

Abstract
Increasing evidence suggests that the calcineurin (CN)-dependent transcription factor NFAT (Nuclear Factor of Activated T cells) mediates deleterious effects of astrocytes in progressive neurodegenerative conditions. However, the impact of astrocytic CN/NFAT signaling on neural function/recovery after acute injury has not been investigated extensively. Using a controlled cortical impact (CCI) procedure in rats, we show that traumatic brain injury is associated with an increase in the activities of NFATs 1 and 4 in the hippocampus at 7 d after injury. NFAT4, but not NFAT1, exhibited extensive labeling in astrocytes and was found throughout the axon/dendrite layers of CA1 and the dentate gyrus. Blockade of the astrocytic CN/NFAT pathway in rats using adeno-associated virus (AAV) vectors expressing the astrocyte-specific promoter Gfa2 and the NFAT-inhibitory peptide VIVIT prevented the injury-related loss of basal CA1 synaptic strength and key synaptic proteins and reduced the susceptibility to induction of long-term depression. In conjunction with these seemingly beneficial effects, VIVIT treatment elicited a marked increase in the expression of the prosynaptogenic factor SPARCL1 (hevin), especially in hippocampal tissue ipsilateral to the CCI injury. However, in contrast to previous work on Alzheimer's mouse models, AAV-Gfa2-VIVIT had no effects on the levels of GFAP and Iba1, suggesting that synaptic benefits of VIVIT were not attributable to a reduction in glial activation per se. Together, the results implicate the astrocytic CN/NFAT4 pathway as a key mechanism for disrupting synaptic remodeling and homeostasis in the hippocampus after acute injury.
SIGNIFICANCE STATEMENT Similar to microglia, astrocytes become strongly “activated” with neural damage and exhibit numerous morphologic/biochemical changes, including an increase in the expression/activity of the protein phosphatase calcineurin. Using adeno-associated virus (AAV) to inhibit the calcineurin-dependent activation of the transcription factor NFAT (Nuclear Factor of Activated T cells) selectively, we have shown that activated astrocytes contribute to neural dysfunction in animal models characterized by progressive/chronic neuropathology. Here, we show that the suppression of astrocytic calcineurin/NFATs helps to protect synaptic function and plasticity in an animal model in which pathology arises from a single traumatic brain injury. The findings suggest that at least some astrocyte functions impair recovery after trauma and may provide druggable targets for treating victims of acute nervous system injury.
Keywords: astrocytes, brain injury, calcineurin, calcium, glia, synapse
Introduction
Astrocytes exhibit striking morphological changes with CNS injury and disease, which are collectively referred to as “astrocyte activation” (Pekny and Nilsson, 2005; Parpura et al., 2012; Anderson et al., 2014). Hypertrophy of astrocyte somata and processes, along with a corresponding increase in the expression of the glial fibrillary acidic protein (GFAP), are the most robust and recognizable biomarkers of activated astrocytes, yet the functional phenotype of these cells appears highly complex. In intact animal models, targeted approaches for inhibiting GFAP expression or for modulating the activity of major astrocytic signaling pathways have demonstrated both protective and deleterious roles of activated astrocytes in neural function depending on the nature of the injury (e.g., chronic neurodegeneration vs acute trauma) and the proximity of astrocytes to the site of injury/pathology, among many other factors (Sofroniew, 2009; Pekny and Pekna, 2014; Pekny et al., 2014; Burda et al., 2015).
Several key morphologic and biochemical features of astrocyte activation have been linked to the expression and/or hyperactivation of the Ca2+/calmodulin-dependent protein phosphatase calcineurin (CN) (Norris et al., 2005; Fernandez et al., 2007; Canellada et al., 2008; Furman et al., 2012; Furman and Norris, 2014). Although weakly expressed in astrocytes in healthy adult nervous tissue, CN can appear at high levels in activated astrocytes associated with aging (Norris et al., 2005), acute injury (Hashimoto et al., 1998), and progressive amyloid pathology (Norris et al., 2005; Celsi et al., 2007; Abdul et al., 2009). In peripheral tissues, including lymphoid tissue, heart, vascular smooth muscle cells, and skeletal muscle, CN is known to strongly regulate the morphological and functional phenotypes of cells through the activation of NFAT (Nuclear Factor of Activated T cells) transcription factors (Crabtree and Olson, 2002; Horsley and Pavlath, 2002; Hogan et al., 2003; Wilkins and Molkentin, 2004; Hassoun et al., 2009). There are four CN-dependent NFAT isoforms (i.e., NFATs 1–4), all of which are expressed in mammalian brain (Nguyen and Di Giovanni, 2008; Vihma et al., 2008). Changes in NFAT expression/activity, especially for the NFAT1 and 4 isoforms, have been observed in astrocytes in progressive neurodegenerative conditions such as Alzheimer's disease (AD) (Abdul et al., 2009) and Parkinson's disease (Caraveo et al., 2014) and also after acute nervous system injury (Serrano-Pérez et al., 2011; Yan et al., 2014). Blockade of CN/NFAT interactions in astrocytes using adeno-associated virus (AAV) vectors and the CN docking site mimetic peptide VIVIT suppressed glial activation, reduced amyloid pathology, and improved synaptic function in a mouse model of AD (Furman et al., 2012), suggesting that astrocytic CN/NFAT signaling may be detrimental to neural function in chronic neurodegenerative conditions. Whether CN/NFAT plays a similarly deleterious role in activated astrocytes after acute neural injury is unknown.
Here, we investigated the DNA-binding activities of all four CN-dependent NFAT isoforms in rat hippocampus 7 d after a unilateral cortical contusion injury (CCI). Although we found evidence for increased expression of the NFAT1 and 4 isoforms, specific colocalization with GFAP-positive astrocytes was only observed for NFAT4. Intrahippocampal injection of AAV vectors containing the astrocyte-specific promoter Gfa2 and the NFAT inhibitory peptide VIVIT prevented the weakening of basal CA3–CA1 synaptic strength, reduced susceptibility to long-term synaptic depression (LTD), and protected against the loss of several key synaptic proteins. However, AAV-Gfa2-VIVIT did not appreciably alter astrocyte or microglial protein markers (i.e., GFAP and Iba-1, respectively). The results suggest that astrocytic CN/NFAT signaling can impair and/or prevent the recovery of synaptic function after traumatic brain injury without reducing astrocyte activation per se.
Materials and Methods
Animals and TBI.
Adult (∼2-month-old) male Sprague Dawley rats (250–275 g; Harlan Laboratories) were housed 2/cage on a 12 h light/dark cycle, provided access to food and water ad libitum, and used in accordance with the Institutional Animal Care and Use Committee of the University of Kentucky. Stereotaxic surgeries and delivery of traumatic brain injury was performed with slight modifications of our previous methods (Norris and Scheff, 2009; Ansari et al., 2013; Scheff et al., 2013). Briefly, rats were anesthetized with 2% isofluorane and immobilized in a Kopf stereotaxic frame with the incisor bar set at −5. Body temperature for each rat was monitored and maintained at 36°C with a Space Gel heating pad (Braintree Scientific). After a midline incision and retraction of the skin, a 6-mm-diameter craniotomy was made lateral to midline and approximately midway between bregma and lambda with a Michele hand trephine and the skull disk was removed without disturbing the dura. An electronic controlled pneumatic impact device (TBI0310; Precision System) with a hard stop Bimba cylinder was used to deliver a moderate unilateral controlled cortical impact (CCI) injury, which compressed the cortex to a depth of 2.0 mm at a velocity of 3.5 m/s. After the injury, the craniotomy site was sealed with MASCOT adhesive (EMS) and an 8 mm disc formed from clear polyester (0.5 mm; Midwest Products). In some cases, rats were subjected to craniotomy, but did not receive a CCI, and served as sham controls. The surgical procedure was completed in 15–20 min.
Electrophoretic mobility shift assay.
As the EMSA probe, a duplex DNA oligonucleotide labeled on its 5′ ends with IRDye 700 (LI-COR) was obtained from Integrated DNA Technologies. The sequence of the strands were (5′-IRDye700-ACATTGGAAAATTTTATTACACCA-3′ annealed to 5′-IRDye700-TGGTGTAATAAAATTTTCCAATGT-3′) and corresponded to the NFAT binding site from the IL4 promoter, with which previous high-quality EMSAs have been performed by others (Neal and Clipstone, 2001). Probe specificity was assessed using an unlabeled wild-type competitor of the same sequence or a mutant competitor containing an altered NFAT-binding site (TGGAAAA→ CTTTAAA). When indicated, these competitors were included at a 100-fold molar excess in the EMSA-binding mix.
Whole cell extracts from frozen brain tissue at 7 d after CCI were made using a kit from Active Motif following the manufacturer's instructions and protein assays performed using a Bradford assay. The binding reaction used for these experiments contained 10 mm Tris, pH 7.5, 50 mm NaCl, 1 mm EDTA, 1 mm DTT, 0.05% NP40, 5% glycerol, 0.5 μg of poly dIC, 5 μg of BSA, 10% 10× orange loading buffer (LI-COR), 1% protease inhibitor mixture III (EMD Millipore), and 1% phosphatase inhibitor mixture II (EMD Millipore), indicated extract, and 10 femtomoles of IL4 NFAT probe. To induce supershift/blockshift with NFAT antibodies, 3.3 μl of the indicated NFAT antibody was added to the mixture and 3.3 μl of PBS to the negative control. NFAT1 (Abcam, catalog #ab2272), NFAT2 (Santa Cruz Biotechnology, catalog #sc-13033x), NFAT3 (Santa Cruz Biotechnology, catalog #sc-13036x), and NFAT4 (Santa Cruz Biotechnology, catalog #sc-8405x) were used individually. For some conditions, NFAT1 and NFAT4 antibodies were used in combination, as indicated. EMSA samples were incubated on ice for 2 h, covered with foil to protect fluorescent probe from photobleaching, and incubated at room temperature for 10 min before loading on gels. Electrophoresis was performed on chilled 5% nondenaturing polyacrylamide gels in ½× TBE running buffer in a cold room for 1 h and 50 min at 200 V, covering the apparatus with foil and keeping the lights out. The orange dye/free NFAT probe was close to the bottom of the gel at the end of the gel run. Images were obtained on an Odyssey scanner (LI-COR) using the 700 channel at the highest intensity and Image Studio 2.1 software (LI-COR) was used to quantify fluorescence of shifted bands. Protein concentrations used in these studies were optimized from titration assays (data not shown) and fell within the linear detection range of the Odyssey scanner. Control (contralateral side of the hippocampus) and injured (ipsilateral side of the hippocampus) tissue samples were run on the same gel for direct statistical comparisons.
Immunofluorescent labeling of NFAT isoforms in brain sections.
On day 7 of recovery after treatment with cortical contusion injury, rats were deeply anesthetized and perfused with 50 ml of saline followed by 100 ml of a solution of 4% paraformaldehyde, pH 7.4. After perfusion, the brains were removed from the skull, further fixed for an additional 4 h, and then put in a 30% sucrose solution. Coronal brain sections (40 μm) were made and the sections stored in the sucrose solution in a −20 freezer. As needed, sections were removed and stained as free-floating sections in 12-well tissue culture plates using a bent glass pipette to transfer brain section from well to well containing the appropriate solution. Brain sections were rinsed 2× with TBS buffer (100 mm Tris, pH 7.5, 0.85% saline), treated with H2O2 solution (3% H2O2, 10% methanol in TBS) for 60 min, and then rinsed again with TBS buffer. Sections were pretreated with TBS-T (TBS with 0.1% Triton X-100) and blocked with TBS-BSA (2% BSA in TBS-T). First antibodies, the same ones used for the EMSAs (either NFAT1, Abcam, catalog #ab2722, 1:100 dilution, or NFAT4, Santa Cruz Biotechnology, catalog #sc-8405, 1:20 dilution) were diluted in TBS-BSA and brain sections were incubated overnight at 4 C with rotation on a shaker. The next morning, sections were washed 3× in TBS-T and then blocked in TBS-BSA. The secondary antibody (to the mouse primaries) and an Alexa Fluor 488-tyramide from a tyramide signal amplification kit (Invitrogen, catalog #T20912) were used according to the manufacturer's directions. After additional washes in TBS-T, the sections were blocked again in TBS-BSA and incubated with directly conjugated Alexa Fluor 594-monoclonal Ab to GFAP (Cell Signaling Technology, catalog #8152) at a 1:100 dilution overnight at 4°C. After washes in TBS-T, the sections were mounted on gelatin-coated microscope slides and coverslipped with DAPI-containing Prolong Gold Anti-fade Mounting Medium (Invitrogen, catalog #P36931). Fluorescent images were obtained on a confocal microscope (Nikon Eclipse Ti) and contralateral and ipsilateral sides were taken at the same settings so that quantitative comparisons could be made between the two sides. Images shown are single plane projections of focused stacks of images. NFAT4 nuclear/cytosolic localization images (taken with a 100× objective) were obtained from enhanced green fluorescent protein (EGFP)-positive cells in the dendritic layers of CA1 and the dentate gyrus in a Z-series of 0.5 μm steps. Images used for quantification were randomly acquired and measured by personnel blind to the experimental condition. Cytosolic and nuclear NFAT4 fluorescence intensities were quantified by Imaris 8.1.3 software.
