

Abstract
Copy number variations encompassing the gene encoding Cyfip1 have been associated with a variety of human diseases, including autism and schizophrenia. Here we show that juvenile mice hemizygous for Cyfip1 have altered presynaptic function, enhanced protein translation, and increased levels of F-actin. In developing hippocampus, reduced Cyfip1 levels serve to decrease paired pulse facilitation and increase miniature EPSC frequency without a change in amplitude. Higher-resolution examination shows these changes to be caused primarily by an increase in presynaptic terminal size and enhanced vesicle release probability. Short hairpin-mediated knockdown of Cyfip1 coupled with expression of mutant Cyfip1 proteins indicates that the presynaptic alterations are caused by dysregulation of the WAVE regulatory complex. Such dysregulation occurs downstream of Rac1 as acute exposure to Rac1 inhibitors rescues presynaptic responses in culture and in hippocampal slices. The data serve to highlight an early and essential role for Cyfip1 in the generation of normally functioning synapses and suggest a means by which changes in Cyfip1 levels could impact the generation of neural networks and contribute to abnormal and maladaptive behaviors.
SIGNIFICANCE STATEMENT Several developmental brain disorders have been associated with gene duplications and deletions that serve to increase or decrease levels of encoded proteins. Cyfip1 is one such protein, but the role it plays in brain development is poorly understood. We asked whether decreased Cyfip1 levels altered the function of developing synapses. The data show that synapses with reduced Cyfip1 are larger and release neurotransmitter more rapidly. These effects are due to Cyfip1's role in actin polymerization and are reversed by expression of a Cyfip1 mutant protein retaining actin regulatory function or by inhibiting Rac1. Thus, Cyfip1 has a more prominent early role regulating presynaptic activity during a stage of development when activity helps to define neural pathways.
Keywords: 15q11.2, F-actin, Sra1, synaptogenesis, WAVE1, WRC
Introduction
Human genetic studies have identified CYFIP1 as a gene that is dysregulated in a wide variety of developmental brain disorders, including particular forms of Angelman and Prader-Willi syndromes, autism spectrum disorders, and schizophrenia (Butler et al., 2004; Bittel et al., 2006; Sahoo et al., 2006; Horsthemke and Wagstaff, 2008; Leblond et al., 2012). CYFIP1 encodes a cytoplasmic protein named cytoplasmic FMRP interacting protein (Cyfip1), which has two structurally independent and highly conserved functions. In the first, it represses the cap-dependent translation of FMRP target mRNAs by binding to FMRP and serving as a noncanonical initiation factor 4E binding protein (4E-BP) (Napoli et al., 2008). In the second, it regulates the generation of branched actin filaments at the plasmalemma as an integral component of the WAVE regulatory complex (WRC) (Derivery and Gautreau, 2010). Regulation of both cap-dependent translation and actin cytoskeleton is known to be important for synapse assembly, morphology, and plasticity and has been shown to be altered in developmental brain disorders (Marin-Padilla, 1972; Purpura, 1974; Huttenlocher, 1975, 1991; Matus et al., 1982; Jay et al., 1991; Kremer et al., 1991; Irwin et al., 2000; Inoki et al., 2002; Zalfa et al., 2006; Au et al., 2007; Jansen et al., 2008). Thus, it is easy to appreciate why loss of a single copy of CYFIP1 could have broad consequences on normal brain function, but the function of Cyfip1 at developing synapses is not fully understood.
Cyfip1 is essential for mouse development. Mice lacking both copies of Cyfip1 (homozygous-null) die soon after fertilization (Bozdagi et al., 2012; Pathania et al., 2014). Young adult mice hemizygous for Cyfip1 appear grossly normal and show normal baseline synaptic transmission and presynaptic release properties but exhibit an enhanced form of long-term synaptic depression (mGluR-dependent LTD) that is abnormally independent of a requirement for new protein synthesis (Bozdagi et al., 2012). This synapse plasticity alteration is also consistent with abnormalities in postsynaptic dendritic spine morphology observed in neurons from Cyfip1+/− mice (De Rubeis et al., 2013; Pathania et al., 2014). However, Cyfip1 is likely to have additional, earlier actions. It is expressed in cortex and hippocampus beginning in late gestation (Köster et al., 1998; Yoon et al., 2014) and as a regulator of actin polymerization and protein translation would be expected to participate in the generation and function of synapses at earlier stages of development.
Here we used in vivo and in vitro approaches to investigate the impact of reduced levels of Cyfip1 on synapse formation and function. Our data show that Cyfip1 regulates presynaptic terminal size and vesicle release probability primarily by its participation in WRC, highlighting an important role for Cyfip1 during a period when synapses are forming and modulated by experience. That altered Cyfip1 levels can greatly affect the normal function of individual neurons would be expected to contribute significantly to the generation of abnormal and maladaptive behaviors and lends support to the observation that changes in Cyfip1 levels appear to exacerbate several distinct human brain disorders (Chai et al., 2003; Butler et al., 2004; Bittel et al., 2006; Sahoo et al., 2006; Stefansson et al., 2008; Kirov et al., 2009; Leblond et al., 2012).
Materials and Methods
Animals.
All procedures involving mice and rats were performed in strict accordance with National Institutes of Health guidelines and were approved by the Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai, which adheres to these guidelines. Timed pregnant Sprague Dawley rats were obtained from Taconic, and wild-type (WT) and Cyfip1 haploinsufficient mice were bred on site from a line of mice that was generated and backcrossed to C57BL/6NTac as described recently (Bozdagi et al., 2012). Litters were obtained from WT X Cyfip1+/− matings, and animals of both sexes were used in all experiments.
Antibodies, reagents, and cDNA constructs.
Primary antibodies include the following: GFP (Aves Labs), vGlut1/2/3 (Millipore), Synapsin1 (Synaptic Systems), PSD95 (Pierce), Cyfip1 (Millipore 07–531), WAVE1/SCAR (NeuroMab clone K91/36), and GAPDH (Millipore Bioscience Research Reagents 631401). Secondary antibodies include the following: anti-rabbit Alexa-405, anti-chicken Dylight488, anti-mouse Rhodamine Red-X, anti-guinea pig Alexa-647 (Jackson Immunologicals). FM4-64 lipophilic dye (Invitrogen), bafilomycin A1(1 μm, Tocris Bioscience), CNQX (50 μm, Tocris Bioscience), APV (25 μm, Tocris Bioscience), MK-801 (40 μm, Tocris Bioscience), NSC23766 (200 μm, Tocris Bioscience), ADVASEP-7 (0.5 mm, Sigma), and rhodamine phalloidin (Invitrogen).
shCyfip1 is a validated hairpin sequence recognizing both mouse and rat Cyfip1 (Silva et al., 2009) (Dharmacon RNAi: V2LMM_79585, mature antisense: TACATAAAGACAAACATGC); shCyfip1–2 (ttgaaagtgaagatttaacatca; NM_001107517) targets rat Cyfip1 (QIAGEN SureSilence: KR45495G). shCyfip1 was used unless otherwise noted; nonsilencing shRNA construct shCon (Dharmacon, RHS4346).
Human Cyfip1 (hCyfip1), Cyfip1 K743E missense mutation (mutE), and Cyfip1 C terminus truncation (δC/ΔC) expression constructs used in the rescue experiments were generated using CYFIP1TrueORF expression-validated vectors purchased from OriGene (GenBank ID: NM_014608, OriGENE SKU: RC203015), with the respective nucleotide sequences replaced as described in the text and Figure 6A. pcDNA3-SypHluorin 2x (SypH) was kindly provided by Yongling Zhu (Salk Institute) (Zhu and Stevens, 2008).
Figure 6.

Cyfip1-WAVE1 interaction regulates presynaptic function. A, Maps of relevant Cyfip1 binding partners and the mutants that were generated from human Cyfip1 fused in-frame to a N terminus FusionRed fluorescent tag (Evrogen). B, Western blots of whole-cell lysates generated from HEK cells expressing the cDNAs indicated and blotted for the antibodies shown at right. The antigen recognized by anti-Cyfip1 is compromised in the ΔC mutant, but the truncated Cyfip1 mutant can be detected by an anti-tRFP, which recognizes fusion Red. C, Thirty-six hours following transfection, red fluorescence shows that all expression constructs encode fusion protein and that the mutants are expressed as well as the WT human Cyfip1 in neurons in which shCyfip1 (green) is expressed. D, E, Scatter plots show impact of shCyfip1 knockdown alone and together with the indicated mutant and full-length cDNA constructs in 10 DIV rat hippocampal neurons on FM dye labeling. D, Plot of puncta area following uptake. E, Plots of mean (Fuptake − Frelease)/Fuptake. Horizontal black bars are compared with shCon. Gray bars are compared with shCyfip1 alone. F, A cumulative probability plot of uptake intensity reveals that expression of the WT human Cyfip1 (hCy1) along with shCyfip1 shifts the curve leftward relative to control neurons and underscores similarities between control and mute-expressing neurons. Tukey's post test values, *p < 0.05, **p < 0.01, ***p < 0.001.
