogenesis was initiated as in Figure 2. (A) Cultures were treated with Vh (DMSO, 0.1%), rosiglitazone (Rosi), bexarotene (Bex) or TBT (100 nM), cultured for 2–10 days and analyzed for gene expression by RT-qPCR. Data are presented as means ± SE (n=4). Statistically different from day 2, Vh-treated (ˆp<0.05, ˆˆp<0.01, ANOVA, Dunnett’s). Statistically different from Vh-treated on the same day (*p<0.05, **p<0.01, ANOVA, Dunnett’s). (B) Cultures were treated with Vh, rosiglitazone, bexarotene or TBT (100 nM) and cultured for 5 days. ABCA1 expression was determined in whole cell lysates by immunoblot. Data are representative of 4 independent bone marrow preparations. (C) Cultures were treated first with Vh, HX531 (2 μM) or T0070907 (2 μM) and then treated with Vh, rosiglitazone (100 nM), LG100268 (100 nM), TBT (80 nM) or T0901317 (100 nM), cultured for 2 days and analyzed for gene expression by RT-qPCR. Data are presented as means ± SE (n=5–6). Statistically different from Vh+Vh-treated (ˆp<0.05, ˆˆp<0.01, ˆˆˆp<0.001, ANOVA, Dunnett’s). Statistically different from Vh+agonist-treated (*p<0.05, **p<0.01, ANOVA, Dunnett’s).
DISCUSSION
Osteoporosis is one of the most pervasive and fastest growing chronic health care problems in the industrialized world. Recent studies have shown that therapeutic PPARγ agonists increase fracture risk8 and that bone loss seen with thiazolidinedione treatment is likely due at least in part to the ability to increase marrow adiposity while suppressing osteogenesis.9, 10 These findings point to the need for serious consideration as to whether ubiquitous environmental PPARγ ligands (e.g., phthalates and organotins) also have detrimental effects on bone health. In vitro, we examined the hypotheses that TBT is a modifier of BM-MSC differentiation at concentrations seen in humans and that TBT exposure leads to skewing of differentiation away from osteogenesis. Organotins bind and activate both PPARγ and RXRs;15, 24 therefore, engagement of multiple nuclear receptor pathways could contribute to the effects of these toxicants on bone physiology. Thus, we also investigated the contribution of interaction with RXR to TBT’s biological effects.
We used an ex vivo primary BM-MSC culture model to isolate the molecular pathways that are recruited and/or disrupted by TBT during osteoblast differentiation. Given TBT’s ability to interact directly with both PPARγ and RXR, we included specific ligands for these receptors (rosiglitazone (PPARγ), bexarotene (RXR), and LG100268 (RXR)) for comparison. Direct activation of PPARγ apparently was required to induce BM-MSC adipogenesis efficiently. Human and mouse PPARγ activation by TBT occurs at similar concentrations; we measured an EC50 of ~10 nM in reporter assays of transactivation of either human or mouse PPARγ’s and an EC50 of 10 nM for stimulation of adipogenesis in a mouse-derived BMS2 cells.25, 33 In addition, binding affinities of rodent and human PPARγ’s for rosiglitazone are similar.52 In contrast, both PPARγ and RXR agonists had anti-osteogenic capabilities.