AAV delivery.
AAV2/5 vectors constructed using rep and cap genes from AAV2 and AAV5 serotypes, respectively (Hildinger et al., 2001), were obtained from the UPENN virus core. Vectors expressed EGFP alone or EGFP fused to the NFAT inhibitory peptide VIVIT. Transgene expression was under the control of the human GFAP promoter (Gfa2) isolated and characterized by Dr. Michael Brenner's laboratory at the University of Alabama–Birmingham (de Leeuw et al., 2006; Lee et al., 2008). Methods for the bilateral delivery of AAV vectors to rat hippocampus were adapted, with slight modifications, from our previous work on mice (Furman et al., 2012). Briefly, rats were placed in a stereotaxic frame under isoflurane anesthesia as described above. After a midline incision, holes were drilled bilaterally and needles attached to separate Hamilton syringes containing AAV vectors (1012 ifu/ml) were lowered into the hippocampus and injected at a rate of 0.2 μl/min (8 μl total) using a stereotaxic microinjector (Stoelting). Syringe needles were left in place for 2 min after completion to limit reflux. Coordinates for injection relative to bregma were +3.8 mm anteroposterior, +1.8 mm mediolateral, and −2.8 mm dorsoventral. Wounds were closed and rats were returned to their home cages for ∼8 weeks before receiving a unilateral CCI injury, as described above.
Astrocyte immunodepletion and AAV specificity.
At 4 weeks after AAV-Gfa2-EGFP injection, some rats were anesthetized under CO2 and decapitated, with hippocampi dissected away and stored in ice-cold Ca2+/Mg2+-free PBS solution. Hippocampal tissue then was minced finely in a Petri dish, treated with 0.125% trypsin-EDTA, and transferred to a conical tube (in a total volume of 2 ml) using a 1000 μl pipet tip. After 15 min, samples were triturated first with a 1000 μl pipet tip, then with a 200 μl tip (placed over the end of a 1000 μl pipet tip). The sample was then diluted with 2 ml of PBS with 0.5% BSA (buffer) and incubated for an additional 15 min to allow larger fragments of undigested tissue to settle out. The supernatant was passed through a 70 μm filter and the cells centrifuged 2× at 300 × g for 10 min (to remove all trypsin) and resuspended in buffer. After the final spin, cells were resuspended in ∼4 ml of buffer. A small sample was diluted 1:10, treated with trypan blue, and examined on a hemocytometer to determine both cell number and the quality of the single-cell suspension. Typical yields using this method were ∼1–5 × 108 cells.
For cell separation, ∼5 × 107 cells from each sample were pelleted at 300 × g for 10 min and taken up in 100 μl buffer with 150 μl of mouse GLAST antibody (Miltenyi Biotec pure GLAST Ab). “Mock” separations (i.e., in which no GLAST antibody was added) were performed in parallel. Cells were then resuspended by pipetting up and down (never vortexed) and incubated for 13 min at 4°C. Cells were pelleted (300 × g for 10 min), washed 3× (to remove excess antibody), resuspended in 200 μl of buffer containing 40 μl of preequilibrated Dynabeads (Invitrogen), and incubated at 4°C for another 15 min. The astrocyte-enriched (AE) fraction bound to the beads was captured to the side of the tube using a chilled Mag-Rack (GE Healthcare) and the astrocyte-depleted (AD) fraction removed. To ensure removal of all Dynabeads, the AD fraction underwent a second round in the Mag-Rack and the suspension was extracted. This fraction was then pelleted to concentrate the cells. The AE fraction was washed 3× in buffer, capturing it in the Mag-Rack each time to maximize astrocyte enrichment. After each wash, the fraction was resuspended and placed in the Mag-Rack to maximize astrocyte enrichment. At the end of the separation procedure, the AD and AE fractions, along with mock-enriched (ME) and mock-depleted (MD) fractions (i.e., fractions not exposed to GLAST antibody) were dissolved in 60–80 μl of 2× sample buffer, heated at 65°C for 20 min, and then transferred to separate lanes of a 4–20% Criterion gradient gel (Bio-Rad) for SDS-PAGE. The gel was then transferred to Immobilon-FL PVDF membranes (Millipore, catalog #IPFL 00010). Western blots were performed using primary antibodies to GFAP (rabbit; Cell Signaling Technology), EGFP (rabbit; Abcam), and β-actin (mouse; Sigma-Aldrich).
Preparation of hippocampal slices.
Methods for preparing hippocampal slices from brain-injured rats are nearly identical to our previous work (Norris and Scheff, 2009). Briefly, at 7 d after CCI, rats were killed under CO2 anesthesia and decapitated. Brains were rapidly removed and placed in ice-cold, oxygenated (95% O2, 5% CO2) artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 10 dextrose, pH 7.4. Hippocampi were bilaterally dissected and sectioned coronally into ∼400–450 μm slices using a McIlwain Tissue Chopper (Stoelting). Slices were then transferred to a custom interface holding chamber (Mathis et al., 2011) and incubated with warmed (32°C) oxygenated ACSF containing 2 mm CaCl until electrophysiological recordings (usually 1.5–5 h).
Electrophysiological analysis of CA1 synaptic function.
Slices were submerged in oxygenated ACSF (32°C) and perfused at a rate of 1–2 ml/min in an RC22 chamber (Warner Instrument). Schaffer collaterals were activated with a bipolar platinum/iridium electrode located in stratum radiatum near the CA3–CA1 border. Stimulus intensity was controlled by a constant current stimulus isolation unit (World Precision Instruments) and stimulus timing was controlled by Clampex 9.2 software (Molecular Devices). Field potentials were recorded in CA1 stratum radiatum using a glass micropipette (1–6 MΩ) and filled with ACSF containing an Ag/AgCl wire. Physiologic activity was amplified 100×, Bessel-filtered at 1 kHz, and digitized at 10 kHz using a Multiclamp 700B amplifier and a Digidata 1320 digitizer (Molecular Devices).
To assess basal synaptic strength, 100 μs stimulus pulses were given at nine intensity levels (range 30–500 μA) at a rate of 0.1 Hz. Five field potentials at each level were averaged, and measurements of fiber volley (FV) amplitude (in millivolts) and EPSP slope (in millivolts/millisecond) were performed offline using Clampfit software (Molecular Devices). FV amplitudes were plotted against stimulation intensity to estimate the relative level of presynaptic excitability and EPSP slope measures were plotted against their corresponding FV amplitudes to estimate the relative strength of CA3–CA1 synapses. Synaptic strength curves for each hemisphere were fit (SigmaPlot 12; Systat Software) with a sigmoidal equation of the form:
 |
Where a equals the maximal amplitude of the curve, b equals the curve slope, x equals the FV amplitude, and x0 equals the FV amplitude required for half-maximal EPSP amplitude. Synaptic strength was also estimated by averaging the EPSP/FV ratio across the top three stimulus levels of the input output curve.
Methods for inducing and measuring LTD are similar to those used in our previous work (Norris et al., 1996; Sama et al., 2012; Norris et al., 2016). After input/output curves, single-stimulus pulses were delivered at a rate of 0.033 Hz to establish a baseline. Although the ½ maximal stimulus intensity from the input/output curve is often used for setting the baseline and LTD stimulation levels, we instead used the stimulus intensity necessary to elicit an ∼1 mV EPSP. This procedure was implemented to minimize possible hemispheric differences in the level of postsynaptic depolarization achieved during LTD induction (see discussion in Norris and Scheff, 2009; Mathis et al., 2011). It is well established that the induction of both LTD and long-term potentiation (LTP) are strongly dependent on postsynaptic depolarization levels (Malinow and Miller, 1986; Wigstrom and Gustafsson, 1986; Mulkey and Malenka, 1992). We have found that, relative to the contralateral hemisphere, slices from the ipsilateral hippocampus typically show reduced EPSP amplitudes (less depolarization) in response to the same level of presynaptic stimulation at 7 d aftr CCI (Norris and Scheff, 2009). Using the ½ maximal stimulus intensity in the CCI model would therefore necessarily lead to the elicitation of smaller EPSP amplitudes during LTD stimulation for ipsilateral relative to contralateral slices, which may in turn negatively affect the extent to which postsynaptic plasticity mechanisms are engaged.
After a stable baseline period of no less than 20 min, slices received a 15 min train of 1 Hz stimulation at baseline stimulation intensity, followed by another 60 min baseline period (stimulation rate = 0.033 Hz). Within each hemisphere, EPSP measures from the last 10 min of the post-1 Hz stimulation baseline were averaged across slices and expressed as a percentage of the pre-1 Hz stimulation baseline level. Therefore, for all electrophysiologic biomarkers, n is equal to the number of rats used rather than the number of slices.
Western blot.
On day 7 of recovery from CCI injury, hippocampi (contralateral control and ipsilateral injured) were harvested, put into Microfuge tubes, and snap frozen in liquid N2. Samples were stored at −80°C until needed. Samples were homogenized in PBS with a panel of protease and phosphatase inhibitors as described previously (Ansari et al., 2013). Lowry protein assays were performed to normalize protein levels and 25–75 μg of protein was used per gel well (depending on the antibody used for Western analysis). Proteins were resolved on 4–20% Criterion gradient gels (Bio-Rad) and transferred to Immobilon-FL PVDF membranes (Millipore, catalog #IPFL 00010). After transfer, membranes were blocked in 50% Odyssey protein blocking reagent in PBS and then incubated in primary antibodies in 50% Odyssey protein blocking reagent (LI-COR) in PBS with 0.2% Tween overnight at 4°C on a shaking platform. Westerns were performed with the following primary antibodies: GFAP (Cell Signaling Technology, catalog #3670S), Iba1 (Wako, catalog #019-19741), PSD-95 (Cell Signaling Technology, catalog #3450S), synapsin-1 (Cell Signaling Technology, catalog #6710S), GluR1 (Millipore, catalog #AB1504), NR2A (Millipore 07-632), NR2B (Millipore, catalog #05-920), CNAα (Millipore, catalog #07-1492), GAPDH (Abcam, catalog #ab9484), hevin (R&D Systems, catalog #MAB2836), and SPARC (R&D Systems, catalog #MAB942). Membranes were then washed in PBS/0.1% Tween, incubated in LI-COR 800-conjugated anti-mouse and 680-conjugated anti-rabbit secondary antibodies, washed, and imaged on an Odyssey Sa Imager System (LI-COR) on both the green (800) and red (680) channels. Quantification of bands was performed using Image Studio 2.1 software (LI-COR). Expression was normalized to loading control GAPDH for each sample.
Statistical analyses.
Unless otherwise noted, outcome measures were assessed in both the contralateral (uninjured) and ipsilateral (injured) hemispheres and compared across treatment groups using repeated-measures ANOVA. When appropriate, Fisher's protected least significant difference test was used for post hoc analyses. Significance for all statistical comparisons was set at p ≤ 0.05.
