

Abstract
The E. coli single-stranded DNA-binding protein (SSB) binds to the fork DNA helicases RecG and PriA in vitro. Typically for binding to occur, 1.3 M ammonium sulfate must be present, bringing into question the validity of these data as these are non-physiological conditions. To determine whether SSB can bind to these helicases, we examined binding in vivo. First, using fluorescence microscopy, we show that SSB localizes PriA and RecG to the vicinity of the inner membrane in the absence of DNA damage. Localization requires that SSB be in excess over the DNA helicases and the SSB C-terminus and both PriA and RecG be present. Second, using purification of tagged complexes, our results demonstrate that SSB binds to PriA and RecG in vivo, in the absence of DNA. We propose that this may be the “storage form” of RecG and PriA. We further propose that when forks stall, RecG and PriA are targeted to the fork by SSB which, by virtue of its high affinity for single stranded DNA, allows these helicases to out compete other proteins. This ensures their actions in the early stages of the rescue of stalled replication forks.
INTRODUCTION
The essential, single-stranded DNA-binding protein (SSB) is critical to all aspects of DNA metabolism where it functions to stabilize single-stranded DNA (ssDNA) intermediates generated during DNA processing (Meyer & Laine 1990; Shereda et al. 2008). More recently it has become clear that SSB binds to a group of proteins known as the “SSB-interactome” (Costes et al. 2010). Interactome protein binding by SSB is necessary to facilitate loading of these proteins onto the DNA, using an unknown mechanism, to facilitate replication, recombination or repair (Shereda et al. 2008). The interactome proteins include Exonuclease I, the χ-subunit of DNA polymerase III, DnaG, RecO, uracil deglycosylase, topoisomerase III and the PriA, RecG and RecQ DNA helicases (Sandigursky et al. 1996; Costes et al. 2010; Kozlov et al. 2010b).
The multifunctional SSB exists as a homo-tetramer in bacteria, with an N-terminal domain critical to tetramer formation and ssDNA binding and, a C-terminal tail essential to target protein binding (Meyer & Laine 1990; Curth et al. 1996; Shereda et al. 2008). Although it is clear that the SSB C-terminus is required for binding to interactome proteins in vitro, it is unclear if binding occurs in vivo and if so, when binding occurs. Two models stemming from in vitro work have been proposed in the literature. The first proposes that interactome proteins bind to SSB in solution and then targets the repair enzymes to the DNA (Cadman & McGlynn 2004; Buss et al. 2008). In these studies, a co-precipitation technique that required the presence of 1.3 M ammonium sulfate was used to detect binding. In the absence of this salt, binding was not detectable. The second model suggests that the SSB C-termini are sequestered against the body of the protein in solution (Kozlov et al. 2010a). In this sequestered state, the C-termini are inaccessible to binding by target proteins. However, these termini are proposed to be exposed upon DNA binding to provide a landing site for a protein being targeted to the DNA. Key components distinguishing these models are the requirements for ammonium sulfate and for DNA. Determining which is correct is critical to understanding the mechanism and timing of interactome protein loading onto the DNA. This is of particular importance in the rescue of stalled DNA replication forks.
In E. coli, each fork is thought to stall at least once per cell cycle, requiring enzymes of homologous recombination to facilitate replication restart (Seigneur et al. 1998; Cox et al. 2000; Cox 2001). The types of lesions that could disrupt replication include proteins bound to the DNA ahead of the replication fork, non-coding lesions in the template DNA and either single- or double-strand breaks (Kowalczykowski 2000; McGlynn & Lloyd 2002a; Marians 2004). Each of the blocks could lead to a different type of damage to the DNA and this is highlighted by the various recombination and repair gene requirements for dealing with different types of DNA damaging agents (Marians 2000b; McGlynn & Lloyd 2002b; Marians 2004; Michel et al. 2004). Whatever its source, the block has to be removed or bypassed and replication must be restarted.
In bacteria, stalled replication forks can be reversed or directly restarted (Marians 2000b; Lusetti & Cox 2002; Marians 2004; Michel et al. 2004). Two key SSB interactome proteins play critical roles in stalled DNA replication fork rescue. The first is the 76 kDa DNA helicase, RecG (McGlynn & Lloyd 1999; McGlynn et al. 2000; Singleton et al. 2001). Recent studies support a role for RecG in the early stages of fork rescue, a role that is enhanced and controlled by SSB (Slocum et al. 2007; Buss et al. 2008; Abd Wahab et al. 2013). SSB binds to RecG via its highly conserved C-terminus. Reversal of the stalled fork leads to formation of a Holliday Junction-type structure that is then processed by RuvAB (Buss et al. 2008).