TBT both induced adipocyte differentiation and suppressed bone formation in BM-MSC cultures. Importantly, modulation of BM-MSC differentiation by TBT occurred at a concentration of 10 nM, a concentration well within what has been observed in human blood, liver, and milk.19–23 TBT’s ability to inhibit osteogenic differentiation was evidenced by the suppression of Runx2-driven gene expression pathways and suppression of alkaline phosphatase activity and mineralization. The reduction in bone nodule number suggests that TBT suppressed not only osteoblast activity but also osteoblast differentiation. The concomitant activation of PPARγ-mediated pathways during the suppression of osteogenesis suggests that PPARγ is a significant contributor to the TBT-induced suppression, like in rosiglitazone-induced suppression.53
However, TBT may act distinctly from rosiglitazone. TBT, bexarotene and LG100268 engage multiple nuclear receptors. We have previously shown that TBT and bexarotene induce the LXR target Abca1 in a BM-MSC line, but that rosiglitazone does not.25 Here, we demonstrated that RXR contributes significantly to the activation of PPARγ and LXR gene expression by TBT both in BMS2 cells and primary mouse BM-MSCs. Interestingly, a distinct pattern of expression was seen with an alternate LXR target, Srebf1, which was induced by all three ligand types. The expression of Srebf1 was more strongly correlated with the expression of Lxra than was Abca1 (data not shown), and likely reflects the PPARγ-dependent upregulation of the LXRα pathway.54 RXR ligands have been shown to activate LXR-mediated pathways either by direct activation of RXR/LXR or by indirect activation through PPARγ.55 RXR ligands and TBT appear to engage both LXR activation pathways. The LXR pathway has only recently been investigated for its involvement in bone homeostasis, and it has been suggested to have both pro- and anti-osteogenic as well as modulate osteoblast/osteoclast crosstalk.49, 56, 57 The significance of LXR modulation by TBT remains to be determined.
While the role of PPARγ ligands in modifying BM-MSC differentiation is well known, this is the first report of suppression of osteogenesis by RXR ligands. Binding of bexarotene or LG100268 to RXR may initiate RXR homodimer activation or RXR-driven activation of permissive heterodimers such as LXR and PPARγ.30 Indeed, we show that antagonism of RXR decreases both RXR- and LXR-mediated gene transcription stimulated by bexarotene and LXR-mediated transcription by LG100268. In contrast to our work in a bone marrow stromal cell line25 and others’ work in which NIH 3T3 L1 cells were exposed to RXR ligands,15 bexarotene did not induce lipid accumulation in primary BM-MSC and minimally induced adipogenic gene expression, either in insulin-supplemented media or in osteogenic conditions. Interestingly, it also has been shown that while RXR ligands induce adipogenesis in human, adipose-derived MSCs, they do not induce adipogenesis in mouse, adipose-derived MSCs,28 suggesting that human MSCs may be more sensitive to the effects of RXR ligands. In contrast, both bexarotene and LG100268 significantly suppressed osteogenesis. These results are reminiscent of previous studies in which some PPARγ ligands have anti-osteogenic capabilities without having any pro-adipogenic ones, or vice versa.58 Importantly, RXR recently has been shown to play an essential role in postnatal bone remodeling.59 It remains to be determined if TBT, bexarotene and LG100268 are recruiting RXR homodimer- or RXR heterodimer-mediated pathways to alter osteogenesis.
In summary, we show that TBT can induce bone loss and can modify BM-MSC differentiation leading to suppression of osteogenesis at nM concentrations, similar to human body burdens. Results from these studies, coupled with the facts that a growing number of environmental contaminants are being recognized as PPARγ agonists (e.g. phthalates, tetrabromobisphenol A, triflumizole)43, 60, 61 and that other toxicants (e.g. lead, arsenic, alcohol, dioxin) suppress osteogenesis via PPARγ-independent mechanisms,27, 62–64 support the hypothesis that environmental exposures could contribute significantly to the pathology of osteoporosis. Exposure to multiple toxicants that act through different pathways may be particularly deleterious, as co-exposure to dioxin and TBT was shown to enhance suppression of osteogenic gene expression.27 Furthermore, recent studies suggest that PPARγ ligand exposure not only enhances the risk of fracture but also is deleterious to new bone formation required for healing.65 Finally, the novel finding that RXR ligands also suppress osteogenesis, and that direct interaction of TBT with RXR contributes to its biological effects, suggests that multiple nuclear receptor pathways could be targeted by environmental contaminants to impact bone homeostasis. In vivo exposure studies are needed to assess the ability of environmental toxicants to reach the bone marrow and induce physiological effects in human-relevant exposure scenarios.
Supplementary Material
Acknowledgments
The authors also would like to thank Ms. Faye Andrews for her superb technical assistance.