Results
Upregulation of NFAT expression is isoform and cell-type specific
The role of CN/NFAT signaling in TBI was assessed using a unilateral CCI model, which results in a substantial loss of CA3 neurons (Baldwin et al., 1997) and CA3–CA1 synaptic contacts in stratum radiatum of the ipsilateral hemisphere, followed by the partial reinnervation of CA1 during the first few weeks after injury (Scheff et al., 2005). Significant changes in CN expression and/or activity have also been reported in this and other models of acute injury (Huang et al., 2005; Liu et al., 2005; Shioda et al., 2006; Bales et al., 2010; Rosenkranz et al., 2012), although the nature of the change shows considerable variability from study to study. All end point measures were taken at 7 d after CCI, at which time CA3 presynaptic input into CA1 of the ipsilateral hemisphere has plateaued (and is functionally comparable to the contralateral hemisphere), but CA1 postsynaptic strength is still impaired (Norris and Scheff, 2009; Norris et al., 2016).
Figure 1, A and B, shows the appearance of a 45–48 kDa CN fragment in the hippocampus, ipsilateral to CCI, at 7 d after injury (p < 0.05, n = 5). This fragment is similar in size to the constitutively active CN (aCN) proteolytic fragment expressed in rat primary hippocampal cultures exposed to excitotoxic insults (Liu et al., 2005) or oligomeric Aβ peptides (Mohmmad Abdul et al., 2011) (also see Fig. 1A, 45 kDa aCN lane). Previous work has shown that aCN can appear at high levels in nervous tissue of intact rodents as a result of acute injury (Huang et al., 2005; Liu et al., 2005; Shioda et al., 2006), elevated amyloid levels (Abdul et al., 2009), or progressive neurodegeneration (Wu et al., 2010; Mohmmad Abdul et al., 2011), where it may contribute to synaptic deficits, cell death, and neuroinflammation, among other pathologic processes (Norris, 2014).
Figure 1.
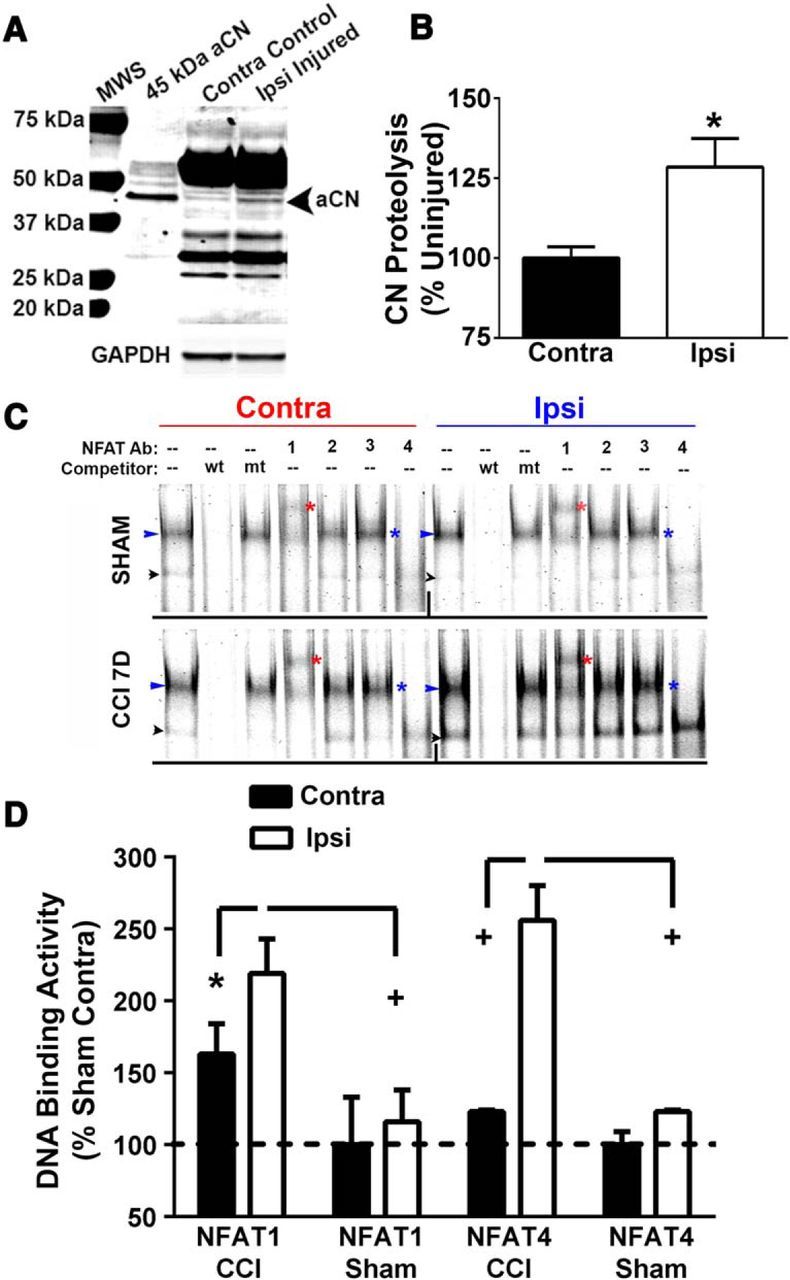
Changes in CN and NFAT signaling properties at 7 d after CCI. A, Representative Western blot for the CN A subunit in rat hippocampal whole-cell lysates from rat contralateral and ipsilateral hemispheres at 7 d after CCI. Whole-cell lysates from primary astrocyte cultures infected with adenovirus (Ad) expressing a constitutively active 45 kDa CN fragment (aCN) were run in parallel. Injury was associated with an increase in the proteolysis of CN to a 45–48 kDa fragment, similar in size to the fragment found in Ad-aCN-infected astrocyte cultures. B, Mean ± SD level (percentage contralateral hemiphere levels) of proteolysis in hippocampal homogenates in contralateral and ipsilateral hemispheres. p < 0.05, paired t test. C, Representative EMSA for hippocampal homogenates from the ipsilateral and contralateral hemispheres of rats exposed to CCI or to sham surgery. NFAT-binding probe was added to homogenates with and without antibodies (Ab) to each of the 4 CN-dependent NFAT isoforms to show supershifts and/or block shifts. Unlabeled WT (wt) and mutant (mt) DNA probe was included in some conditions to demonstrate DNA-binding specificity of the labeled probe. Bands denoted by the blue arrowheads reflect total NFAT binding. Red asterisks point to bands that were supershifted in response to inclusion of the NFAT1 antibody. Blue asterisks appear next to lanes where the NFAT4 antibody was included with the DNA-binding probe and illustrate a clear block shift. The black arrowheads point to bands that were relatively insensitive to antibody treatment conditions and likely reflect nonspecific binding. No gel shifts or block shifts were seen after inclusion of the NFAT 2 and 3 antibodies, suggesting that these isoforms did not significantly contribute to total NFAT binding under these conditions. D, Mean ± SD DNA-binding activity expressed as percentage of sham contralateral condition levels. NFAT1 was assessed from the supershifted bands obtained with the NFAT1 antibody (see red asterisks in C). NFAT 4 was assessed from the bands associated with blue arrowheads in C. n.s., Nonsignificant ipsilateral versus contralateral; *p < 0.05; + p < 0.01 ipsilateral versus contralateral, Fisher's PLSD test.
To investigate possible changes in the CN-dependent transcription factor NFAT, we used an electrophoretic mobility shift assay (EMSA) based on a well characterized NFAT-binding oligonucleotide probe derived from the IL4 enhancer region (Neal and Clipstone, 2001). Using IRDye-labeled probe, we observed a prominent protein-DNA complex (blue arrowheads, Fig. 1C) and a more variable and faint complex (black arrowheads, Fig. 1C) in hippocampal tissue fractions from both hemispheres of injured (n = 8) and sham (n = 5) rats at 7 d after injury. Both of these bands were displaced when EMSAs were performed in the presence of unlabeled wild-type probe, but not with a mutant probe, confirming that observed bands reflect NFAT-specific binding. The intensity of the higher-molecular-weight complex (blue arrowheads) was increased in the ipsilateral relative to the contralateral hemisphere of injured, but not sham-operated, rats at 7 d after CCI (p < 0.05). Supershift/blockshift experiments were then performed to identify which CN-dependent NFAT isoform(s) (NFATs 1–4) gave rise to the DNA-protein complexes. The NFAT1 antibody caused a clear supershift, indicated by the red asterisk in Figure 1C. The NFAT2 and 3 antibodies did not perceptibly alter the basic EMSA, indicating that they were not part of the complex. Finally, the NFAT4 antibody completely blocked the formation of the DNA–protein complex (see blue asterisk in Fig. 1C for where the complex normally migrates). For purposes of quantification, each “pair” of contralateral control and ipsilateral hippocampal homogenates was assayed on one gel. Because the NFAT4 antibody entirely blocked the formation of the DNA-protein complex (indicated by the blue arrow), this band was quantified and used to estimate total NFAT4 binding. For NFAT1, the supershifted band, indicated by the red asterisks, was quantified for each sample set. The quantitative data are shown in Figure 1D. There was a clear and significant increase in the DNA-binding activities of NFATs 1 and 4 in the ipsilateral hippocampi of the CCI animals, with little change observed across hemispheres in the shams.
Previous work has shown that NFATs 1 and 4 can appear in astrocytes as the result of injury or disease (Abdul et al., 2009; Serrano-Pérez et al., 2011; Caraveo et al., 2014). To determine whether the expression differences for NFATs 1 and 4 revealed by EMSA analyses (Fig. 1C,D) are associated with activated astrocytes after CCI, we colabeled coronal sections from injured rats (7 d after CCI) with NFAT and GFAP antibodies and imaged sections using confocal microscopy. As shown in Figure 2, we observed a striking increase in GFAP labeling (shown in red) in the hippocampus of the ipsilateral hemisphere, consistent with robust injury-dependent astrocyte activation. Although both NFAT isoforms exhibited diffuse labeling in the hippocampus of the contralateral hemisphere, there were clear differences in the distribution of the two isoforms in regard to cell type expression and injury. NFAT1 showed very little colocalization with GFAP-positive astrocytes in any region of the hippocampus examined regardless of hemisphere (Fig. 2A). Interhemispheric differences in NFAT1 labeling were characterized primarily by a shift to a punctate labeling pattern in the ipsilateral hippocampus and may be associated with proliferating microglia or infiltrating peripheral cells. In contrast, NFAT4 showed extensive colocalization with GFAP-positive astrocytes, especially in CA1 (Fig. 2B). In the ipsilateral hippocampus, the labeling intensity for NFAT4 was generally increased in proportion to the increase in GFAP labeling intensity. There was little evidence for the expression of either NFAT isoform in neuronal cell bodies, particularly in pyramidal neurons.
Figure 2.