Neuron cultures.
Rats and mice were killed at embryonic day 18 (E18) or postnatal day 3 (P3), respectively. Hippocampi were isolated, dissociated, and plated at 12,000 cells cm2 on poly-L-lysine-coated coverslips (1 mg/ml; Sigma) followed by laminin (13 μg/ml; Sigma) for all assays. Neurons were maintained in Neurobasal Media (Invitrogen) containing NS21 supplements (Chen et al., 2008) in an incubator at 5% CO2, 37°C. cDNAs were introduced into 9–11 DIV neurons using Lipofectamine 2000 (Invitrogen) (Hsiao et al., 2014). Right before transfection, culture media was removed (and reserved) and replaced with 1.5 ml of fresh Neurobasal. 3.75 μg of endo-free cDNA or shRNA were mixed with 4 μl of Lipofectamine 2000 reagent in OPTI-MEM for 10 min at room temperature, according to the manufacturer's instructions, and then added drop-wise onto neurons. After 30 min, fresh culture media was added to transfected cells; and 1.5 h later, three-fourths of the media was replaced with the reserved conditioned culture media. Constructs were expressed 18–24 h before live-cell imaging, fixing, or lysing at 10–12 DIV.
Live-cell imaging.
Hippocampal neurons plated on 35 mm bottom glass dishes (MatTek) were transfected with cDNA and/or shRNA at 9 DIV ∼16 h before imaging. Media was replaced with preequilibrated HEPES-buffered saline extracellular solution (Burrone et al., 2006), and neurons were imaged using a 40× objective on a Zeiss LSM780 inverted confocal microscope in a controlled microenvironment maintained at 35°C, 5% CO2.
For SypHluorin experiments, transfected cells were identified by faint GFP fluorescence under nondepolarizing conditions. All axons imaged were in the vicinity of dendrites. Fluorescence images were collected at 3.803 pixels per μm with 488 ex/505 em filter sets before and after depolarization in the presence of 1 μm bafilomycin A1 to inhibit the vesicular H+ ATPase.
For FM-dye experiments, presynaptic boutons were labeled with FM4-64 (Invitrogen) by 120 s incubation in high potassium Tyrode's solution (90 mm KCl, 31.5 mm NaCl) containing 10 μm FM dye, followed by a 60 s incubation in normal Tyrode's solution with 10 μm FM dye. AP5 (50 μm) and CNQX (25 μm) were added to the stimulation solution to block glutamate receptors to prevent recurrent excitation as well as changes in network properties due to postsynaptic activity. Neurons were then washed in Ca2+-free Tyrode's solution containing ADVASEP-7 (0.1 mm) for 1 min before imaging. Destaining was performed by 45 mm KCl hyperkalemic stimulation. Transfected axons (in the vicinity of dendrites) or dendrites were imaged before and after hyperkalemic stimulation (two time points at multiple sites), or every 20 s throughout the duration of the stimulation (at a single site) (Hsiao et al., 2014).
For all live imaging experiments, images from the same site, at different time-points, were aligned using ImageJ (StackReg) and labeled sites in transfected axons were analyzed using SynD, a semiautomated image analysis routine run in MATLAB (Schmitz et al., 2011) (see Immunocytochemistry, image acquisition, quantification, and analysis; Fig. 3D). Vesicle clusters exhibiting lateral movement were excluded from analysis. For presentation, brightness and contrast were adjusted in Photoshop. In instances showing the same field at different times, the same settings were used across all images.
Figure 3.
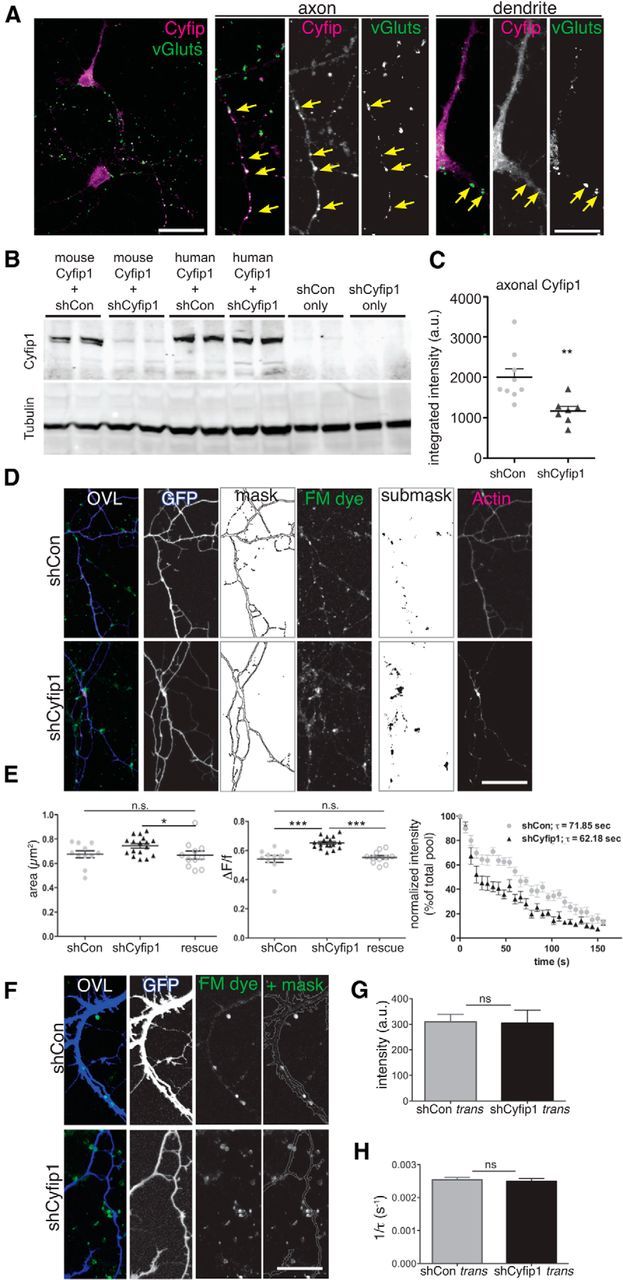
Targeted reduction of presynaptic Cyfip1 increases SV recycling pool size. A, Confocal images represent distribution of endogenous Cyfip1 (magenta) and vGluts1/2/3 (green) in cultured rat hippocampal neurons at 7 DIV. Axonal and somatodendritic regions are shown at higher magnification in panels at right. In axons, Cyfip1 concentrates at vGlut-labeled vesicle clusters (arrows, axon) and, in dendrites, is modestly enriched opposite vGlut clusters as well (arrows, dendrite). Scale bar, 20 μm. Scale bar: inset, 5 μm. B, Western blots of whole-cell lysates of Neuro2A cells expressing either mouse or human Cyfip1 (hCyfip1) together with shCon (nontargeting control short-hairpin vector) or shCyfip1. shCyfip1 reduces only mouse and not hCyfip1. Tubulin was used as a loading control. C, Scatter plot represents mean integrated intensity of Cyfip1 immunofluorescence in axons identified by GFP labeling and expressing either shCon or shCyfip1 (9 shCon and 7 shCyfip1 images). **p = 0.006 (t test). D, Images represent data and quantification strategy: FM4-64 dye-loaded terminals (green in overlay) in transfected axons (GFP, blue in overlay) that are innervating untransfected dendrites in rat hippocampal neurons at DIV 10. Presynaptic actin in this example was visualized by coexpression of eCFP-actin (magenta in overlay). FM-dye labeled sites (or other synaptic markers) were analyzed within a mask that was generated from the image of GFP-expressing axons; other labels, such as CFP-actin, were analyzed within a submask defined by the FM-dye label. Scale bar, 20 μm. E, Scatter plots compare mean (± SEM) and distribution of area (left) and change in intensity (middle) of FM4-64 clusters: shCon (gray), shCyfip1 (black), rescue (white circles; shCyfip1 + hCyfip1); *p = 0.04; ***p = 0.001; FM dye destaining kinetics (right): fluorescence intensity normalized to baseline level of FM dye uptake at axonal terminals as 100% versus time. Each data point indicates an average of five separate experiments, each of which included measurements of ∼20 FM dye-labeled puncta, performed in two cultures transiently transfected with shCon (gray), shCyfip1 (black) short hairpin expression constructs. F, Images of dendrites expressing shCon or shCyfip1 (blue) contacted by FM4-64 labeled (green) clusters at DIV 10 that are contained within a mask. G, Bar graph compares mean FM4-64 dye intensity or destaining rate (H) (± SEM) of FM4-64 puncta-innervating neurons expressing shCon (gray) or shCyfip1 (black). N = 4 separate experiments. ns, Not significant (t test). Scale bar, 30 μm.