FUNDING INFORMATION
This work was supported by the Superfund Research Program at the National Institutes of Health [P42ES007381] to JJS and by a Ruth L. Kirschstein National Research Service Award [F30ES019394] to AHB.
ABBREVIATIONS
- ABCA1
ATP-binding cassette, sub-family A, member 1
- ALPL
alkaline phosphatase
- BGLAP
bone gamma-carboxyglutamate protein
- BM-MSC
bone marrow multipotent mesenchymal stromal cells
- CEBPA
CCAAT/enhancer binding protein α
- DMP1
dentin matrix phosphoprotein
- ETS1
E26 avian leukemia oncogene
- FABP4
fatty acid binding protein 4
- LXR
liver X receptor
- MMP13
matrix metallopeptidase 13
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- OSX
osterix
- PLIN1
perilipin
- pNPP
p-nitrophenyl phosphate
- PPARγ
peroxisome proliferator-activated receptor γ
- RAR
retinoic acid receptor
- RFUs
relative fluorescence units
- RN18S
18s ribosomal RNA
- RUNX2
runt-related transcription factor 2
- RXR
retinoid X receptor
- SREBF1
steroid regulatory element binding transcription factor 1
- TGM2
transglutaminase 2
- TZD
thiazolidinedione
Footnotes
SUPPORTING INFORMATION
The Supporting Information contains catalog numbers and references for the primers used in qPCR analyses. Data include analyses of toxicity, of experiments investigating mixed exposures of agonists and antagonists and of retinoic acid receptor-dependent gene expression. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARγ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wan Y, Chong LW, Evans RM. PPARγ regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- 3.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 4.Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA. 2005;102:3324–3329. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeon MJ, Kim JA, Kwon SH, Kim SW, Park KS, Park SW, Kim SY, Shin CS. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J Biol Chem. 2003;278:23270–23277. doi: 10.1074/jbc.M211610200. [DOI] [PubMed] [Google Scholar]
- 6.Moldes M, Zuo Y, Morrison RF, Silva D, Park BH, Liu J, Farmer SR. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/β-catenin signalling during adipogenesis. Biochem J. 2003;376:607–613. doi: 10.1042/BJ20030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazoladinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARγ) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 8.Bilik D, McEwen LN, Brown MB, Pomeroy NE, Kim C, Asao K, Crosson JC, Duru OK, Ferrara A, Hsiao VC, Karter AJ, Lee PG, Marrero DG, Selby JV, Subramanian U, Herman WH. Thiazolidinediones and fractures: Evidence from translating research into action for diabetes. J Clin Endocrinol Metab. 2010;95:4560–4565. doi: 10.1210/jc.2009-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 10.Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148:2669–2680. doi: 10.1210/en.2006-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ackert-Bicknell CL, Shockley KR, Horton LG, Lecka-Czernik B, Churchill GA, Rosen CJ. Strain-specific effects of rosiglitazone on bone mass, body composition, and serum insulin-like growth factor-I. Endocrinology. 2009;150:1330–1340. doi: 10.1210/en.2008-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sardone LD, Renlund R, Willett TL, Fantus IG, Grynpas MD. Effect of rosiglitazone on bone quality in a rat model of insulin resistance and osteoporosis. Diabetes. 2011;60:3271–3278. doi: 10.2337/db10-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gimble JM, Robinson CE, Wu X, Kelly KA, Rodriguez BR, Kliewer SA, Lehmann JM, Morris DC. Peroxisome proliferator-activated receptor-gamma activation by thiazolidinediones induces adipogenesis in bone marrow stromal cells. Mol Pharmacol. 1996;50:1087–1094. [PubMed] [Google Scholar]
- 14.Benvenuti S, Cellai I, Luciani P, Deledda C, Baglioni S, Giuliani C, Saccardi R, Mazzanti B, Dal Pozzo S, Mannucci E, Peri A, Serio M. Rosiglitazone stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells. J Endocrinol Invest. 2007;30:RC26–30. doi: 10.1007/BF03350807. [DOI] [PubMed] [Google Scholar]