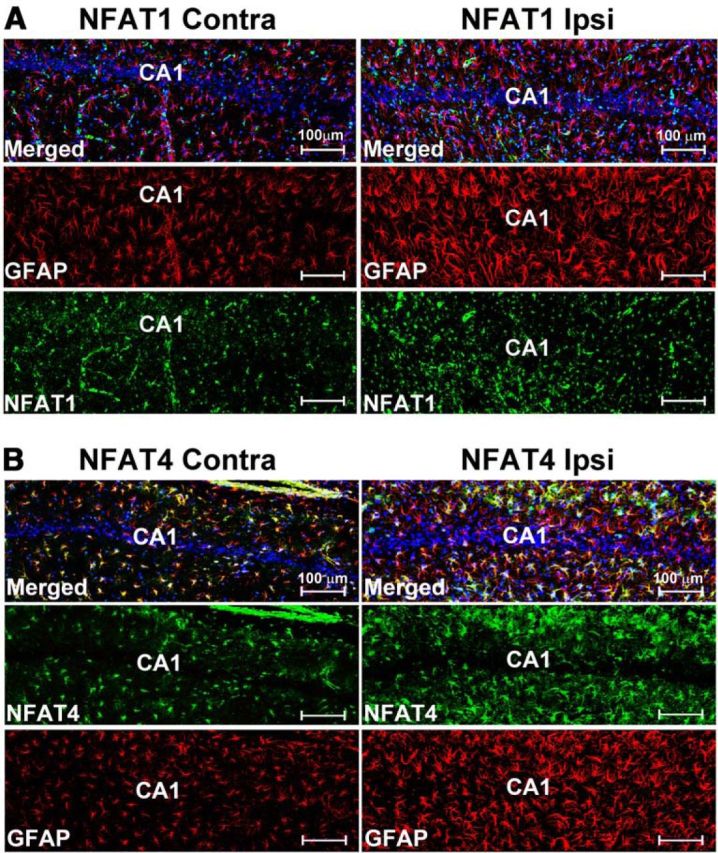
Confocal microscopic images showing the expression of NFATs 1 and 4 in the hippocampus at 7 d after CCI. A, B, NFAT and GFAP labeling shown separately and together (merged) in the CA1 region of the hippocampus in contralateral and ipsilateral hemispheres at 7 d after CCI. For each NFAT isoform, images were obtained from the same coronal plane of the same animal and are shown with identical brightness and contrast settings. Both NFATs are expressed throughout stratum radiatum and stratum oriens in the contralateral and ipsilateral hemispheres. NFAT1 (A) exhibited a more punctate labeling pattern in the ipsilateral hemisphere, but there was little to no colocalization with GFAP-positive astrocytes regardless of injury. In contrast, NFAT4 (B) was extensively colocalized to GFAP-positive astrocytes (see numerous yellow/orange cells in merged images) in both hemispheres and was upregulated as a result of injury.
Interestingly, NFAT4/GFAP colocalization showed marked heterogeneity across different regions of the hippocampus (Fig. 3). NFAT4 colocalization with GFAP was extensive in the stratum lacunosum/moleculare, stratum radiatum, and stratum oriens of CA1 (Fig. 3A–C), as well as the molecular layers and hilar regions of the dentate gyrus (Fig. 3M,N), but was much less apparent in the neuronal cell body layers (Fig. 3, cf. H vs I, N vs O). Moreover, in contrast to an earlier study that used a kainic acid insult (Serrano-Pérez et al., 2011), we did not observe extensive colocalization of NFAT4 in astrocytes of CA3 even though numerous activated astrocytes were found in this region after injury (Fig. 3B,J–L). This discrepancy may be attributable to differences in the type of cellular degeneration occurring after kainic acid treatment versus CCI. Regardless, these observations suggest possible phenotype differences between astrocytes in different hippocampal subregions.
Figure 3.

Heterogenous expression of NFAT4 in hippocampus. A, B, Low-power confocal micrographs of the same coronal slice (ipsilateral hemisphere) showing GFAP (A) and NFAT4 (B) labeling at 7 d after CCI. C–F, Low- (C) and high-powered (D–F) confocal images of NFAT4 and GFAP shown alone (E, F) or merged (C, D) in area CA1 of the hippocampus of the ipsilateral hemisphere. Micrographs illustrate extensive colocalization of NFAT4 throughout astrocyte somata, nuclei and major processes. G–O, NFAT4 and GFAP labeling in CA1 (G–I), CA3 (J–L), and the dentate gyrus (M–O) of the ipsilateral hemisphere. In CA1 and the DG, NFAT4 is found in most astrocytes. The exception is for those astrocytes found immediately adjacent to neuronal cell body layers, many of which are apparently devoid of NFAT4 (e.g., see arrowheads in G–I). Although numerous GFAP-positive astrocytes were found throughout the CA3 region, relatively few of these cells exhibited NFAT4 colabeling.
Use of AAV-Gfa2-VIVIT to inhibit astrocytic CN/NFAT signaling selectively in intact rats
To investigate the impact of astrocytic CN/NFAT signaling on functional outcome after TBI, we used an AAV-mediated gene delivery approach. Adult rats received bilateral hippocampal injections of AAV2/5 vectors expressing the NFAT inhibitor VIVIT (coupled to EGFP) or EGFP alone (control, CT). Some rats received identical injections of vehicle as an additional control. At approximately 8 weeks after AAV treatment, rats were subjected to a moderate CCI injury (Fig. 4A). To limit transgene expression to astrocytes, AAV vectors expressed the human GFAP promoter (Gfa2) (de Leeuw et al., 2006; Lee et al., 2008). As shown in our earlier work, a single injection (8 μl) of high titer (1012 ifu/ml) AAV-Gfa2 vectors into the hippocampus results in wide spread infection across all hippocampal subregions (Fig. 4B). Because CN is ubiquitously expressed and found at particularly high levels in hippocampal neurons, it was important to verify that AAV-Gfa2-mediated transgene expression occurred exclusively in astrocytes. Confocal microscopy was used to inspect the possible colocalization of AAV-dependent EGFP expression with neurons (MAP2b; Fig. 4E,F) and astrocytes (GFAP; Fig. 4D,G). As shown in Figure 4G, EGFP expression in CA1 strongly colocalized with GFAP (Fig. 4G,H), but there was no overlap between EGFP and MAP2b (Fig. 4F,H). Nearly identical observations were made in other hippocampal subregions as well (data not shown).
Figure 4.
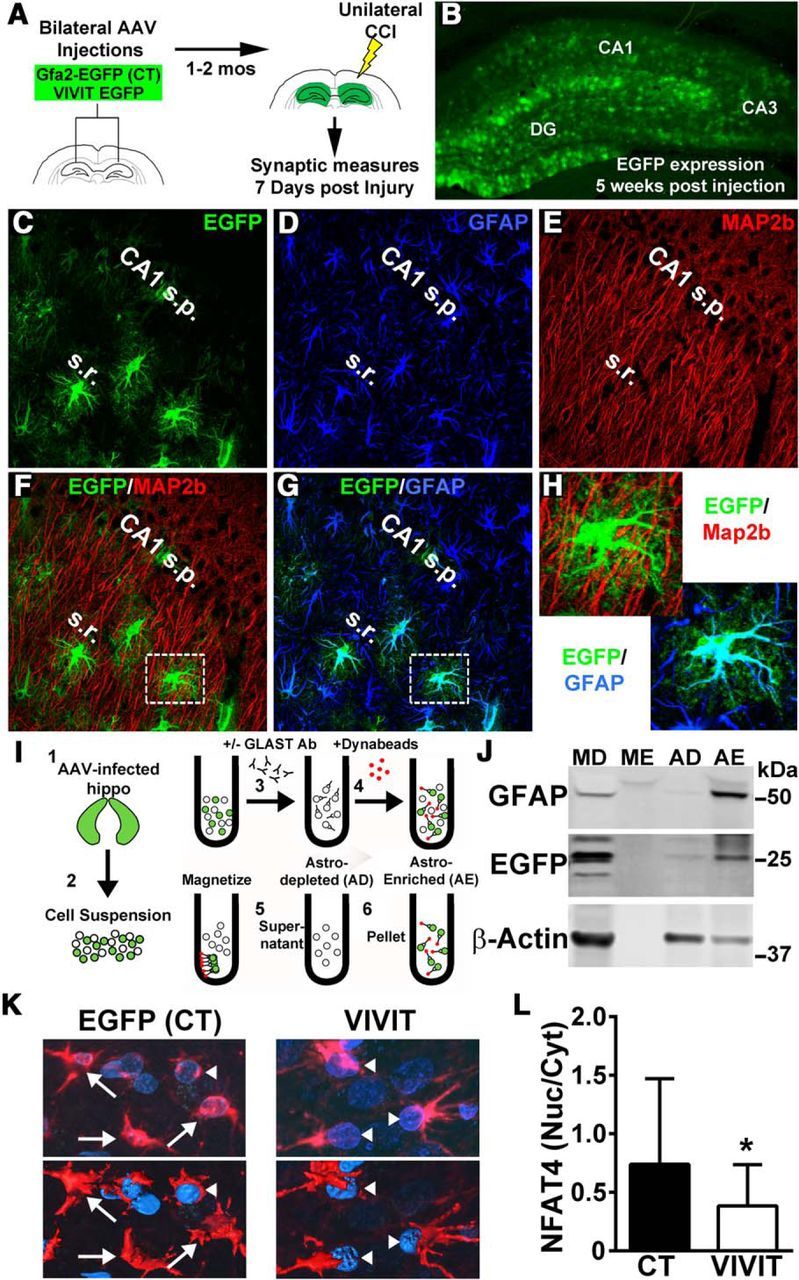
AAV-Gfa2 vectors for targeting astrocytic CN/NFAT signaling. A, AAV-Gfa2 vectors expressing EGFP alone (CT) or EGFP fused with the NFAT-inhibitory peptide VIVIT were bilaterally injected into the hippocampus of adult rats. At 1–2 months after injection, rats received a unilateral CCI and biomeasures were taken at 7 d after injury. B, Extensive EGFP expression throughout the molecular layers of the rat hippocampus at 5 weeks after injection. C–H, Confocal micrographs showing labeling patterns for EGFP (C, F), GFAP (D, G), and the neuronal marker MAP2B (E, F) from an AAV-Gfa2-EGFP-infected rat. Higher-magnification images of the regions shown in F and G are shown in H. There was extensive colocalization between EGFP and GFAP, but nearly no colocalization between EGFP and MAP2b (F, H). I, Diagram illustration of the protocol used to immunodeplete astrocytes from intact hippocampal tissue of AAV-Gfa2-EGFP infected rats. Single-cell suspensions were prepared from hippocampal tissue (Steps 1 and 2) and then added to a Microfuge tube. GLAST antibody was added (Step 3) to tag astrocytes. Metal Dynabeads were then added (Step 4) and tubes were placed in a magnetic rack. The supernatant containing unbound cells was collected (Step 5) and are referred to as the AD fraction, whereas the antibody-bound fraction contained in the pellet (Step 6) is referred to as the AE fraction. J, Representative Western blot showing the expression of GFAP, EGFP, and β-actin loading control in AD and AE fractions. MD and ME fractions (no GLAST antibody added) were run in parallel as controls. EGFP appears in the AE fraction, but not in the AD fraction, confirming that AAV-Gfa2 targets astrocytes selectively. K, Confocal micrographs showing the cellular localization of NFAT4 (red) in EGFP-expressing cells in rats infected with AAV-Gfa2-EGFP (CT) or AAV-Gfa2-VIVIT-EGFP. Note that astrocyte nuclei are labeled blue (DAPI). Top panels are 2D confocal images and the bottom panels are 3D-rendered images generated from Z stacks. Note that, for clarity, the EGFP signal is not shown. Arrows point to cells where NFAT4 is found distributed across both the cytosolic and nuclear compartments. Arrowheads point to cells where NFAT4 is largely excluded from the nucleus. L, Mean ± SD for the nuclear-to-cytosolic ratio for NFAT4 in rats treated with AAV-Gfa2-EGFP or AAV-Gfa2-VIVIT (n = 3 rats per group). VIVIT-treated rats show a significant reduction in the nuclear localization of NFATs. *p < 0.01, Student's t test.
To further confirm that AAV-Gfa2 vectors selectively targeted astrocytes, we performed immunodepletion experiments (Fig. 4I). Hippocampi from AAV-Gfa2-EGFP-infected rats were extracted and triturated into single-cell suspensions, which were then placed in a Microfuge tube and treated with GLAST antibodies (to tag astrocytes) coupled to metal Dynabeads. Tubes were then placed in a magnetic rack to pull out bead-coupled cells from the suspension. The fraction bound to the beads is referred to as AE, whereas the unbound fraction is referred to as AD. Some suspensions were passed through the same protocol, but were not treated with GLAST antibodies and served as MD and ME control fractions. Western blots were then performed to determine which fraction contained EGFP.