Immunocytochemistry, image acquisition, quantification, and analysis.
Cultured hippocampal neurons (10–12 DIV) were fixed using 4% PFA and 4% sucrose, washed and permeabilized using 0.2% Triton X-100, and incubated with relevant antibodies at 25°C for 2 h, or overnight at 4°C. Immunocytochemical and other labeling were imaged on a Zeiss LSM 780.
Images were analyzed using MATLAB (The MathWorks) and ImageJ (National Institutes of Health). In brief, confocal image stacks (134.63 μm × 134.63 μm × 2.32 μm, 6 planes; 3.803 pixels per μm; 16 bits per pixel) were average projected into 2D images and imported into SynD (Schmitz et al., 2011). A threshold defining GFP labeling was used to generate a mask of transfected axons and/or dendrites. Within the area defined by the mask, a second threshold defining labeling for vGlut1 or FM4-64 (1 SD above the mean intensity) was used to generate a submask. Minimum size for fluorescent clusters was set at 0.78 × 0.78 μm2. Intensity and area at 35–100 ROIs from transfected neurons were measured and averaged and the mean data from several neurons were used to compare groups.
For tissue sections, P10 mice (Cyfip1+/− and WT) were perfused with 4% PFA, brains were removed, postfixed for 6 h, cryoprotected in 30% sucrose, and sectioned at 30 μm on a freezing microtome. Sections were immunolabeled with anti-vGlut1. Single 1024 × 1024 optical sections (three images per section, three sections per brain, three mice per genotype) through CA1 stratum radiatum were captured at a magnification of 100× by an investigator blinded to genotype. Images were analyzed in ImageJ (analyze particles). Results were exported to Excel, decoded, and statistical analyses were performed using Prism (GraphPad).
Western blots and metabolic labeling.
Dense hippocampal cultures (7.5E5 neurons per well in a six-well plate) were used to obtain cell lysates for Western blots. Forty-eight hours following transfection (see above), two wells of cells were lysed directly in 100 μl 2× Laemmli sample buffer. Equal amounts of cell lysates were loaded per lane, resolved by SDS-PAGE, blotted onto PVDF membrane, immunolabeled, and imaged using LiCor Odyssey far-red scanner. For presentation, images were inverted, converted to grayscale and brightness and contrast were adjusted in Photoshop.
Primary cortical cultures from Cyfip1+/− and WT littermates were deprived of methionine for 30 min and then incubated with AHA (L-azidohomoalanine, a methionine substitute) (Dieterich et al., 2007, 2010). Copper-free Click-it reaction with DBCO-Seta650 (Seta Biomedicals) and SDS-PAGE electrophoresis was performed as instructed by manufacturer. Gel was imaged using LiCor and grayscale was inverted for presentation. For in situ labeling of de novo synthesized protein with AHA, primary Cyfip1+/− and WT hippocampal cultures were deprived of methionine for 30 min and then incubated with AHA for 3 h. Cells were thoroughly washed to remove residual AHA labeling reagent and a click chemistry reaction with DBCO-Seta650 was performed on paraformaldehyde fixed cells.
Electrophysiology.
Acute hippocampal slices (350 μm) were prepared from brains of P10 to 4-week-old Cyfip1+/− mice and WT littermates for extracellular and intracellular recordings as described previously (Bozdagi et al., 2012). Slices were maintained in extracellular recording solution containing the following (in mm): 125.0 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 26.2 NaHCO3, 2.5 CaCl2, and 11.0 glucose. Briefly, the field EPSPs (fEPSPs) were evoked by stimulation of the Schaffer collateral-commissural afferents. Paired-pulse responses were measured with two stimuli delivered with interstimulus intervals of 20, 50, and 100 ms, for 1 min period during the baseline or drug application, and were expressed as the ratio of the averaged responses from the second stimulation pulses (FP2) to those from the first (FP1). For recording miniature EPSCs (mEPSCs), CA1 pyramidal neurons were held at −70 mV, in the presence of tetrodotoxin (0.5 μm) and picrotoxin (50 μm). The recording patch pipettes were filled with the following (in mm): 120 cesium methane sulfonate, 5 NaCl, 10 HEPES, 1.1 EGTA, 4 Mg-ATP, and 0.3 Na-GTP. Rate of decay of NMDA receptor-mediated EPSCs was assessed following repetitive presynaptic stimulation at 0.1 Hz in the presence of MK-801 (40 μm) at a holding potential of −40 mV (Rosenmund et al., 1993).
F-actin/G-actin assay.
F-actin/G-actin ratios were assayed in mouse hippocampal lysates using the G-actin/F-actin In vivo Assay Kit (Cytoskeleton) according to the manufacturer's instructions. Briefly, hippocampus was dissected from Cyfip1+/− mice and WT littermates, homogenized, and lysed in a detergent-based lysis buffer that stabilizes and maintains the G- and F-forms of cellular actin. Centrifugation at 100,000 × g was used to separate F-actin from the soluble G-actin. Samples of supernatant (including the G-actin fraction) and pellet (including the F-actin fraction) were then separated by SDS-PAGE, immunoblotted for actin, and imaged using LiCor. For presentation, image was inverted and brightness and contrast were adjusted in Photoshop.
Statistical comparisons.
For all experiments, group sizes are indicated in the text and/or figure legends. Both males and females were included in all of the assays. Statistical comparisons between groups were done by unpaired Student's t test or by one way ANOVA for more than two groups. Sources of differences in ANOVA were identified using Tukey's or Dunn's post hoc test. A detailed table of the data, statistics and p values used for the figures can be found here: Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.nt60p).
Results
Cyfip1 restrains presynaptic vesicle recycling specifically in immature synapses
To explore the role of Cyfip1 at developing synapses, we first asked whether immature mice (P10) lacking a single copy of Cyfip1 showed detectable differences in glutamatergic synapse structure or function in hippocampus. In sections taken through CA1, vGlut1-immunolabeled terminals were enlarged and their density was modestly increased in Cyfip1+/− mice compared with WT littermate controls (Fig. 1A–C). Whole-cell recordings of CA1 pyramidal cells showed increased mEPSC frequency in Cyfip1+/− neurons, but no change in amplitude compared with WT [Fig. 1D; mEPSC frequency, 0.8 ± 0.2 Hz (WT) vs 1.2 ± 0.18 Hz (Cyfip1+/−), amplitude, 12 ± 0.9 pA (WT) vs 11 ± 1.1 pA (Cyfip1+/−)], and by P21, mEPSC frequency and amplitude were similar between genotypes (Fig. 1D; P21 WT: mEPSC frequency, 3.58 ± 0.3 Hz, amplitude, 13.14 ± 1.2 pA; P21 Cyfip1+/−: 3.63 ± 0.4 Hz, amplitude, 13.48 ± 0.98 pA). To further explore the possibility that presynaptic function was altered at P10, we measured paired-pulse facilitation (PPF) at Schaeffer collateral synapses. Two presynaptic pulses were delivered at intervals ranging from 20 to 100 ms, and the ratio of the two responses (FP2/FP1) was compared. Cyfip1+/− mice had significantly decreased PPF that was most pronounced at shorter intervals compared with WT (Fig. 1E), an effect that is no longer evident at P21 (Bozdagi et al., 2012). To test whether decreased PPF reflected altered release probability, we measured rate of decay of NMDA receptor-mediated EPSCs during repetitive presynaptic stimulation in the presence of MK-801, a use-dependent channel blocker (Rosenmund et al., 1993). EPSCs from P10 Cyfip1+/− mice were blocked more rapidly consistent with a higher release probability (Fig. 1F). Together, these data indicate that, in developing Cyfip1+/− hippocampus, excitatory presynaptic terminals are enlarged and show a higher release probability than in WT mice.
Figure 1.