Once the fork has been processed into a DNA structure that permits the resumption of replication, the SSB interactome protein PriA (Primosomal protein A) takes over (Gabbai & Marians 2010). This 82 kDa protein binds with high affinity to D-loops and to fork structures via its N-terminus (Jones & Nakai 1999; Nurse et al. 1999). Once bound to a fork, PriA displays two types of activities. A 3’→5’ helicase activity responsible for unwinding any lagging-strand DNA present at the fork thereby generating single-stranded DNA for the replicative helicase, DnaB (Jones & Nakai 1999). The second activity is loading of DnaB onto the lagging-strand template (Masai & Arai 1996; Marians 1999, 2000a). Once DnaB has been loaded, a new replisome forms, leading to the resumption of DNA replication (Marians 1999, 2004). Critical to the function of PriA is the binding of SSB which like RecG, occurs via the C-terminus of the ssDNA binding protein (Cadman & McGlynn 2004). Also similar to RecG, SSB plays a key role in regulating PriA function at forks.
Critical to the roles of SSB, RecG and PriA in fork rescue, is a clear understanding of the timing and mechanism of their loading onto the DNA. Specifically, must a single-stranded DNA region at the fork be available for SSB to bind first, providing a landing site for each enzyme? If this model is correct and since SSB is present at 1 to 2,000 tetramers in the cell, how do the 7 to 70 molecules of RecG or PriA locate fork-bound tetramers in the milieu of SSBs present in the cell? Alternatively, could an SSB-helicase complex exist in the cell functioning as both a storage form of the helicase providing a means to rapidly and efficiently load RecG or PriA onto exposed regions of ssDNA when needed, thereby circumventing the remaining SSB tetramers in the cell?
Our approach to address this issue was first to image helicases in vivo. Results show that in the absence of exogenous DNA damage, SSB localizes both PriA and RecG to the cell periphery, possibly to the inner membrane. Localization requires the C-terminus of SSB as SSBΔC8 cannot substitute for wild type. In addition, results also suggest that SSB localizes these enzymes to sites of fork stalling, the active locations of PriA and RecG.
As the localization results implied that indeed SSB is binding to these helicases, we next attempted to purify the SSB-RecG and SSB-PriA complexes. Successful purification was demonstrated using differential tagging purifications, in which we were able to purify the SSB-RecG, SSB-PriA and RecG-SSB-PriA complexes in the absence of ammonium sulfate. Subsequent analysis reveals that these complexes did not contain DNA. More importantly, we were also able to purify the PriA-SSB-RecG complex. This is the first demonstration of binding between SSB and both of these interactome partners in vivo and our work clarifies the mechanism of SSB-mediated helicase loading onto the DNA to facilitate stalled replication fork rescue.
RESULTS
Plasmid construction and protein activity
To facilitate the studies described below, both high- and low-copy number expression vectors were constructed. First, N-terminal histidine tagged constructs were made in either pET28 (recG) or pET15 (ssb and separately, priA). Cloning was done using Infusion technology as described in the Experimental Procedures. Then, the mcherry or GFP genes were inserted into the NdeI site of pET28-his-recG and pET15-his-priA, respectively. Following this, the entire cloning region including the promoter and lacI gene was sub-cloned into low copy number vectors: pWKS130 (recG and priA) or pDUET (ssb). To perform expression studies, all experiments were done in Tuner (DE3) cells which are essentially BL21 (DE3) cells with a mutation in lacY. This allows for carefully controlled levels of expression by varying the concentration of IPTG and also provides more uniform expression throughout the culture as determined by visual inspection in the fluorescence microscope.
To determine whether the addition of an autofluorescent protein affected helicase activity, separate purifications of his-RecG, his-mcherry-RecG, his-PriA and his-GFP-PriA were done. Then, the DNA-dependent ATPase activity of RecG was assayed on M13 ssDNA while PriA was measured in the presence of φX174 DNA. The results show that when RecG is histidine tagged, activity decreases by 20% relative to wild type (Figure S1 in Supporting Information). When the mcherry protein is fused in-frame to RecG, the ATPase activity decreases further to 52% of wild type or to 65% of the his-tagged protein. In contrast, PriA is not significantly affected by the addition of GFP.