As shown in Figure 4J, the astrocyte-specific marker GFAP was found in the MD fraction (as expected) and also in the AE fraction, but not in the AD fraction. The prominent appearance of β-actin (which is expressed across all cell types) in the AD fraction demonstrates that the lack of GFAP expression was not attributable to a simple loss of total protein. These observations validate the successful immunodepletion of astrocytes in this protocol. If transgene expression after AAV-Gfa2 treatment is selective to astrocytes, then EGFP should show a similar expression pattern to GFAP, which is exactly what was observed: EGFP appeared in MD and AE fractions, but not in AD fractions. Essentially the same results were observed in four separate animals. Combined with the confocal images shown in Figure 4, C–H, these data demonstrate that AAV-Gfa2 vectors selectively target astrocytes.
The VIVIT peptide, expressed by AAV-Gfa2 vectors, is derived from a critical endogenous CN-docking site (PxIxIT) found in the regulatory subregion of NFATs 1–4 (Aramburu et al., 1999). Numerous studies have shown that VIVIT inhibits the nuclear localization or the transcriptional activity of NFATs in diverse tissues and cell types (Yu et al., 2007). Previously, we showed that VIVIT inhibited NFAT activity in primary astrocytes treated with proinflammatory or cytotoxic factors (Sama et al., 2008; Abdul et al., 2009). Confocal micrographs in Figure 4K show the cellular localization of NFAT4 (red) in EGFP- or VIVIT-expressing astrocytes of AAV-infected rats. Note that nuclei are labeled blue (DAPI) and that the EGFP signal (green) has been removed for clarity. In EGFP-expressing astrocytes, NFAT4 is mostly present in the cytosol, but also appears throughout the nucleus (blue) in many cells (Fig. 4K, arrows). In contrast, NFAT4 expression in VIVIT-expressing astrocytes is largely excluded from the nucleus (Fig. 4K, arrowheads), consistent with NFAT inhibition. The average nuclear-to-cytosolic ratios for NFAT4 measured from 32–42 cells per treatment condition across three animals per group (10–16 cells per rat) are shown in Figure 4L. Therefore, AAV-Gfa2 vectors not only provide selective targeting of astrocytes (Fig. 4C–J), they provide an effective way to limit the nuclear localization and activation of NFATs in these cells.
AAV-Gfa2-VIVIT protects synaptic function in ipsilateral hippocampus
The rat unilateral CCI model is associated with extensive loss of synaptic contacts in the CA1 region of the ipsilateral hippocampus (Scheff et al., 2005). Although there is a remarkable degree of structural and functional recovery with time after injury, deficits in basal synaptic strength are still evident at 7 d after injury in this model (Norris and Scheff, 2009), providing a reasonable time point to investigate the impact of astrocytic CN/NFAT signaling on synaptic function. One advantage of the rat CCI model, compared with mouse models or fluid-percussion injury models, is that moderate injuries to the ipsilateral hemisphere in adult rats (as investigated here) cause very few measurable changes in the contralateral hemisphere. For instance, previous work on oxidative stress markers after unilateral CCI in rats found no changes in the contralateral hemisphere of injured rats compared with either hemisphere of sham-operated rats or even to naive rats (Gilmer et al., 2009). Multiple presynaptic and postsynaptic protein markers, found at reduced levels in the ipsilateral hippocampus of rats after CCI, are also unchanged in the contralateral hemisphere compared with sham-operated controls (Ansari et al., 2013; Scheff et al., 2013). Similarly, our previous work using electrophysological measures of synaptic function taken at 2, 7, and 14 d after CCI showed comparable levels of basal synaptic strength and plasticity in the contralateral hemisphere of injured and sham-operated rats (Norris and Scheff, 2009). These results support the validity of using the contralateral hemisphere as a within-animal control for synaptic measures after unilateral CCI in rats.
To determine the extent to which astrocytic CN/NFAT signaling influences synaptic function after CCI, we measured basal hippocampal synaptic strength in slices from ipsilateral and contralateral hippocampi of AAV-treated rats (n = 6–8 rats/group) at 7 d after injury (Fig. 5A–D). Field potentials were recorded in stratum radiatum of area CA1 in response to electrical stimulation of CA3 Schaffer collaterals. Synaptic strength curves were generated by plotting EPSP slope values against FV amplitudes across increasing stimulus intensities. As shown in Table 1, all synaptic strength curve parameters investigated here were statistically comparable across the contralateral hemisphere of injured, sham-operated, and naive adult (∼2-month-old) Sprague Dawley rats.
Figure 5.
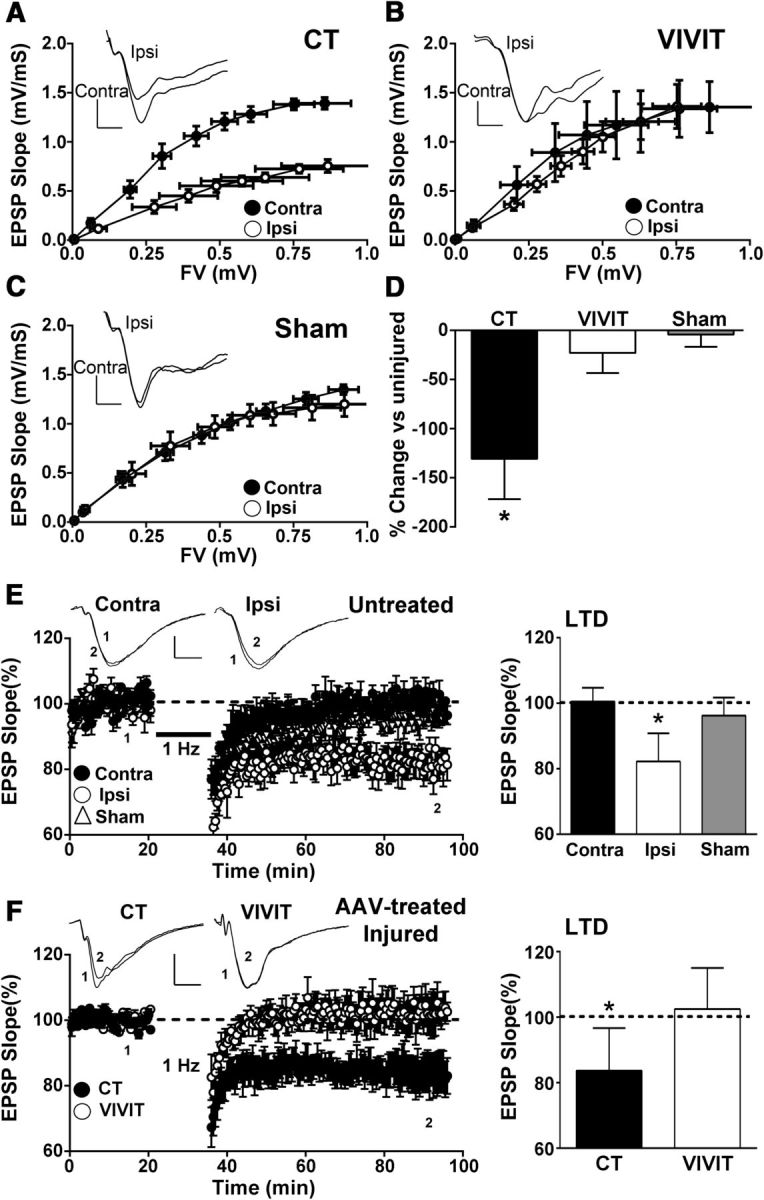
Effects of AAV-Gfa2-VIVIT on synaptic strength and plasticity. A, B, CA1 synaptic strength curves constructed from the mean ± SEM EPSP slope versus the mean ± SEM FV amplitude for hippocampal slices from the contralateral and ipsilateral hemispheres of rats treated with control (CT) AAV-Gfa2-EGFP (A) or with AAV-Gfa2-VIVIT (B). Slices from contralateral and ipsilateral hemispheres of sham-operated rats (C) are also shown for comparison. Insets in A–C show representative EPSP waveforms in each hemisphere matched on the basis of FV amplitude. Note, that for AAV-Gfa2-EGFP CT rats (A), the synaptic strength curve exhibited a marked downward shift in the ipsilateral relative to the contralateral hemisphere. In contrast, synaptic strength curves across hemispheres are qualitatively and quantitatively similar in sham rats (C) and rats treated with AAV-Gfa2-VIVIT (B). D, Mean ± SD of the EPSP-to-FV ratio in the ipsilateral hemisphere of AAV-treated and sham rats expressed as percentage change from the contralateral hemisphere. A reduction in the EPSP-to-FV ratio is only observed for AAV-Gfa2-EGFP CT rats.*p < 0.01 ipsilateral versus contralateral, Fisher's PLSD test. E, Left, Time plots showing mean ± SEM EPSP slope values (% baseline) from slices collected from the contralateral and ipsilateral hemispheres of untreated rats (i.e., no AAV) and the ipsilateral hemisphere of sham-operated rats. EPSPs were recorded before and for 60 min after delivery of a 15 min train of 1 Hz stimulation (bar). Representative waveforms in slices from each hemisphere of CCI rats measured before (1) and 60 min after (2) 1 Hz stimulation are shown in the inset. Calibration bars are 0.5 mV/5 ms. E, Right, Mean ± SD EPSP slope at 60 min after 1 Hz stimulation in contralateral and ipsilateral hemispheres and also in the ipsilateral hemisphere of sham-operated rats. Significant LTD was only observed in slices from the ipsilateral hemisphere of injured rats, *p < 0.05 Fisher's PLSD test. F, Left, LTD time plots (mean ± SEM EPSP slopes) and bar graphs (right) showing mean ± SD EPSP values (% baseline) at 60 min after 1 Hz stimulation in slices collected from the ipsilateral (injured) hemispheres of AAV-treated rats. Insets show representative waveforms as described for E. Note that LTD is present in the ipsilateral hemisphere of rats pretreated with AAV-Gfa2-EGFP control vector (CT), but not in rats treated with AAV-Gfa2-VIVIT. *p < 0.05, Fisher's PLSD test.
Table 1.
CA3–CA1 synaptic strength curve parameters (mean ± SD) for naive rats, sham-operated rats, and AAV-treated rats 7 d after CCI
| Condition | EPSP/FV ratio | Curve amplitude | Curve slope | Curve ½ max |
|---|---|---|---|---|
| Naive | 1.71 ± 0.72 | 1.31 ± 0.25 | 0.15 ± 0.05 | 0.35 ± 0.11 |
| Sham contra | 1.49 ± 0.27 | 1.20 ± 0.26 | 0.11 ± 0.03 | 0.28 ± 0.07 |
| Sham ipsi | 1.58 ± 0.19 | 1.34 ± 0.16 | 0.13 ± 0.03 | 0.33 ± 0.11 |
| EGFP/CT contra | 1.87 ± 0.30 | 1.40 ± 0.12 | 0.11 ± 0.03 | 0.29 ± 0.07 |
| EGFP/CT ipsi | 1.15 ± 0.65* | 0.78 ± 0.22* | 0.12 ± 0.06 | 0.30 ± 0.12 |
| VIVIT contra | 1.66 ± 0.23 | 1.42 ± 0.64 | 0.12 ± 0.05 | 0.32 ± 0.14 |
| VIVIT ipsi | 1.81 ± 0.43 | 1.41 ± 0.70 | 0.12 ± 0.06 | 0.37 ± 0.13 |
n = 6–10 rats/group.
*p < 0.05 versus EGFP/CT contra, Fisher's PLSD.
Hippocampal slices from the ipsilateral hemisphere of AAV-Gfa2-EGFP control rats showed a downward shift in the synaptic strength curve relative to the contralateral hemisphere within the same animals (Fig. 5A). Moreover, the EPSP-FV ratio for the ipsilateral hemisphere was reduced in AAV-Gfa2-EGFP control rats by ∼130% relative to the contralateral side (p < 0.01), indicative of reduced synaptic strength (Fig. 5D). In contrast, synaptic strength curves were qualitatively and quantitatively similar across hemispheres in rats pretreated with AAV-Gfa2-VIVIT-EGFP (Fig. 5B) and strongly resembled synaptic strength curves recorded in untreated sham rats (Fig. 5C) and naive rats (Table 1). The EPSP-FV ratio in AAV-Gfa2-VIVIT-EGFP rats showed only a 23% reduction in the ipsilateral hemisphere, which was similar to the ipsilateral hemisphere of sham rats (∼4% reduction) and significantly less (p < 0.05) than the reduction found in AAV-Gfa2-EGFP control rats (Fig. 5D).