Presynaptic function in Cyfip1+/− neurons is altered in hippocampus. A, Confocal images of vGlut1-labeled single optical sections of CA1 in P10 mice of the indicated genotypes. B, Bar graphs represent an increase in size of vGlut-labeled puncta (t test, 0.011, n = 3 mice each) and density (C) (t test, p = 0.015). Scale bar, 10 μm. D, In whole-cell recordings, mEPSC frequency is increased in hippocampal slices from Cyfip1+/− mice compared with WT at P10 but is not different at P21. Top, Representative traces from whole-cell voltage-clamp recordings of mEPSCs in hippocampal slices from WT and Cyfip1+/− mice. Calibration: 200 ms, 20 pA. Bottom, Bar graphs representing mean amplitude and frequency data (± SEM) for mEPSCs in Cyfip1+/− and WT mice (n = 6 mice/genotype). *p < 0.01 (t test). E, Graph of PPF plots the mean ratio (FP2/FP1) at different interpulse intervals (IPI) in WT and Cyfip1+/− littermates at P10. Cyfip1+/− neurons (black) showed decreased ratio compared with WT (gray) (n = 6 mice/genotype, 1 or 2 slices/animal). **p < 0.001 (t test). *p < 0.01 (t test). Inset, Representative fEPSP traces for PPF (50 ms IPI) from Cyfip1+/− (black) and WT (gray) mouse, with traces normalized to the first response for comparison. Calibration: 0.5 mV, 20 ms. F, Graph of NMDA receptor-mediated EPSCs evoked at 0.1 Hz in the presence of 40 μm MK-801 in Cyfip1+/− (black) and WT (gray) (N = 4 mice/genotype). p = 0.035 (one-way ANOVA). Inset, Representative EPSC traces at the first (1) and the last (2) stimulus in a slice from a Cyfip1+/− mouse. Calibration, same as E.
To investigate the mechanism, we first established that hippocampal neurons grown in culture from Cyfip1+/− and WT littermate mice displayed phenotypes consistent with what is observed in hippocampal slices and in vivo. To visualize synaptic vesicle (SV) pools, we transfected neurons with SypHluorin (SypH), a modified synaptophysin having 2 copies of pH-sensitive pHluorin in its SV lumen-facing loop. SypH shows increased fluorescence intensity as vesicles exocytose (pH 5.5–7.2) (Miesenböck et al., 1998; Zhu and Stevens, 2008). To estimate recycling pool size, neurons 10–12 DIV were imaged at rest, depolarized with 90 mm KCl in the presence of bafilomycin to prevent vesicle reacidification, and imaged again (Fig. 2A), and fold change in fluorescence between resting and stimulated states (FS/FR) was determined (Fig. 2B). To estimate total pool size, neurons were exposed to 50 mm NH4Cl to alkalize vesicles (and unquench all SypH molecules) and fold change in fluorescence between NH4Cl exposure and resting status (FNH4Cl/FR) was measured. The data show a significant increase in the recycling SV pool in Cyfip1+/− neurons compared with WT that appears to be driven in part by a larger total pool (Fig. 2B), consistent with the larger terminal sizes observed in vivo.
Figure 2.

Cyfip1+/− regulates presynaptic function in cultured neurons. A, Confocal images of SypH-transfected hippocampal neurons at rest, after hyperkalemic depolarization with 90 mm KCl for 3 min (stim) and following NH4Cl application to visualize the total SypH-labeled pool. Scale bar, 20 μm. B, Bar graph compares mean fold change in intensity (FSTIM/FREST ± SEM) for recycling and total vesicle pools in Cyfip1+/− (black) and WT (gray) neurons (WT: 7 fields and 674 puncta; Cyfip1+/−: 5 fields and 561 puncta). *p ≤ 0.04 (t test). C, Schematic outlines procedure used for FM4-64 uptake and release experiment. D, FM dye destaining kinetics: fluorescence intensity normalized to baseline level of FM dye uptake at axonal terminals as 100% versus time. Each data point indicates an average of two separate experiments. E, Scatter plots compare mean (± SEM) change in intensity of FM4-64 clusters (Fuptake − Fend of release)/Fuptake, in WT (gray) and Cyfip1+/− (black) neurons. ****p < 0.0001 (t test).
SV dynamics were monitored using the styryl dye FM4-64 (Betz et al., 1996). Neurons 10–12 DIV were first depolarized in the presence of FM4-64 to label the recycling pool of SVs When SVs fuse with the presynaptic plasmalemma, their exposed lumenal surface incorporates FM4-64; and following internalization, vesicle clusters were imaged (uptake). Consistent with the SypH data, fluorescence intensity of the recycling pool in boutons on Cyfip1+/− neurons was greater than that in WT neurons (Fig. 2C; mean intensity FM puncta: Cyfip1+/−, 388.9 ± 15.41; WT, 161.2 ± 11.58, in arbitrary units; t test, p < 0.001; n = 3 independent experiments). Vesicle release kinetics were then measured by recording fluorescence traces of depolarization-induced FM dye destaining. Cyfip1+/− terminals showed a more rapid loss of FM dye fluorescence compared with WT (Fig. 2C,D; mean time constants of synaptic FM4-64 destaining, τWT = 87.06 ± 4.135 s, n = 56 synapses; τCyfip+/− = 67.40 ± 4.338 s, n = 45 synapses; t test, p < 0.005; two independent experiments). A comparison of dye loss during activity-dependent discharge showed enhanced exocytosis in Cyfip1+/− terminals (Fig. 2E; mean fractional release ΔF/f, WT, 0.56 ± 0.006; Cyfip1+/−, 0.64 ± 0.007, in arbitrary units; t test, p < 0.0001). Together, the data support that there are more SVs in Cyfip1+/− terminals that are more readily released upon stimulation, suggesting that Cyfip1 restrains the size and release of the recycling pool.
Cyfip1 mediates its effects on presynaptic terminals directly
The presynaptic phenotypes observed in Cyfip1+/− neurons could be caused by decreased Cyfip1 levels in presynaptic terminals. However, it is also possible that Cyfip1 haploinsufficiency sets in motion a number of developmental changes that indirectly affect presynaptic function. Cyfip1 can regulate postsynaptic properties in more mature neurons in culture and in ex vivo slice preparations, making it also plausible that the effects we observed occur retrograde to a postsynaptic mechanism (Bozdagi et al., 2012; De Rubeis et al., 2013; Pathania et al., 2014). To distinguish between direct and indirect effects, we first asked where Cyfip1 was localized in developing (7 DIV) WT neurons. Immunolabeling for Cyfip1 was diffusely localized in cell bodies and dendrites and additionally showed a clustered distribution in axons and dendrites (Fig. 3A). Such clusters were commonly, but not exclusively, aligned with vGlut-labeled presynaptic terminals (Fig. 3A, arrows), consistent with ultrastructural observations (De Rubeis et al., 2013).
To directly assess the impact of Cyfip1 on presynaptic function, we transfected RNA interference constructs expressing Cyfip1 shRNA (shCyfip1) (Silva et al., 2009) or control shRNA (shCon) and GFP into 8 DIV rat hippocampal neurons. We confirmed that shCyfip1 reduces Cyfip1 expression in axons by ∼40% (Fig. 3B,C). The sparse transfection of primary neurons generates mosaic cultures in which untransfected neurons are innervated by shCyfip1 or shCon expressing axons and vice versa. This experimental design allows the isolation of cell-autonomous phenotypes. After 36 h, FM4-64 was used to label and compare SV recycling pools in transfected axons innervating untransfected neurons (cis- or direct effect) and vice versa (trans-synaptic or retrograde effect). In cis-, destaining kinetics and fraction released during exocytosis (ΔF/f) showed increased FM dye discharge (Fig. 3D,E) and a steeper fluorescence decay (mean time constants of synaptic FM4-64 destaining, τshCon = 71.85 ± 3.711 s; τCyfip1+/− = 62.18 ± 2.421 s; t test, p = 0.0381; two independent experiments). A second shRNA targeting Cyfip1 produced similar results (data not shown), and effects of shCyfip1 could be rescued by coexpressing human Cyfip1 (hCyfip1), which is resistant to shRNA knockdown (Fig. 3B,E). In contrast, reducing Cyfip1 postsynaptically in dendrites (in trans) showed no trans-synaptic effect on SV pool size or exocytosis on untransfected axonal terminals compared with controls (Fig. 3F–H).
In 10–12 DIV neurons, not all recycling sites are opposite postsynaptic densities, leaving open the possibility that the changes observed were principally at nonsynaptic sites. We used immunolabeling for PSD95 and vGlut1 to identify synapses on transfected axons. The data show that PSD95-labeled puncta were increased in size opposite terminals expressing shCyfip1 compared with shCon (Fig. 4A–D), suggesting that postsynaptic sites enlarge commensurate with presynaptic changes. Consistent with this idea, vGlut1 labeling intensity was increased in transfected boutons apposed to PSD95 (Fig. 4A–C,E,F). Coexpression of hCyfip1 with shCyfip1 successfully rescued the aberrant synaptic phenotypes (Fig. 4C–F).
Figure 4.