- 15.Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, Gardiner DM, Kanno J, Iguchi T, Blumberg B. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 16.Cornelissen G, Pettersen A, Nesse E, Eek E, Helland A, Breedveld GD. The contribution of urban runoff to organic contaminant levels in harbour sediments near two norwegian cities. Mar Pollut Bull. 2008;56:565–573. doi: 10.1016/j.marpolbul.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Fromme H, Mattulat A, Lahrz T, Ruden H. Occurrence of organotin compounds in house dust in berlin (germany) Chemosphere. 2005;58:1377–1383. doi: 10.1016/j.chemosphere.2004.09.092. [DOI] [PubMed] [Google Scholar]
- 18.Kannan K, Takahashi S, Fujiwara N, Mizukawa H, Tanabe S. Organotin compounds, including butyltins and octyltins, in house dust from Albany, New York, USA. Arch Environ Contam Toxicol. 2010;58:901–907. doi: 10.1007/s00244-010-9513-6. [DOI] [PubMed] [Google Scholar]
- 19.Kannan K, Senthilkumar K, Giesy JP. Occurence of butyltin compounds in human blood. Environ Sci Technol. 1999;33:1776–1779. [Google Scholar]
- 20.Mino Y, Amano F, Yoshioka T, Konishi Y. Determination of organotins in human breast milk by gas chromatography with flame photometric detection. J Health Sci. 2008;54:224–228. [Google Scholar]
- 21.Takahashi S, Mukai H, Tanabe S, Sakayama K, Miyazaki T, Masuno H. Butyltin residues in livers of humans and wild terrestrial mammals and in plastic products. Environ Pollut. 1999;106:213–218. doi: 10.1016/s0269-7491(99)00068-8. [DOI] [PubMed] [Google Scholar]
- 22.Lo S, Allera A, Albers P, Heimbrecht J, Jantzen E, Klingmuller D, Steckelbroeck S. Dithioerythritol (DTE) prevents inhibitory effects of triphenyltin (tpt) on the key enzymes of the human sex steroid hormone metabolism. J Steroid Biochem Mol Biol. 2003;84:569–576. doi: 10.1016/s0960-0760(03)00074-8. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen JB, Strand J. Butyltin compounds in human liver. Environ Res. 2002;88:129–133. doi: 10.1006/enrs.2001.4321. [DOI] [PubMed] [Google Scholar]
- 24.le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, Balaguer P, Bourguet W. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 2009;10:367–373. doi: 10.1038/embor.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yanik SC, Baker AH, Mann KK, Schlezinger JJ. Organotins are potent activators of PPARγ and adipocyte differentiation in bone marrow multipotent mesenchymal stromal cells. Toxicol Sci. 2011;122:476–488. doi: 10.1093/toxsci/kfr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carfi M, Croera C, Ferrario D, Campi V, Bowe G, Pieters R, Gribaldo L. TBTC induces adipocyte differentiation in human bone marrow long term culture. Toxicology. 2008;249:11–18. doi: 10.1016/j.tox.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 27.Koskela A, Viluksela M, Keinanen M, Tuukkanen J, Korkalainen M. Synergistic effects of tributyltin and 2,3,7,8-tetrachlorodibenzo-ρ-dioxin on differentiating osteoblasts and osteoclasts. Toxicol Appl Pharmacol. 2012;263:210–217. doi: 10.1016/j.taap.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Kirchner S, Kieu T, Chow C, Casey S, Blumberg B. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24:526–539. doi: 10.1210/me.2009-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsukamoto Y, Ishihara Y, Miyagawa-Tomita S, Hagiwara H. Inhibition of ossification in vivo and differentiation of osteoblasts in vitro by tributyltin. Biochem Pharmacol. 2004;68:739–746. doi: 10.1016/j.bcp.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 30.Szeles L, Poliska S, Nagy G, Szatmari I, Szanto A, Pap A, Lindstedt M, Santegoets SJ, Ruhl R, Dezso B, Nagy L. Research resource: Transcriptome profiling of genes regulated by RXR and its permissive and nonpermissive partners in differentiating monocyte-derived dendritic cells. Mol Endocrinol. 