We next investigated the extent to which astrocytic CN/NFAT signaling influences synaptic plasticity after TBI. Although LTP is commonly measured in injury and disease model systems, our previous work with the rat CCI model did not reveal LTP deficits in the ipsilateral hemisphere at 7 d after CCI (Norris and Scheff, 2009). In contrast, we recently found that the induction of LTD in CA1 using a 15 min/1 Hz stimulus train is significantly enhanced in the ipsilateral hemisphere at both 7 and 14 d after injury (Norris et al., 2016). To confirm these observations, we used a similar 1 Hz stimulus paradigm to induce LTD in the ipsilateral and contralateral hemispheres of injured rats (7 d after CCI) and sham-operated rats in the absence of AAV treatment. As shown in Figure 5E, slices from sham rats and slices from the contralateral hemisphere of injured rats showed no significant depression (96% and 101% of baseline, respectively) after 1 Hz stimulation. In contrast, the same stimulus paradigm caused a modest (82% of baseline) but significant (p < 0.01) depression in ipsilateral slices from injured rats, confirming that CCI increases the susceptibility to LTD in the ipsilateral hemisphere. Similar to these results, ipsilateral slices from AAV-Gfa2-EGFP-treated rats showed significant LTD (∼83% of baseline, p < 0.05) after 1 Hz stimulation (Fig. 5F). In contrast, LTD was completely prevented in ipsilateral slices from rats treated with AAV-Gfa2-VIVIT (103% of baseline). These results suggest that the astrocytic CN/NFAT pathway helps to drive synapse dysfunction in the ipsilateral hippocampus after CCI injury.
AAV-Gfa2-VIVIT prevents the loss of synapse-related proteins in ipsilateral hippocampus
In addition to impaired synaptic function and altered plasticity, CCI is also associated with the loss of several key synaptic proteins in the ipsilateral hippocampus within the first week after injury, including the postsynaptic anchoring protein PSD-95, the presynaptic vesicle docking protein synapsin 1, AMPA/kainate-type glutamate receptor isoforms (e.g., GluR1), and NMDA-type glutamate receptor isoforms (e.g., NR2A and NR2B) (Ansari et al., 2013; Scheff et al., 2013). To determine whether these changes are sensitive to astrocytic CN/NFAT signaling, we harvested contralateral and ipsilateral hippocampus at 7 d after CCI from sham rats (n = 4) and rats treated with AAV-Gfa2-VIVIT vectors (n = 6) and used Western blot to quantify synaptic protein levels. For controls, some rats (n = 6) received bilateral injections of AAV-Gfa2 expressing EGFP alone, whereas other rats (n = 6) received vehicle injections. Because the two control groups showed no significant differences on any of the outcome measures, statistical comparisons were made only for the AAV-Gfa2-EGFP and AAV-Gfa2-VIVIT treatment groups.
For AAV-Gfa2-EGFP-treated rats, the postsynaptic protein PSD-95 and the GluR1 AMPA receptor subunit were each significantly reduced, by 56% and 82%, respectively, in the ipsilateral, relative to the contralateral, hippocampus (Fig. 6A,D and C,F). However, the loss of PSD-95 and GluR1 protein was comparably small (19% and 27%, respectively) for rats treated with AAV-Gfa2-VIVIT. Although levels for synapsin 1 were not reduced in the ipsilateral hippocampus of AAV-Gfa2-EGFP at 7 d after CCI, the AAV-Gfa2-VIVIT group showed a significant 110% increase in the ipsilateral hemisphere (p < 0.05) (Fig. 6B,E). Similar to PSD-95 and GluR1, we also observed a significant reduction in NR2A (−26 ± 57% mean ± SD, p < 0.05) and a small, nonsignificant reduction in NR2B levels (−20 + 35% mean ± SD, p > 0.05) in AAV-Gfa2-EGFP rats (data not shown). However, these changes were not significantly altered by AAV-Gfa2-VIVIT (NR2A −26 ± 27% mean ± SD, p < 0.05; NR2B −23 ± 24% mean ± SD, p > 0.05). Therefore, together with measures of basal synaptic strength (Fig. 5A–D), the results show that AAV-Gfa2-VIVIT protects against the loss of several key synaptic proteins after CCI.
Figure 6.

AAV-Gfa2-VIVIT protects against the loss of synaptic proteins. A–C, Representative Western blots and mean ± SD levels (D–F) for hippocampal synaptic proteins in the contralateral and ipsilateral hemispheres of rats treated with AAV-Gfa2-EGFP (CT) or AAV-Gfa2-VIVIT. Protein levels in D–F are from the ipsilateral hemisphere and are expressed as the percentage change from the contralateral hemisphere. The percentage change in the ipsilateral versus contralateral hemisphere of sham controls are also provided. Levels for PSD95 and GluR1 showed significant reductions in the ipsilateral hemisphere of AAV-Gfa2-EGFP rats, *p < 0.05 ipsilateral versus contralateral, Fisher's PLSD test, but not in the ipsilateral hemisphere of VIVIT-treated rats. Although Syn1 was not significantly reduced with injury, protein levels in the ipsilateral hemisphere were significantly increased by pretreatment with AAV-Gfa2-VIVIT. *p < 0.05 ipsilateral versus contralateral, Fisher's PLSD test, n = 5–6 rats per group.
AAV-Gfa2-VIVIT does not reduce glial activation after CCI, but does modulate astrocyte-related proteins involved in synaptic remodeling
Our work, and work from others, has shown that the CN/NFAT pathway is an important regulator of the activated phenotypes of both astrocytes (Norris et al., 2005; Fernandez et al., 2007; Sama et al., 2008; Serrano-Pérez et al., 2011; Furman et al., 2012) and microglia (Kataoka et al., 2009; Nagamoto-Combs and Combs, 2010; Shiratori et al., 2010). We previously showed that AAV-Gfa2-VIVIT protects synaptic function in a chronic, progressive disease model (APP/PS1 mice) in parallel with reductions in GFAP and Iba1 (a microglial protein). As shown in Figure 7, A–D, we performed similar measures on GFAP and Iba1 protein levels in the rat CCI model. Although we did observe significant injury-related elevations for both proteins, consistent with robust glial activation, neither protein marker was sensitive to AAV-Gfa2-VIVIT treatment. These results suggest that the synaptoprotective effects of AAV-Gfa2-VIVIT after acute injury do not require a reduction in glial activation.
Figure 7.
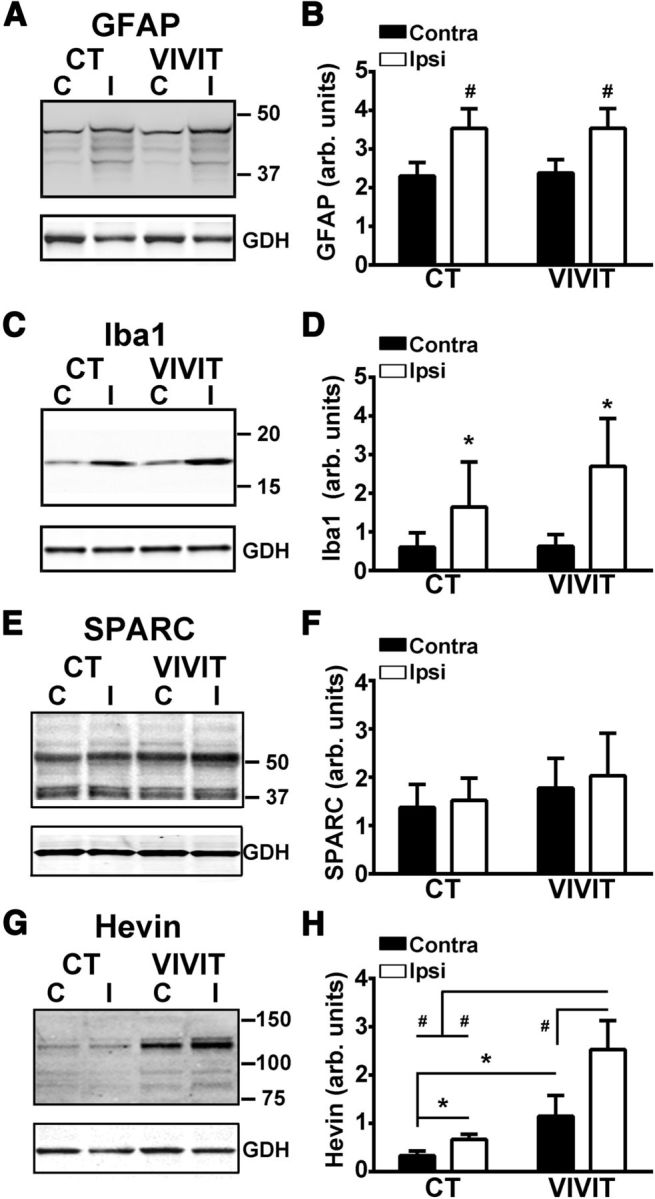
AAV-Gfa2-VIVIT does not alter GFAP or Iba1 levels, but causes an increase in hevin levels. Representative Western blots (A, C) and mean ± SD. GFAP and Iba1 protein levels (B, D) in the contralateral and ipsilateral hippocampus of AAV-treated rats at 7 d after CCI. Note that both glial markers showed a significant increase in the hippocampus of the ipsilateral hemisphere, but were not significantly altered by pretreatment with AAV-Gfa2-VIVIT. #p < 0.001 ipsilateral versus contralateral, Fisher's PLSD. n = 5–6 rats, group. E–H, Representative Western blots (E, G) and mean ± SD SPARC and hevin protein levels (F, H) in the contralateral and ipsilateral hippocampus of AAV-treated rats at 7 d after CCI. No virus or injury-dependent effects were observed for SPARC. In contrast, hevin was sensitive to both injury and AAV treatment. In both AAV groups, hevin was elevated in the ipsilateral relative to the contralateral hemisphere. Overall hevin levels were greater in the VIVIT-treated group regardless of hemisphere, but were highest in the injured hemisphere. *p < 0.05; #p < 0.001 ipsilateral versus contralateral, Fisher's LSD, n = 5–6 rats.
We next tested whether AAV-Gfa2-VIVIT modulates other glial-based mechanisms directly involved in synaptic remodeling. SPARC and SPARCL1 (hevin) are secreted matricellular factors that modulate synaptogenesis during development and possibly after neural injury (Jones et al., 2011; Kucukdereli et al., 2011; Albrecht et al., 2012; Blakely et al., 2015). Hevin has been shown to increase the number and size of synapses between developing neurons in culture, and SPARC appears to counter these actions (Kucukdereli et al., 2011). Several studies have reported an increase in SPARC and hevin levels in activated astrocytes after injury (Jones et al., 2014). Moreover, a microarray study from our group observed significant changes in hevin and SPARC gene transcripts in primary neuron/astrocyte cultures as a result of CN overexpression (Norris et al., 2005).
Western blots of ipsilateral and contralateral hippocampus from rats at 7 d after CCI revealed differential sensitivities of SPARC and hevin to injury and AAV treatment (Fig. 7E–H). Although SPARC was readily detected in both hemispheres, it was relatively unaffected by either injury or AAV treatment (Fig. 7E,F). In contrast, hevin showed very low expression in the contralateral hemisphere, but exhibited increased levels in the ipsilateral hippocampus (p < 0.05) regardless of virus treatment, indicative of a putative compensatory reaction to synapse loss with injury (Fig. 7G,H). Remarkably, hevin was also significantly affected by AAV-Gfa2-VIVIT treatment. Among the proteins investigated in this study, hevin was the only one to show a change in expression in the contralateral hemisphere across virus treatment groups, with significantly greater levels appearing in rats treated with AAV-Gfa2-VIVIT (p < 0.05). Moreover, the effects of injury and AAV-Gfa2-VIVIT were synergistic, such that the highest levels of hevin were found in the ipsilateral hemisphere of AAV-Gfa2-VIVIT-treated rats (p < 0.001). These results suggest that AAV-Gfa2-VIVIT may increase and/or promote synaptic recovery after injury by driving the expression of the prosynaptogenesis factor hevin.