Presynaptic Cyfip1 regulates synapse size. A–C, Confocal images of 10 DIV neurons expressing shCon, shCyfip1, or shCyfip1 + hCyfip1 together (rescue) and immunolabeled as indicated. Low-magnification views represent GFP (white), PSD95 (magenta), and vGlut1 (green) as an overlay. High-magnification views of areas within dotted lines represent a mask of GFP labeling (gray) used to demarcate axons; PSD95 (magenta) and vGLUT1 (green) labeling is shown within the GFP mask. Yellow arrows indicate sites with both labels. Scale bars: overlay, 25 μm; inset, 15 μm. Bar graphs compare sizes of PSD95-immunolabeled clusters within transfected axons (D; ANOVA, p = 0.003) and vGlut1 labeling intensity at PSD95 sites (E, F; ANOVA, p = 0.0006). Differences observed with shCyfip1 are restored to controls conditions by coexpression of hCyfip1 (rescue). *p < 0.05; **p < 0.01. ns, Not significant.
Collectively, the data strongly support that the presynaptic phenotype observed in Cyfip1+/− neurons is exerted locally within axons at synapses and occurs as a direct consequence of diminished Cyfip1 levels.
Reduced Cyfip1 enhances protein translation and F-actin polymerization
Cyfip1 is a key component of the WRC, and consistent with this, previous studies have shown that decreasing Cyfip1 has an impact on F-actin, but the direction of change has been difficult to predict. For example, 15-day-old mouse cortical neurons with decreased Cyfip1 levels have longer dendritic protrusions with reduced F-actin levels (De Rubeis et al., 2013), whereas neuromuscular junction presynaptic terminals in Drosophila cyfip mutants show enhanced F-actin assembly (Zhao et al., 2013). To compare F-actin levels in axons, Texas Red phalloidin labeling intensity was quantified in transfected axons within a submask defined by SynapsinI-labeled presynaptic terminals (Fig. 5A–C). The data show that F-actin labeling intensity was increased in axons expressing shCyfip1 compared with shCon and reduced to control levels when shCyfip1 was coexpressed with hCyfip1. (Fig. 5D,E). To confirm these findings and to clearly distinguish presynaptic from postsynaptic actin, we cotransfected CFP-actin with shCyfip1 or shCon. CFP-actin showed discrete puncta or varicosities that often corresponded to dye recycling sites labeled with fixable FM4-64 in the transfected axons (Fig. 3D, rightmost panel) similar to what has been described (Morales et al., 2000). CFP-actin fluorescence intensity in presynaptic terminals is ∼1.6-fold higher in shCyfip1 boutons compared with shCon consistent with the increased presynaptic F-actin detected with phalloidin labeling.
Figure 5.
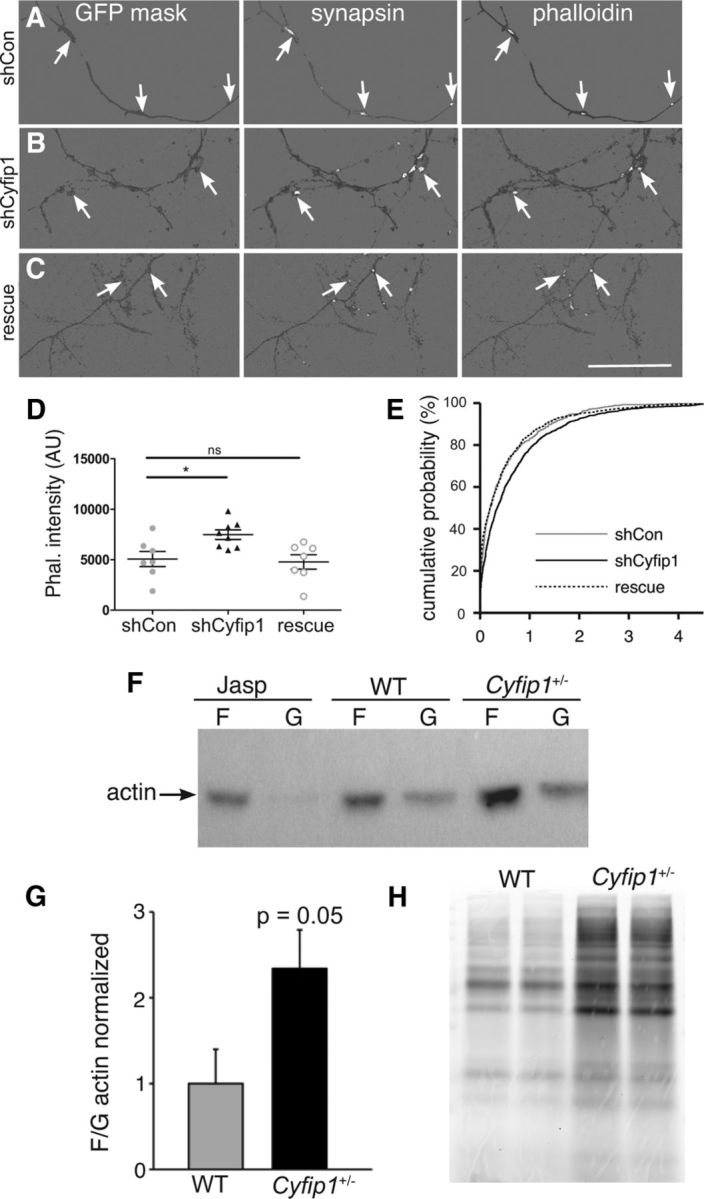
Presynaptic Cyfip1 regulates actin polymerization. A–C, Images of 10 DIV neurons expressing shCon, shCyfip1, or shCyfip1 + hCyfip1 together (rescue), immunolabeled for GFP and synapsin I, and stained with rhodamine-phalloidin. A mask of GFP labeling (dark gray) was used to demarcate axons; synapsin and phalloidin labeling (white) is shown within the mask. Background was lightened to make the labeling easier to see. Scale bar, 50 μm. D, Scatter plot shows that intensity of rhodamine-phalloidin labeling at sites defined by presynaptic labeling is increased in axons expressing shCyfip1 compared with shCon or rescue (shCyfip1 + human Cyfip1 cotransfection) (mean ± SEM). p = 0.013 (ANOVA). E, Cumulative probability plot of presynaptic phalloidin-associated fluorescence in shCon (gray), shCyfip1 (black), and rescue (dotted). F, Western blots reveal higher F-/G-actin in Cyfip1+/− brain tissues in 1-week-old mice compared with WT; Jasplakinolide (Jasp)-treated hippocampal lysate served as a positive control. G, Quantification of F-/G-actin ratio in the hippocampus from 1- and 4-week-old WT and Cyfip1+/− mice (n = 10 mice/genotype). p = 0.05 (t test). H, Image of a gel from a metabolic labeling experiment in which newly synthesized proteins in primary cortical neurons cultured from Cyfip1+/− and WT littermates were labeled by a click chemistry reaction. *p< 0.05. ns, Not significant.
To determine whether or not this effect was more generalized, we used Western blots to compare polymerized (F-) and soluble (G-) actin fractions from whole hippocampal lysates taken from Cyfip1+/− and WT littermate mice. Cyfip1+/− mice showed a consistent increase in F- to G-actin ratio relative to WT (Fig. 5F,G) (WT: 1.5 ± 0.3 vs Cyfip1+/−: 3.5 ± 1.1 arbitrary intensity units; n = 10 each, p = 0.05).
Because work in vitro shows that Cyfip1 can repress protein translation (Napoli et al., 2008), we compared levels of baseline protein translation in neurons cultured from Cyfip1+/− and WT littermate mice using FUNCAT (fluorescent noncanonical amino acid tagging) (Dieterich et al., 2007, 2010) to label newly synthesized proteins. There was a notable increase in overall levels of protein translation in lysates from Cyfip1+/− neurons compared with WT (Fig. 5H). Similar experiments performed in situ also showed an increase in FUNCAT-labeling intensity (WT: 37.2 ± 12.4 vs Cyfip1+/−: 166.7 ± 13.2 arbitrary intensity units).
Cyfip1 regulates presynaptic function via its actions on the WAVE1 complex
The crystal structure of WRC and FRET experiments indicates that Cyfip1 cannot interact with eIF4E and WRC simultaneously (Chen et al., 2010; De Rubeis et al., 2013). Thus, to determine which Cyfip1 role is most relevant to presynaptic function, we used a Cyfip1 knockdown + mutant rescue approach. Previous work has shown that a point mutation in the eIF4E-binding region of Cyfip1 (mut E) prevents translation initiation factor eIF4E binding (Napoli et al., 2008; Di Marino et al, 2015) but does not prevent Nap1 binding (De Rubeis et al., 2013) (Fig. 6A). A C-terminal Cyfip1 truncation mutant (ΔC) lacks both Nap1- and eIF4E-binding regions (Fig. 6A). Full-length and mutant Cyfip1 proteins, all of which are shCyfip1-insensitive, were cloned in-frame to an N terminus fusionRed tag to verify their expression and stability (Fig. 6B,C). Neurons were cotransfected with shCyfip1 together with mut E, ΔC, or full-length hCyfip1 and compared with neurons transfected with shCon or shCyfip1 alone. The data show that both full-length Cyfip1 and mut E fully restored the size of the FM dye-labeled vesicle pool and reduced release compared with Cyfip1 knockdown alone, indicating that Cyfip1's eIF4E binding activities are unlikely to play a major role in presynaptic function. By contrast, neurons expressing the ΔC mutant (with Nap1 binding sites removed) showed no recovery (Fig. 6D,E). A cumulative distribution plot of the intensity data additionally reveal a leftward shift in axons expressing full-length Cyfip1 (along with Cyfip1 shRNA), consistent with the idea that Cyfip1 restrains vesicle recycling (Fig. 6F).