2010;24:2218–2231. doi: 10.1210/me.2010-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamann LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR, Jr, Heyman RA. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 32.Boehm MF, Zhang L, Badea BA, White SK, Mais DE, Berger E, Suto CM, Goldman ME, Heyman RA. Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J Med Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- 33.Watt J, Schlezinger JJ. Structurally-diverse, ppargamma-activating environmental toxicants induce adipogenesis and suppress osteogenesis in bone marrow mesenchymal stromal cells. Toxicology. 2015;331:66–77. doi: 10.1016/j.tox.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pietrangeli C, Hayashi SI, Kincade P. Stromal cell lines which support lymphocyte growth: Characterization, sensitivity to radiation and responsiveness to growth factors. Eur J Immunol. 1988;18:863–872. doi: 10.1002/eji.1830180606. [DOI] [PubMed] [Google Scholar]
- 35.Padovani AM, Molina MF, Mann KK. Inhibition of liver X receptor/retinoid X receptor-mediated transcription contributes to the proatherogenic effects of arsenic in macrophages in vitro. Arterioscler Thromb Vasc Biol. 2010;30:1228–1236. doi: 10.1161/ATVBAHA.110.205500. [DOI] [PubMed] [Google Scholar]
- 36.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ebisawa M, Umemiya H, Ohta K, Fukasawa H, Kawachi E, Christoffel G, Gronemeyer H, Tsuji M, Hashimoto Y, Shudo K, Kagechika H. Retinoid X receptor-antagonistic diazepinylbenzoic acids. Chem Pharm Bull (Tokyo) 1999;47:1778–1786. doi: 10.1248/cpb.47.1778. [DOI] [PubMed] [Google Scholar]
- 38.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. MPPARγ 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 40.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc Natl Acad Sci USA. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm MF, Zhang L, Zhi L, McClurg MR, Berger E, Wagoner M, Mais DE, Suto CM, Davies JA, Heyman RA, et al. Design and synthesis of potent retinoid X receptor selective ligands that induce apoptosis in leukemia cells. J Med Chem. 1995;38:3146–3155. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- 42.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, Tudor C, Anghel SI, Grosdidier A, Lathion C, Engelborghs Y, Michielin O, Wahli W, Desvergne B. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282:19152–19166. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- 44.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266:11341–11346. [PubMed] [Google Scholar]
- 45.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 46.Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- 47.Rahman S, Czernik PJ, Lu Y, Lecka-Czernik B. Beta-catenin directly sequesters adipocytic and insulin sensitizing activities but not osteoblastic activity of PPARγ2 in marrow mesenchymal stem cells. PLoS One. 2012;7:e51746. doi: 10.1371/journal.pone.0051746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamauchi T, Waki H, Kamon J, Murakami K, Motojima K, Komeda K, Miki H, Kubota N, Terauchi Y, Tsuchida A, Tsuboyama-Kasaoka N, Yamauchi N, Ide T, Hori W, Kato S, Fukayama M, Akanuma Y, Ezaki O, Itai A, Nagai R, Kimura S, Tobe K, Kagechika H, Shudo K, Kadowaki T. Inhibition of RXR and PPARγ ameliorates diet-induced obesity and type 2 diabetes. J Clin Invest. 2001;108:1001–1013. doi: 10.1172/JCI12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prawitt J, Beil FT, Marshall RP, Bartelt A, Ruether W, Heeren J, Amling M, Staels B, Niemeier A. Short-term activation of liver X receptors inhibits osteoblasts but long-term activation does not have an impact on murine bone in vivo. Bone. 2010;48:339–346. doi: 10.1016/j.bone.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 50.Laurencikiene J, Ryden M. Liver X Receptors and fat cell metabolism. Int J Obes (Lond) 2012;36:1494–1502. doi: 10.1038/ijo.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lammi J, Perlmann T, Aarnisalo P. Corepressor interaction differentiates the permissive and non-permissive retinoid X receptor heterodimers. Arch Biochem Biophys. 2008;472:105–114. doi: 10.1016/j.abb.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Young PW, Buckle DR, Cantello BC, Chapman H, Clapham JC, Coyle PJ, Haigh D, Hindley RM, Holder JC, Kallender H, Latter AJ, Lawrie KW, Mossakowska D, Murphy GJ, Roxbee Cox L, Smith SA. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J Pharmacol Exp Ther. 1998;284:751–759. [PubMed] [Google Scholar]