Discussion
Astrocyte activation and the CN/NFAT pathway after acute injury
Recent advances in genetic approaches, including the use of cell-type-specific promoters to selectively target astrocyte signaling pathways, have greatly increased our understanding of the functional impact of activated astrocytes. Several targeted approaches to limiting astrocyte activation have been reported, including the suppression of astrocyte proliferation (Bush et al., 1999), the deletion of astrocyte intermediate filament proteins (Pekny et al., 1999; Lundkvist et al., 2004), and the knock-down/inhibition of key inflammatory signaling pathways (Okada et al., 2006; Herrmann et al., 2008; Wanner et al., 2013). These manipulations have revealed many apparently protective functions of activated astrocytes after acute CNS trauma; for example, the formation of protective glial scars, support of the blood–brain barrier, myelin preservation, and/or the protection of neuronal viability. In contrast, other reports have suggested that activated astrocytes have a detrimental impact on nervous tissue. For instance, suppression of astrocyte activation via the combined knock-down of GFAP and vimentin led to improved synaptic recovery in the outer molecular layer of the dentate gyrus after an entorhinal cortex lesion (Wilhelmsson et al., 2004). These observations highlight the complexity and possibly the heterogeneity of astrocyte activation during injury and disease.
The present study sought to inhibit astrocyte activation in the rat CCI model by selectively inhibiting the CN/NFAT pathway in astrocytes. In many cell types, CN/NFATs are key transcriptional regulators capable of driving striking changes in cellular phenotype (Crabtree and Olson, 2002; Horsley and Pavlath, 2002; Hogan et al., 2003; Wilkins and Molkentin, 2004; Hassoun et al., 2009). Although less attention has focused on CN/NFAT signaling in the CNS, mounting evidence suggests that CN/NFATs are associated with phenotype changes (i.e., activation) in astrocytes (Fernandez et al., 2007; Sama et al., 2008; Serrano-Pérez et al., 2011; Furman et al., 2012) and microglia (Kataoka et al., 2009; Nagamoto-Combs and Combs, 2010; Shiratori et al., 2010). In primary cultures, CN/NFAT induces numerous immune/inflammatory pathways, many of which are suppressed by the NFAT inhibitor VIVIT (Sama et al., 2008; Nagamoto-Combs and Combs, 2010). Previously, we showed that VIVIT expression in astrocytes over the course of 10–11 months using AAV-Gfa2 vectors was associated with a reduction in biochemical and morphological markers of glial activation in a mouse model (APP/PS1) of AD (Furman et al., 2012). Similar to cell culture studies, the effects of AAV-Gfa2-VIVIT in APP/PS1 mice were beneficial in nature and included improved synaptic function and cognition, suggesting that astrocyte activation can exacerbate neural dysfunction, at least in the context of progressive amyloid pathology.
Together, these studies provided seemingly strong rationale for using AAV-Gfa2-VIVIT to test the impact of activated astrocytes on neural function in an intact animal model of TBI. To our surprise, however, AAV-Gfa2-VIVIT had little to no effect on GFAP or Iba1 levels in brain-injured rats. If anything, AAV-Gfa2-VIVIT precipitated an increase in Iba1 levels in ipsilateral hippocampus, consistent with greater microglial activation. This apparent discrepancy with earlier findings in APP/PS1 mice could indicate fundamental differences in the activated glial phenotype(s) associated with acute injury versus progressive neuropathology (i.e., amyloid deposition). Indeed, there is strong evidence to suggest that multiple phenotypic characteristics of activated astrocytes, including transcriptional profile and proliferative capacity, can differ significantly depending on the nature of the insult and/or underlying pathology (Zamanian et al., 2012; Sirko et al., 2013). It is also possible that the relative contribution of astrocytes and CN/NFAT signaling to glial activation, and neuroinflammation in general, is greater in chronic disease conditions relative to acute injury. Regardless, the synaptoprotective effects of AAV-Gfa2-VIVIT, in the face of robust glial activation, suggests that the astrocytic CN/NFAT pathway influences neural function through mechanisms that are independent of classic neuroinflammatory signaling mediators.
Effects of AAV-Gfa2-VIVIT on synaptic function markers
Although the impact of AAV-Gfa2-VIVIT on glial activation appeared markedly different in the rat CCI model compared with APP/PS1 mice, the beneficial effects of VIVIT treatment on synaptic function were highly similar. The large reduction in synaptic strength measured in ipsilateral slices from AAV-Gfa2-EGFP control rats was essentially absent in AAV-Gfa2-VIVIT-treated rats. AAV-Gfa2-VIVIT also prevented the loss (or hastened the recovery) of several synaptic proteins and reduced susceptibility to LTD in injured hippocampus. Although the specific mechanisms linking astrocytic CN/NFATs to impaired synaptic function remain unsettled, Western blots suggested a possibly important role for hevin, an astrocyte-secreted protein that regulates physical interactions between cells and the extracellular matrix in a variety of tissues (Sullivan et al., 2004; Jones and Bouvier, 2014). The dynamic interactions between hevin and SPARC have been proposed as a critical mechanism for fine tuning synaptic networks during development (Kucukdereli et al., 2011) and possibly after neural injury (Blakely et al., 2015). Similar to previous reports (Lively and Brown, 2008; Lively et al., 2011), we observed an injury-related increase in hevin levels in the rat CCI model. Perhaps more remarkably, hevin expression was strongly induced by VIVIT, an effect that was potentiated with injury. It is possible that elevated NFAT activity in activated astrocytes keeps hevin expression somewhat in check through direct transcriptional suppression. Conversely, NFATs may limit the activity/expression of other transcription factors that stimulate hevin induction.
In addition to hevin, there are many other possible astrocyte-based mechanisms that could link CN/NFAT signaling to synaptic function. For instance, complement components such as C3 are associated with astrocyte activation (Tomimoto et al., 1997; Cardinaux et al., 2000) and synapse loss (Stevens et al., 2007) and are sensitive to CN activity (Norris et al., 2005). Astrocytes also protect neuronal viability and fine tune synaptic transmission by removing excess glutamate from the extracellular milieu (Sattler and Rothstein, 2006), a process that is protected by VIVIT in primary culture models of neuroinflammation and amyloid toxicity (Sama et al., 2008; Abdul et al., 2009). Alternatively, astrocytic CN/NFAT signaling may stimulate the release of excitotoxic factors that disrupt neuronal Ca2+ dysregulation, which in turn lead to synapse dysfunction, particularly in regard to the expression of synaptic plasticity. LTD, for example, is normally absent in hippocampal slices from healthy adult rodents when the bath Ca2+/Mg2+ concentrations are at a one-to-one ratio (O'Dell and Kandel, 1994; Norris et al., 1996; Vouimba et al., 2000; Foster and Kumar, 2007; Shankar et al., 2008; Dumas, 2012), but is much easier to induce in slices under conditions that promote Ca2+ dysregulation such as aging, injury, and elevated amyloid levels (Norris et al., 1996; Vouimba et al., 2000; Foster and Kumar, 2007; Shankar et al., 2008; Li et al., 2009; Ma et al., 2012; Sama et al., 2012; Norris et al., 2016). Clearly, future studies will be needed to fully characterize NFAT interactions with hevin and multiple other astrocyte-based signaling mechanisms and their roles in synaptic recovery and repair.
NFAT isoforms and astrocyte signaling after CCI
Similar to peripheral tissues, changes in glial signaling may hinge upon the expression of specific NFAT isoforms. NFATs 1, 2, and 4 have each been implicated in the production of proinflammatory cytokines and chemokines in glial cells (Canellada et al., 2008; Perez-Ortiz et al., 2008; Nagamoto-Combs and Combs, 2010; Neria et al., 2013). Previously, we observed elevated levels of NFAT1 activation in astrocytes in human hippocampus at early stages of cognitive decline associated with AD (Abdul et al., 2009). Other groups have observed an increase in the localization of NFAT4 to hippocampal astrocytes, particularly after acute injury (Serrano-Pérez et al., 2011; Neria et al., 2013; Yan et al., 2014). Consistent with these reports, the present study found that NFAT–DNA interactions were increased in the hippocampus after unilateral CCI and attributable largely to the activities of NFATs 1 and 4. However, confocal micrographs showed that the expression of NFAT4 is far more selective for astrocytes. Unlike our work on human tissue, we found little to no evidence linking NFAT 1 to activated astrocytes in brain-injured rats. It's possible that NFAT1 is primarily associated with microglia and/or infiltrating immune cells in rodents or that its activity is strongly stimulated in astrocytes at earlier time points after injury, but then recedes with time. There may also be significant differences in the expression and function of different NFAT isoforms across rodents and humans. Further work using biochemical and fluorescence microscopy techniques will be needed to evaluate these possibilities and to identify the cell types that express NFAT1 after injury.
Injury-dependent elevations in activity, coupled with intense astrocyte-specific expression, suggest that NFAT4 is the most likely target of VIVIT's synaptoprotective effects. Interestingly, the colocalization of NFAT4 with GFAP exhibited considerable heterogeneity across the hippocampus, characterized by extensive labeling in CA1 and the dentate and little labeling in CA3. Within the CA1 and dentate regions, NFAT4 was found in the dendritic–axonal layers, but was largely excluded from the cell body layers. The dendrite/axonal layers of CA1 and the dentate are where much of the loss (and subsequent recovery) of synapses take place in the hippocampus after unilateral CCI (Scheff et al., 2005; Winston et al., 2013), exactly where NFAT4 upregulation appeared in the present study. These results suggest that astrocytic NFAT4 is intimately associated with excitatory synaptic contacts (which are largely excluded from neuronal cell body layers), where it appears to robustly respond to changes in the functional status and/or viability of these connections. Although our findings suggest that NFAT4 may negatively affect synaptic recovery after injury by limiting the expression of hevin (or other protective factors), the role of NFAT4 in synaptic function during nondisease/injury conditions is unknown. Previously, we found few alterations in basal synaptic strength and LTP in wild-type mice treated for >10 months with AAV-Gfa2-VIVIT, suggesting that astrocytic NFAT4 does not dramatically alter fundamental mechanisms of synaptic transmission in adult animals. Nonetheless, further work will be necessary to clarify the impact of NFAT4 on synapses under both normal and pathological conditions.
Conclusion
Activated astrocytes have been shown to play both protective and deleterious roles in acute injury and chronic neurodegenerative disease. Along with several other earlier reports, the present study suggests that the deleterious actions of activated astrocytes—in particular the synaptotoxic effects of astrocytes—may hinge upon the functional status of the CN/NFAT pathway. The results of this study may offer important clues into how astrocytes affect neuronal circuit rewiring after acute injury and establish astrocyte-based signaling pathways as possible targets for preserving or improving neural function after acute head trauma.
Footnotes
This work was supported by grants the National Institutes of Health (Grant AG027297 to C.M.N., Grant AG000242 to P.S., and Grant AG047762 to M.M.P.); the Kentucky Spinal Cord and Head Injury Research trust (Grant 12-10A to C.M.N. and Grant 12-16A to S.W.S.); the PhRMA Foundation (graduate fellowships to J.L.F. and M.M.P.); the Irene and Eric Simon Brain Research Foundation (undergraduate fellowship to E.J.P.); and the Hazel Embry Research Trust. We thank Linda Simmerman and Adam Bachstetter for technical assistance.