Rac1 inhibition restores presynaptic function
The data in cultured neurons showed that reduced Cyfip1 levels increase synaptic vesicle recycling pool size by dysregulating WRC. One possibility was that decreased Cyfip1 alters WRC function by triggering a compensatory decrease in WAVE1, a mechanism that has been observed in dividing cells (Kunda et al., 2003; Grove et al., 2004; Schenck et al., 2004; Yoon et al., 2014). However, in neurons expressing shCyfip1, WAVE1 immunostaining intensity was similar to those expressing shCon (Fig. 7A,B). Similarly, WAVE1 levels measured in Western blots of whole hippocampal lysates were similar in Cyfip1+/− mice compared with WT littermate controls (Cyfip1+/−: 1.85 ± 1.3 arbitrary intensity units, WT: 1.47 ± 1.3 arbitrary intensity units, n = 8/genotype, t test, p = 0.2).
Figure 7.

Pharmacological rescue of presynaptic phenotype in synapses with reduced Cyfip1. A, Confocal images (top) show immunolabeling for WAVE1 (red) and vGlut (green) along with axons expressing shCon or shCyfip1 (GFP mask, gray). Inverted mask (bottom) shows only WAVE1 labeling within the mask. Arrows point to sites of WAVE1 concentration. Scale bar, 15 μm. B, Intensity levels of WAVE1 immunostaining in axons are not detectably altered by shCyfip1 expression compared with shCon. C, D, Scatter plots represent that brief exposure of 10 DIV cultured rat hippocampal neurons to Rac inhibitor NSC23766 restores SV recycling parameters in axons expressing shCyfip1 to vehicle-treated control values. The same inhibitor had no effect on shCon-expressing neurons. Tukey's post test values, ***p < 0.001; **p < 0.01; ns, not significant. E, Cumulative distribution plot of mean FM dye uptake intensity suggests that Rac inhibition overshoots control values similar to Cyfip1 overexpression (Fig. 5F). F, In hippocampal slices, Rac1 inhibition (200 μm, 30 min) normalized PPR in Cyfip1+/− mice at P10 (n = 6 mice/genotype). *p < 0.05 (t test). Inset, Representative fEPSP traces for PPF (20 ms IPI) from Cyfip1+/− mouse in the presence of the vehicle (black) and NSC23766 (red), with traces normalized to the first response for comparison. G, Representative mEPSC traces recorded in CA1 pyramidal neurons in the presence of TTX from WT and Cyfip1+/− mice in the presence of vehicle or NSC23766 at P10. Calibration: 200 ms, 20 pA. H, Cumulative plot of mEPSC frequencies in WT and Cyfip1+/− mice in the presence of the vehicle and NSC23766 shows a reversal effect of the Rac1 inhibitor on the increased in mEPSCs in Cyfip1+/− mice.
Another possibility was that abnormal presynaptic function lay downstream of Rac1, which activates WRC. To test this, neurons expressing shCyfip1 or shCon were exposed to a Rac1 inhibitor, NSC23766, or vehicle for 30 min, and the impact on recycling pools was assessed. The data show that NSC2366 restored the size of recycling and releasable vesicle pools in neurons expressing shCyfip1, although it had no effect on either parameter in neurons expressing shCon (Fig. 7C–E). Significantly, Rac1 inhibition also restored responses in Cyfip1+/− hippocampal slices. When P10 hippocampal slices from Cyfip1+/− and WT littermate control mice were exposed to NSC23766 or vehicle for 30 min, the decreased PPF observed in Cyfip1+/− mutants was restored to WT levels. Vehicle treatment showed no effect (Fig. 7F,H vs Fig. 1E). Similarly, mEPSC frequency in P10 Cyfip1+/− mice was restored to WT levels in slices exposed to NSC23766 (mEPSC frequency (vehicle), 1.32 ± 0.21 Hz; (NSC23766), 0.91 ± 0.24) (Fig. 7G). These data indicate that reductions in Cyfip1 increase vesicle pool size and release probability downstream of Rac1.
Discussion
Our data show that Cyfip1 regulates presynaptic terminal development, function, and plasticity and that it acts directly within presynaptic neurons at a time when synapses are forming and maturing. While Cyfip1 regulates both protein translation and WRC function, its roles in restraining vesicle pool size and release are mediated principally by the WRC and are consistent with the observed increases in levels of presynaptic F-actin. The impact of Cyfip1 haploinsufficiency is rescued acutely by treatment with Rac1 inhibitors, indicating that altered function is not the consequence of an earlier developmental defect, but rather the outcome of an ongoing process of dysregulated cytoskeletal organization downstream of Rac1. The strong effects of reduced Cyfip1 on presynaptic function during development would be anticipated to alter the normal course of synapse assembly, maturation, and pruning, which require the coordinated activity of neural circuits and precise regulation of the actin cytoskeleton and could contribute to the role that Cyfip1 plays in a variety of developmental brain disorders.
Cyfip1, WAVE1, and F-actin
The data indicate that the presynaptic dysfunction observed in Cyfip1 mutants is due principally to the loss of Cyfip1's WRC-related function and not to its 4EBP/translation repression function. In neurons having reduced levels of Cyfip1, abnormal presynaptic terminal size and function were rescued by a Cyfip1 eIF4E binding mutant as effectively as with full-length Cyfip1, and mutants lacking the Nap1/WAVE1 interaction domain failed to rescue either parameter. Conversely, the increased protein translation observed in Cyfip1+/− mice did not appear to alter baseline presynaptic function.
Defective neuromuscular junction development in Drosophila cyfip mutants suggests that Cyfip's participation in WRC-dependent actin dynamics could play conserved roles in synapse assembly (Schenck et al., 2003; Zhao et al., 2013); and consistent with our findings, cyfip mutations enhance F-actin polymerization at presynaptic terminals (Schenck et al., 2003; Zhao et al., 2013). A normally regulated, dynamic actin cytoskeleton is critical for the generation of synapses (Allison et al., 1998; Zhang and Benson, 2001) and supports normally functioning presynaptic terminals (Morales et al., 2000; Sun and Bamji, 2011; Waites et al., 2011). Several actin-anchored cell adhesion molecules influencing synapse assembly and maturation, including protocadherins, cadherins, and Ig superfamily members, have a WRC binding interface generated by interactions between Cyfip and Abi2 that could be used to trigger actin polymerization at sites of synapse adhesion (Nakao et al., 2008; Chen et al., 2014; Yoon et al., 2014). Interestingly, as the influence of Cyfip1 on presynaptic activity declines over the course of maturation (Fig. 1D), F-actin also transitions from an essential role anchoring presynaptic to postsynaptic appositions to alternate roles in the regulation of synapse plasticity and dendritic spine shape (Allison et al., 1998; Fischer et al., 1998; Zhang and Benson, 2001; Bozdagi et al., 2012).
A simple interpretation of our findings would be that Cyfip1 normally exerts an inhibitory action on WAVE that is relieved by activated Rac1. There are some data to support this idea (Eden et al., 2002), but more recent work suggests that a more nuanced mechanism is at work. The most widely accepted model of WRC function posits that Cyfip1 shields the VCA domain in WAVE and prevents the complex from promoting constitutive Arp2/3-mediated actin polymerization. Rac1 binding triggers a conformational change in Cyfip1 that exposes the VCA domain without dissociation (Ismail et al., 2009; Chen et al., 2010; Derivery and Gautreau, 2010). Because our data show that Rac1 inhibition restores normal presynaptic function (Fig. 6), it is most likely that decreased Cyfip1 levels trigger indirect modulation of WRC by Rac1 or promote activation of an alternate Rac-activated pathway. For example, Rac can regulate actin polymerization via IQGAP or by inhibiting cofilin (Govek et al., 2005; Brandt and Grosse, 2007; Pertz, 2010). Rac1 may also regulate translation, which is increased in Cyfip1+/− mice, but restoring Cyfip1's eIF4E binding alone failed to restore presynaptic phenotypes, suggesting that increased translation of Cyfip1 targets does not impact the phenotypes measured here.