- 53.Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARγ2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 2009;106:232–246. doi: 10.1002/jcb.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A ppar gamma-lxr-abca1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 55.Nishimaki-Mogami T, Tamehiro N, Sato Y, Okuhira K, Sai K, Kagechika H, Shudo K, Abe-Dohmae S, Yokoyama S, Ohno Y, Inoue K, Sawada J. The RXR agonists PA024 and HX630 have different abilities to activate LXR/RXR and to induce ABCA1 expression in macrophage cell lines. Biochem Pharmacol. 2008;76:1006–1013. doi: 10.1016/j.bcp.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Kim WK, Meliton V, Tetradis S, Weinmaster G, Hahn TJ, Carlson M, Nelson SF, Parhami F. Osteogenic oxysterol, 20(s)-hydroxycholesterol, induces notch target gene expression in bone marrow stromal cells. J Bone Miner Res. 2010;25:782–795. doi: 10.1359/jbmr.091024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kleyer A, Scholtysek C, Bottesch E, Hillienhof U, Beyer C, Distler JH, Tuckermann JP, Schett G, Kronke G. Liver X receptors orchestrate osteoblast/osteoclast crosstalk and counteract pathologic bone loss. J Bone Miner Res. 2012;27:2442–2451. doi: 10.1002/jbmr.1702. [DOI] [PubMed] [Google Scholar]
- 58.Lazarenko OP, Rzonca SO, Suva LJ, Lecka-Czernik B. Netoglitazone is a PPARγ ligand with selective effects on bone and fat. Bone. 2006;38:74–84. doi: 10.1016/j.bone.2005.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Menendez-Gutierrez MP, Roszer T, Fuentes L, Nunez V, Escolano A, Redondo JM, De Clerck N, Metzger D, Valledor AF, Ricote M. Retinoid X receptors orchestrate osteoclast differentiation and postnatal bone remodeling. J Clin Invest. 2015;125:809–823. doi: 10.1172/JCI77186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riu A, Grimaldi M, le Maire A, Bey G, Phillips K, Boulahtouf A, Perdu E, Zalko D, Bourguet W, Balaguer P. Peroxisome proliferator-activated receptor gamma is a target for halogenated analogs of bisphenol A. Environ Health Perspect. 2011;119:1227–1232. doi: 10.1289/ehp.1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Pham HT, Janesick AS, Blumberg B. Triflumizole is an obesogen in mice that acts through peroxisome proliferator activated receptor gamma (PPARγ) Environ Health Perspect. 2012;120:1720–1726. doi: 10.1289/ehp.1205383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu CT, Lu TY, Chan DC, Tsai KS, Yang RS, Liu SH. Effects of arsenic on osteoblast differentiation in vitro and on bone mineral density and microstructure in rats. Environ Health Perspect. 2014;122:559–565. doi: 10.1289/ehp.1307832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Lumpkin CK, Badger TM, Ronis MJ. Inhibition of NADPH oxidases prevents chronic ethanol-induced bone loss in female rats. J Pharmacol Exp Ther. 2011;336:734–742. doi: 10.1124/jpet.110.175091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beier EE, Maher JR, Sheu TJ, Cory-Slechta DA, Berger AJ, Zuscik MJ, Puzas JE. Heavy metal lead exposure, osteoporotic-like phenotype in an animal model, and depression of wnt signaling. Environ Health Perspect. 2013;121:97–104. doi: 10.1289/ehp.1205374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu L, Aronson J, Lecka-Czernik B. Rosiglitazone disrupts endosteal bone formation during distraction osteogenesis by local adipocytic infiltration. Bone. 2013;52:247–258. doi: 10.1016/j.bone.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.