The authors declare no competing financial interests.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht D, López-Murcia FJ, Pérez-González AP, Lichtner G, Solsona C, Llobet A. SPARC prevents maturation of cholinergic presynaptic terminals. Mol Cell Neurosci. 2012;49:364–374. doi: 10.1016/j.mcn.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Anderson MA, Ao Y, Sofroniew MV. Heterogeneity of reactive astrocytes. Neurosci Lett. 2014;565:23–29. doi: 10.1016/j.neulet.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. Dose- and time-dependent neuroprotective effects of Pycnogenol following traumatic brain injury. J Neurotrauma. 2013;30:1542–1549. doi: 10.1089/neu.2013.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J Neurotrauma. 1997;14:385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- Bales JW, Ma X, Yan HQ, Jenkins LW, Dixon CE. Expression of protein phosphatase 2B (calcineurin) subunit A isoforms in rat hippocampus after traumatic brain injury. J Neurotrauma. 2010;27:109–120. doi: 10.1089/neu.2009.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely PK, Hussain S, Carlin LE, Irani DN. Astrocyte matricellular proteins that control excitatory synaptogenesis are regulated by inflammatory cytokines and correlate with paralysis severity during experimental autoimmune encephalomyelitis. Front Neurosci. 2015;9:344. doi: 10.3389/fnins.2015.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2015 doi: 10.1016/j.expneurol.2015.03.020. pii: S0014-4886(15)00089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/S0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Canellada A, Ramirez BG, Minami T, Redondo JM, Cano E. Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1–4 and cyclooxygenase-2 as NFAT target genes. Glia. 2008;56:709–722. doi: 10.1002/glia.20647. [DOI] [PubMed] [Google Scholar]
- Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, Yan X, Ben-Johny M, Soste M, Picotti P, Kim H, Caldwell KA, Caldwell GA, Sulzer D, Yue DT, Lindquist S. Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proc Natl Acad Sci U S A. 2014;111:E3544–3552. doi: 10.1073/pnas.1413201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinaux JR, Allaman I, Magistretti PJ. Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia. 2000;29:91–97. [PubMed] [Google Scholar]
- Celsi F, Svedberg M, Unger C, Cotman CW, Carrì MT, Ottersen OP, Nordberg A, Torp R. Beta-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol Dis. 2007;26:342–352. doi: 10.1016/j.nbd.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–S79. doi: 10.1016/S0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- de Leeuw B, Su M, ter Horst M, Iwata S, Rodijk M, Hoeben RC, Messing A, Smitt PS, Brenner M. Increased glia-specific transgene expression with glial fibrillary acidic protein promoters containing multiple enhancer elements. J Neurosci Res. 2006;83:744–753. doi: 10.1002/jnr.20776. [DOI] [PubMed] [Google Scholar]
- Dumas TC. Postnatal alterations in induction threshold and expression magnitude of long-term potentiation and long-term depression at hippocampal synapses. Hippocampus. 2012;22:188–199. doi: 10.1002/hipo.20881. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci. 2007;27:8745–8756. doi: 10.1523/JNEUROSCI.1002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, Kumar A. Susceptibility to induction of long-term depression is associated with impaired memory in aged Fischer 344 rats. Neurobiol Learn Mem. 2007;87:522–535. doi: 10.1016/j.nlm.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Norris CM. Calcineurin and glial signaling: neuroinflammation and beyond. J Neuroinflammation. 2014;11:158. doi: 10.1186/s12974-014-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease. J Neurosci. 2012;32:16129–16140. doi: 10.1523/JNEUROSCI.2323-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmer LK, Roberts KN, Joy K, Sullivan PG, Scheff SW. Early mitochondrial dysfunction after cortical contusion injury. J Neurotrauma. 2009;26:1271–1280. doi: 10.1089/neu.2008.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Kawamata T, Saito N, Sasaki M, Nakai M, Niu S, Taniguchi T, Terashima A, Yasuda M, Maeda K, Tanaka C. Isoform-specific redistribution of calcineurin A alpha and A beta in the hippocampal CA1 region of gerbils after transient ischemia. J Neurochem. 1998;70:1289–1298. doi: 10.1046/j.1471-4159.1998.70031289.x. [DOI] [PubMed] [Google Scholar]
- Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildinger M, Auricchio A, Gao G, Wang L, Chirmule N, Wilson JM. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J Virol. 2001;75:6199–6203. doi: 10.1128/JVI.75.13.6199-6203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol. 2002;156:771–774. doi: 10.1083/jcb.200111073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta JB, Dobberfuhl A, Filippopolous T, Guo Y, Kwon G, Grosskreutz CL. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci U S A. 2005;102:12242–12247. doi: 10.1073/pnas.0505138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EV, Bouvier DS. Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast. 2014;2014:321209. doi: 10.1155/2014/321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EV, Bernardinelli Y, Tse YC, Chierzi S, Wong TP, Murai KK. Astrocytes control glutamate receptor levels at developing synapses through SPARC-beta-integrin interactions. J Neurosci. 2011;31:4154–4165. doi: 10.1523/JNEUROSCI.4757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka A, Tozaki-Saitoh H, Koga Y, Tsuda M, Inoue K. Activation of P2X7 receptors induces CCL3 production in microglial cells through transcription factor NFAT. J Neurochem. 2009;108:115–125. doi: 10.1111/j.1471-4159.2008.05744.x. [DOI] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011;108:E440–E449. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Lively S, Brown IR. Localization of the extracellular matrix protein SC1 coincides with synaptogenesis during rat postnatal development. Neurochem Res. 2008;33:1692–1700. doi: 10.1007/s11064-008-9606-z. [DOI] [PubMed] [Google Scholar]
- Lively S, Moxon-Emre I, Schlichter LC. SC1/hevin and reactive gliosis after transient ischemic stroke in young and aged rats. J Neuropathol Exp Neurol. 2011;70:913–929. doi: 10.1097/NEN.0b013e318231151e. [DOI] [PubMed] [Google Scholar]
- Lundkvist A, Reichenbach A, Betsholtz C, Carmeliet P, Wolburg H, Pekny M. Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J Cell Sci. 2004;117:3481–3488. doi: 10.1242/jcs.01221. [DOI] [PubMed] [Google Scholar]
- Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1(9–36) amide rescues synaptic plasticity and memory deficits in Alzheimer's disease model mice. J Neurosci. 2012;32:13701–13708. doi: 10.1523/JNEUROSCI.2107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Miller JP. Postsynaptic hyperpolarization during conditioning reversibly blocks induction of long-term potentiation. Nature. 1986;320:529–530. doi: 10.1038/320529a0. [DOI] [PubMed] [Google Scholar]
- Mathis DM, Furman JL, Norris CM. Preparation of acute hippocampal slices from rats and transgenic mice for the study of synaptic alterations during aging and amyloid pathology. J Vis Exp. 2011;49:2330. doi: 10.3791/2330. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Baig I, Levine H, 3rd, Guttmann RP, Norris CM. Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Abeta in primary cell culture. Aging Cell. 2011;10:103–113. doi: 10.1111/j.1474-9726.2010.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-C. [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells) J Neurosci. 2010;30:9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem. 2001;276:3666–3673. doi: 10.1074/jbc.M004888200. [DOI] [PubMed] [Google Scholar]
- Neria F, del Carmen Serrano-Pérez M, Velasco P, Urso K, Tranque P, Cano E. NFATc3 promotes Ca(2+)-dependent MMP3 expression in astroglial cells. Glia. 2013;61:1052–1066. doi: 10.1002/glia.22494. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Di Giovanni S. NFAT signaling in neural development and axon growth. Int J Dev Neurosci. 2008;26:141–145. doi: 10.1016/j.ijdevneu.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM. Calpain interactions with the protein phosphatase calcineurin in neurodegeneration. Advances in Biochemistry in Health and Disease. 2014;8:17–45. [Google Scholar]
- Norris CM, Scheff SW. Recovery of afferent function and synaptic strength in hippocampal CA1 following traumatic brain injury. J Neurotrauma. 2009;26:2269–2278. doi: 10.1089/neu.2009.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Korol DL, Foster TC. Increased susceptibility to induction of long-term depression and long- term potentiation reversal during aging. J Neurosci. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Sompol P, Roberts KN, Ansari M, Scheff SW. Pycnogenol protects CA3–CA1 synaptic function in a rat model of traumatic brain injury. Exp Neurol. 2016;276:5–12. doi: 10.1016/j.expneurol.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Kandel ER. Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn Mem. 1994;1:129–139. [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF, Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94:1077–1098. doi: 10.1152/physrev.00041.2013. [DOI] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallén A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–38. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- Pérez-Ortiz JM, Serrano-Pérez MC, Pastor MD, Martín ED, Calvo S, Rincón M, Tranque P. Mechanical lesion activates newly identified NFATc1 in primary astrocytes: implication of ATP and purinergic receptors. Eur J Neurosci. 2008;27:2453–2465. doi: 10.1111/j.1460-9568.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- Rosenkranz K, May C, Meier C, Marcus K. Proteomic analysis of alterations induced by perinatal hypoxic-ischemic brain injury. J Proteome Res. 2012;11:5794–5803. doi: 10.1021/pr3005869. [DOI] [PubMed] [Google Scholar]
- Sama DM, Mohmmad Abdul H, Furman JL, Artiushin IA, Szymkowski DE, Scheff SW, Norris CM. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS One. 2012;7:e38170. doi: 10.1371/journal.pone.0038170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Rothstein JD. Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol. 2006;175:277–303. doi: 10.1007/3-540-29784-7_14. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Ansari MA, Roberts KN. Neuroprotective effect of Pycnogenol(R) following traumatic brain injury. Exp Neurol. 2013;239:183–191. doi: 10.1016/j.expneurol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pérez MC, Martin ED, Vaquero CF, Azcoitia I, Calvo S, Cano E, Tranque P. Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia. 2011;59:94–107. doi: 10.1002/glia.21079. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda N, Moriguchi S, Shirasaki Y, Fukunaga K. Generation of constitutively active calcineurin by calpain contributes to delayed neuronal death following mouse brain ischemia. J Neurochem. 2006;98:310–320. doi: 10.1111/j.1471-4159.2006.03874.x. [DOI] [PubMed] [Google Scholar]
- Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem. 2010;114:810–819. doi: 10.1111/j.1471-4159.2010.06809.x. [DOI] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M, Fischer IR, Plesnila N, Staufenbiel M, Haass C, Snapyan M, Saghatelyan A, Tsai LH, Fischer A, Grobe K, Dimou L, Götz M. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog [corrected] Cell Stem Cell. 2013;12:426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Sullivan MM, Sage EH. Hevin/SC1, a matricellular glycoprotein and potential tumor-suppressor of the SPARC/BM-40/Osteonectin family. Int J Biochem Cell Biol. 2004;36:991–996. doi: 10.1016/j.biocel.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Wakita H, Suenaga T, Nakamura S, Kimura J. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer's disease patients. Acta Neuropathol. 1997;94:146–152. doi: 10.1007/s004010050686. [DOI] [PubMed] [Google Scholar]
- Vihma H, Pruunsild P, Timmusk T. Alternative splicing and expression of human and mouse NFAT genes. Genomics. 2008;92:279–291. doi: 10.1016/j.ygeno.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouimba RM, Foy MR, Foy JG, Thompson RF. 17beta-estradiol suppresses expression of long-term depression in aged rats. Brain Res Bull. 2000;53:783–787. doi: 10.1016/S0361-9230(00)00377-4. [DOI] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigstrom H, Gustafsson B. Postsynaptic control of hippocampal long-term potentiation. J Physiol (Paris) 1986;81:228–236. [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, Burns MP. Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J Neurotrauma. 2013;30:1966–1972. doi: 10.1089/neu.2013.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HQ, Shin SS, Ma X, Li Y, Dixon CE. Differential effect of traumatic brain injury on the nuclear factor of activated T Cells C3 and C4 isoforms in the rat hippocampus. Brain Res. 2014;1548:63–72. doi: 10.1016/j.brainres.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, van Berkel TJ, Biessen EA. Therapeutic potential of VIVIT, a selective peptide inhibitor of nuclear factor of activated T cells, in cardiovascular disorders. Cardiovasc Drug Rev. 2007;25:175–187. doi: 10.1111/j.1527-3466.2007.00011.x. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]