Presynaptic versus postsynaptic actions of Cyfip1
Our data show that, in 10–12 DIV neurons in culture and in P10 hippocampus and hippocampal slices, reduced Cyfip1 levels increase size and function of synaptic vesicle pools, with no detectable impact on postsynaptic strength, measured by mEPSC amplitude. By the third week of development, presynaptic function in Cyfip1+/− mice appears similar to WT, a time when postsynaptic responses are aberrant: Cyfip1+/− mice show enhanced mGluR-dependent LTD that is also atypically independent of mTOR activation and protein translation (Bozdagi et al., 2012), hippocampal neurons have abnormal dendritic spine shapes (Pathania et al., 2014); and in cultured cortical neurons, Cyfip1 knockdown prevents normal postsynaptic morphological responses to BDNF or NMDA (De Rubeis et al., 2013; Pathania et al., 2014). The strong similarity between the exaggerated LTD phenotype of Cyfip1+/− mice and that of Fmr1 knock-out mice strongly suggests that this effect is mediated by Cyfip1's actions as a translational suppressor with FMRP (Napoli et al., 2008), whereas spine shape appears to be controlled by both Cyfip1 roles (De Rubeis et al., 2013; Chazeau et al., 2014). It will be important in future work to further parse the actions of Cyfip1 with respect to developmental epoch and synaptic locus, as well as to address whether some effects of Cyfip1 reflect the shuttling of Cyfip1 molecules between WRC and eIF4E pools, as has been suggested by work in cultured neurons (De Rubeis et al., 2013) or may be complemented by actions of Cyfip2 (Han et al., 2015).
Relevance of Cyfip1 to human disease
Our findings in mice are likely to have relevance to human disease. Copy number variations (CNVs) in 15q11.2, the region that contains CYFIP1, are replicated risk factors for schizophrenia, epilepsy, intellectual disability, developmental delay, and autism (Horsthemke and Wagstaff, 2008; Kirov, 2010; Tam et al., 2010; van der Zwaag et al., 2010; Kirov et al., 2012; Chaste et al., 2014; Fromer et al., 2014; Vanlerberghe et al., 2015). For example, in two large studies of schizophrenia, recurrent CNVs in the 15q11.2 region were associated with 2-fold to 4-fold increased risk (Chai et al., 2003; Stefansson et al., 2008; Kirov et al., 2009). Type I deletions of Prader-Willi and Angelman syndromes that include CYFIP1 have been associated with more severe manifestations, compared with Type II deletions that do not include this interval (Butler et al., 2004; Bittel et al., 2006; Sahoo et al., 2006), and microdeletions including CYFIP1 in the absence of these syndromes have been associated with delayed speech and motor development, and behavioral problems including autism and obsessive-compulsive disorder (Doornbos et al., 2009). Additional results suggest that this same region and, in particular, CYFIP1 deletion (Leblond et al., 2012), increases risk for autism, likely in the presence of other genetic risk factors (Murthy et al., 2007; Doornbos et al., 2009). However, either increased or decreased CYFIP1 dosage is likely to be detrimental (Oguro-Ando et al., 2015). A recent study of human controls shows that 15q11.2(BP1-BP2) deletion carriers have reduced volume of gray and white matter in key brain areas compared with noncarriers that are increased in a reciprocal manner in duplication carriers, and both CNVs show cognitive outcomes that track between normal and schizophrenic, providing evidence for allele dose-dependent effects of CNVs (Stefansson et al., 2014).
Because Cyfip1 is common to several disorders and its dysregulation appears to exacerbate symptoms, therapies targeting Cyfip1 function may prove beneficial for a variety of human diseases.
Footnotes
This work was supported by the Beatrice and Samuel A. Seaver Foundation to O.B.-G., Simons Foundation to D.L.B., and National Institute of Mental Health to D.L.B. and O.B.-G. We thank Roxana Mesias for generating the primary neuronal cultures; Frances Williams for technical assistance; and Dr. Silvia De Rubeis and Dr. George Huntley for their critical comments on the manuscript.
The authors declare no competing financial interests.
References
- Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, Wheless JW, Baumgartner JE, Roa BB, Wilson CM, Smith-Knuppel TK, Cheung MY, Whittemore VH, King TM, Northrup H. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9:88–100. doi: 10.1097/GIM.0b013e31803068c7. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Curr Opin Neurobiol. 1996;6:365–371. doi: 10.1016/S0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Bittel DC, Kibiryeva N, Butler MG. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics. 2006;118:e1276–e1283. doi: 10.1542/peds.2006-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Dorr N, Pilorge M, Takahashi N, Buxbaum JD. Haploinsufficiency of Cyfip1 produces a Fragile X-like phenotype in mice. PLoS One. 2012;7:e42422. doi: 10.1371/journal.pone.0042422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt DT, Grosse R. Get to grips: steering local actin dynamics with IQGAPs. EMBO Rep. 2007;8:1019–1023. doi: 10.1038/sj.embor.7401089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Li Z, Murthy VN. Studying vesicle cycling in presynaptic terminals using the genetically encoded probe synaptopHluorin. Nat Protoc. 2006;1:2970–2978. doi: 10.1038/nprot.2006.449. [DOI] [PubMed] [Google Scholar]
- Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. 2004;113:565–573. doi: 10.1542/peds.113.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai JH, Locke DP, Greally JM, Knoll JH, Ohta T, Dunai J, Yavor A, Eichler EE, Nicholls RD. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003;73:898–925. doi: 10.1086/378816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaste P, Sanders SJ, Mohan KN, Klei L, Song Y, Murtha MT, Hus V, Lowe JK, Willsey AJ, Moreno-De-Luca D, Yu TW, Fombonne E, Geschwind D, Grice DE, Ledbetter DH, Lord C, Mane SM, Martin DM, Morrow EM, Walsh CA, et al. Modest impact on risk for autism spectrum disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res. 2014;7:355–362. doi: 10.1002/aur.1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazeau A, Mehidi A, Nair D, Gautier JJ, Leduc C, Chamma I, Kage F, Kechkar A, Thoumine O, Rottner K, Choquet D, Gautreau A, Sibarita JB, Giannone G. Nanoscale segregation of actin nucleation and elongation factors determines dendritic spine protrusion. EMBO J. 2014;33:2745–2764. doi: 10.15252/embj.201488837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, Rosen MK. The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell. 2014;156:195–207. doi: 10.1016/j.cell.2013.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: redefined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Borek D, Padrick SB, Gomez TS, Metlagel Z, Ismail AM, Umetani J, Billadeau DD, Otwinowski Z, Rosen MK. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. doi: 10.1038/nature09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E, Gautreau A. Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. BioEssays. 2010;32:119–131. doi: 10.1002/bies.200900123. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, Pasciuto E, Li KW, Fernández E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJ, Komiyama NH, Grant SG, Poujol C, Choquet D, Achsel T, Posthuma D, Smit AB, Bagni C. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron. 2013;79:1169–1182. doi: 10.1016/j.neuron.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Lee JJ, Link AJ, Graumann J, Tirrell DA, Schuman EM. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marino D, D'Annessa I, Tancredi H, Bagni C, Gallicchio E. A unique binding mode of the eukaryotic translation inititiation factor 4E for guiding the design of novel peptide inhibitors. Protein Sci. 2015;24:1370–1382. doi: 10.1002/pro.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CA, Dijkhuizen T, Bijlsma EK, Gijsbers AC, Hilhorst-Hofstee Y, Hordijk R, Verbruggen KT, Kerstjens-Frederikse WS, van Essen T, Kok K, van Silfhout AT, Breuning M, van Ravenswaaij-Arts CM. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur J Med Genet. 2009;52:108–115. doi: 10.1016/j.ejmg.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/S0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Grove M, Demyanenko G, Echarri A, Zipfel PA, Quiroz ME, Rodriguiz RM, Playford M, Martensen SA, Robinson MR, Wetsel WC, Maness PF, Pendergast AM. ABI2-deficient mice exhibit defective cell migration, aberrant dendritic spine morphogenesis, and deficits in learning and memory. Mol Cell Biol. 2004;24:10905–10922. doi: 10.1128/MCB.24.24.10905-10922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Chen H, Gennarino VA, Richman R, Lu HC, Zoghbi HY. Fragile X-like behaviors and abnormal cortical dendritic spines in cytoplasmic FMR1-interacting protein 2-mutant mice. Hum Mol Genet. 2015;24:1813–1823. doi: 10.1093/hmg/ddu595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A. 2008;146:2041–2052. doi: 10.1002/ajmg.a.32364. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Bozdagi O, Benson DL. Axonal cap-dependent translation regulates presynaptic p35. Dev Neurobiol. 2014;74:351–364. doi: 10.1002/dneu.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Synaptic and dendritic development and mental defect. In: Buchwald NA, Brazier MAB, editors. Brain mechanisms in mental retardation; Proceedings of a Conference in the Series on Mental Retardation Held at Oxnard; January 1974; California. New York: Academic; 1975. pp. 123–140. [Google Scholar]
- Huttenlocher PR. Dendritic and synaptic pathology in mental retardation. Pediatr Neurol. 1991;7:79–85. doi: 10.1016/0887-8994(91)90001-2. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Ismail AM, Padrick SB, Chen B, Umetani J, Rosen MK. The WAVE regulatory complex is inhibited. Nat Struct Mol Biol. 2009;16:561–563. doi: 10.1038/nsmb.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen FE, Braams O, Vincken KL, Algra A, Anbeek P, Jennekens-Schinkel A, Halley D, Zonnenberg BA, van den Ouweland A, van Huffelen AC, van Nieuwenhuizen O, Nellist M. Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology. 2008;70:908–915. doi: 10.1212/01.wnl.0000280578.99900.96. [DOI] [PubMed] [Google Scholar]
- Jay V, Becker LE, Chan FW, Perry TL., Sr Puppet-like syndrome of Angelman: a pathologic and neurochemical study. Neurology. 1991;41:416–422. doi: 10.1212/WNL.41.3.416. [DOI] [PubMed] [Google Scholar]
- Kirov G. The role of copy number variation in schizophrenia. Expert Rev Neurother. 2010;10:25–32. doi: 10.1586/ern.09.133. [DOI] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayés A, Fernandez E, Olason PI, Böttcher Y, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, Craddock N, Owen MJ, O'Donovan MC. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster F, Schinke B, Niemann S, Hermans-Borgmeyer I. Identification of shyc, a novel gene expressed in the murine developing and adult nervous system. Neurosci Lett. 1998;252:69–71. doi: 10.1016/S0304-3940(98)00531-X. [DOI] [PubMed] [Google Scholar]
- Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13:1867–1875. doi: 10.1016/j.cub.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G, Konyukh M, Chaste P, Ey E, Rastam M, Anckarsäter H, Nygren G, Gillberg IC, Melke J, Toro R, Regnault B, Fauchereau F, Mercati O, Lemière N, Skuse D, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8:e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M. Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 1972;44:625–629. doi: 10.1016/0006-8993(72)90324-1. [DOI] [PubMed] [Google Scholar]
- Matus AI, Ackermann M, Pehling G, Randolph Byers H, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Morales M, Colicos MA, Goda Y. Actin-dependent regulation of neurotransmitter release at central synapses. Neuron. 2000;27:539–550. doi: 10.1016/S0896-6273(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Murthy SK, Nygren AO, El Shakankiry HM, Schouten JP, Al Khayat AI, Ridha A, Al Ali MT. Detection of a novel familial deletion of four genes between BP1 and BP2 of the Prader-Willi/Angelman syndrome critical region by oligo-array CGH in a child with neurological disorder and speech impairment. Cytogenet Genome Res. 2007;116:135–140. doi: 10.1159/000097433. [DOI] [PubMed] [Google Scholar]
- Nakao S, Platek A, Hirano S, Takeichi M. Contact-dependent promotion of cell migration by the OL-protocadherin-Nap1 interaction. J Cell Biol. 2008;182:395–410. doi: 10.1083/jcb.200802069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, Witke W, Costa-Mattioli M, Sonenberg N, Achsel T, Bagni C. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134:1042–1054. doi: 10.1016/j.cell.2008.07.031. [DOI] [PubMed] [Google Scholar]
- Oguro-Ando A, Rosensweig C, Herman E, Nishimura Y, Werling D, Bill BR, Berg JM, Gao F, Coppola G, Abrahams BS, Geschwind DH. Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR. Mol Psychiatry. 2015;20:1069–1078. doi: 10.1038/mp.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, Davenport EC, Muir J, Sheehan DF, Lopez-Doménech G, Kittler JT. The autism and schizophrenia associated gene CYFIP1 is critical for the maintenance of dendritic complexity and the stabilization of mature spines. Transl Psychiatry. 2014;4:e374. doi: 10.1038/tp.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz O. Spatio-temporal Rho GTPase signaling: where are we now? J Cell Sci. 2010;123:1841–1850. doi: 10.1242/jcs.064345. [DOI] [PubMed] [Google Scholar]
- Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science. 1974;186:1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Sahoo T, Peters SU, Madduri NS, Glaze DG, German JR, Bird LM, Barbieri-Welge R, Bichell TJ, Beaudet AL, Bacino CA. Microarray based comparative genomic hybridization testing in deletion bearing patients with Angelman syndrome: genotype-phenotype correlations. J Med Genet. 2006;43:512–516. doi: 10.1136/jmg.2005.036913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/S0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- Schenck A, Qurashi A, Carrera P, Bardoni B, Diebold C, Schejter E, Mandel JL, Giangrande A. WAVE/SCAR, a multifunctional complex coordinating different aspects of neuronal connectivity. Dev Biol. 2004;274:260–270. doi: 10.1016/j.ydbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, Cornelisse LN, Toonen RF, Veldkamp W. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods. 2011;195:185–193. doi: 10.1016/j.jneumeth.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Silva JM, Ezhkova E, Silva J, Heart S, Castillo M, Campos Y, Castro V, Bonilla F, Cordon-Cardo C, Muthuswamy SK, Powers S, Fuchs E, Hannon GJ. Cyfip1 is a putative invasion suppressor in epithelial cancers. Cell. 2009;137:1047–1061. doi: 10.1016/j.cell.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir GA, Doyle OM, Tost H, Grimm O, Kristjansdottir S, Snorrason H, Davidsdottir SR, Gudmundsson LJ, Jonsson GF, Stefansdottir B, Helgadottir I, Haraldsson M, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–366. doi: 10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- Sun Y, Bamji SX. beta-Pix modulates actin-mediated recruitment of synaptic vesicles to synapses. J Neurosci. 2011;31:17123–17133. doi: 10.1523/JNEUROSCI.2359-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam GW, van de Lagemaat LN, Redon R, Strathdee KE, Croning MD, Malloy MP, Muir WJ, Pickard BS, Deary IJ, Blackwood DH, Carter NP, Grant SG. Confirmed rare copy number variants implicate novel genes in schizophrenia. Biochem Soc Trans. 2010;38:445–451. doi: 10.1042/BST0380445. [DOI] [PubMed] [Google Scholar]
- van der Zwaag B, Staal WG, Hochstenbach R, Poot M, Spierenburg HA, de Jonge MV, Verbeek NE, van't Slot R, van Es MA, Staal FJ, Freitag CM, Buizer-Voskamp JE, Nelen MR, van den Berg LH, van Amstel HK, van Engeland H, Burbach JP. A co-segregating microduplication of chromosome 15q11.2 pinpoints two risk genes for autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2010;153:960–966. doi: 10.1002/ajmg.b.31055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, Holder-Espinasse M, Delobel B, Duban B, Vallee L, Cuisset JM, Lemaitre MP, Vantyghem MC, Pigeyre M, Lanco-Dosen S, Plessis G, Gerard M, Decamp M, Mathieu M, Morin G, et al. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 2015;58:140–147. doi: 10.1016/j.ejmg.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Waites CL, Leal-Ortiz SA, Andlauer TF, Sigrist SJ, Garner CC. Piccolo regulates the dynamic assembly of presynaptic F-actin. J Neurosci. 2011;31:14250–14263. doi: 10.1523/JNEUROSCI.1835-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim NS, Wen Z, Makri G, Nauen D, Shin JH, Park Y, Chung R, Pekle E, Zhang C, Towe M, Hussaini SM, Lee Y, Rujescu D, St Clair D, Kleinman JE, Hyde TM, et al. Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell. 2014;15:79–91. doi: 10.1016/j.stem.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Achsel T, Bagni C. mRNPs, polysomes or granules: FMRP in neuronal protein synthesis. Curr Opin Neurobiol. 2006;16:265–269. doi: 10.1016/j.conb.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang D, Wang Q, Rodal AA, Zhang YQ. Drosophila cyfip regulates synaptic development and endocytosis by suppressing filamentous actin assembly. PLoS Genet. 2013;9:e1003450. doi: 10.1371/journal.pgen.1003450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Stevens CF. Probing synaptic vesicle fusion by altering mechanical properties of the neuronal surface membrane. Proc Natl Acad Sci U S A. 2008;105:18018–18022. doi: 10.1073/pnas.0809714105. [DOI] [PMC free article] [PubMed] [Google Scholar]