Next, we wanted to determine whether the fluorescent tagged DNA helicases retained binding to SSB. To do this in vitro, co-precipitation assays were done in the absence of DNA using purified proteins. The results show that mcherry and GFP had minimal effects on binding of helicases to SSB (Figure S2 in Supporting Information). As the fluorescent tagged helicases retained significant levels of ATPase activity and were able to bind to SSB in vitro, we proceeded with our in vivo studies.
SSB localizes PriA and RecG to the vicinity of the inner cell membrane
Studies in B. subtilus showed that SSB, RecG, and PriA formed foci (Lecointe et al. 2007). Previously, we showed that E. coli SSB and RecG colocalized in a similar manner (Liu et al. 2011). However, in both studies, the elevated levels of expression of the fluorescent tagged proteins lead to high background outside of the foci and precluded acquisition of details as to how proteins might be targeted to forks. Therefore, to more accurately visualize cellular localization, we used fluorescence microscopy and separate, low copy number vectors (6–8 copies per cell; (Wang & Kushner 1991)).
Tuner cells were transformed with the his-mcherry-RecG, his-GFP-PriA and SSB low copy number plasmids in various combinations, grown to mid-log phase in LB media in the absence of IPTG, processed and imaged. In all experiments, expression was verified using SDS-PAGE and bulk fluorescence measurements in a fluorometer (data not shown).
First, GFP-PriA and mcherry-RecG were imaged separately, with only the chromosomal SSB present. Under these conditions helicase levels are in excess over that of SSB. The microscopy images show that in both cases, cells exhibited a largely uniform fluorescence pattern (Fig. 1A and B, central images). To quantitate localization, cells were analyzed using 3-D surface plots. In these plots, the luminance of the image is interpreted as height (z-axis), with the x- and y-axes representing coordinates within each image. The results show that for PriA, the fluorescence signal is spread throughout the cells and that there is a 25% higher signal in the interior than at the periphery (compare the pink and dark blue colors corresponding to 33,000 and 25,000 luminance units, respectively; Fig. 1A, third panel). A similar pattern was observed for RecG with the signal being 25% higher in the interior where 40,000 luminance units were observed with 30,000 at the periphery (Fig. 1B, third panel).
Figure 1. PriA and RecG are distributed throughout the cell in the presence of endogenous levels of SSB.


For each strain, proteins were expressed from low copy number vectors. Here, overnight cultures from a single colony from which expression had been verified, were diluted 1:100 into 5ml of fresh LB containing antibiotics and grown for 4 hours in the absence of IPTG. Cells were harvested by centrifugation, resuspended into 10 mM MgSO4, attached to poly-L-lysine coated coverslips and imaged as described in Experimental Procedures. Representative microscopy images are presented showing the distribution of PriA and RecG in the presence of chromosomal SSB. The magnification in each microscope image is 400x. (A), GFP-PriA; (B), mcherry-RecG and (C), GFP-PriA and mcherry RecG co-expressed. In Figure 1A and B, panel 1 is the differential interference contrast (DIC) image; panel 2 is the image captured using the corresponding fluorescence filter for each protein; and panel 3 is a 3-D surface plot of a single cell in the center panel indicated by the arrow. (C), GFP-PriA and mcherry-RecG were co-expressed in the same cell. In Figure 1C, panel 1 corresponds to the DIC image; panel 2, fluorescence image captured with the GFP filter (PriA); panel 3, a 3-D surface plot of a single cell in panel 2 indicated by the arrow; 4, fluorescence image captured with the mcherry filter (RecG); 5, colocalization image of panels 2 and 4; panel 6, a 3-D surface plot of a single cell in panel 4 indicated by the arrow. The 3-D surface plots were done using ImageJ.
Next we examined cells where GFP-PriA and mcherry-RecG were co-expressed from separate plasmids in the same cell and SSB was again present at endogenous levels (Fig. 1C). In these cells, expression levels were lower and this was observed as a reduction in the overall fluorescence signal relative to cells expressing each helicase alone, with the RecG signal higher than that of PriA (Fig. 1C). Even though helicases were expressed at lower levels they are still in excess of SSB (data not shown). It is evident in the images shown that in the vast majority of cells, the fluorescence signal from both PriA and RecG is spread out in a relatively uniform fashion in the cell interior with quantitation revealing a slight enhancement in the cell center.
In contrast, when wild-type SSB, GFP-PriA and mcherry-RecG were co-expressed from separate vectors in the same cell, localization of the fluorescence signal was quite different (Fig. 2). In these images, the fluorescence signal was no longer uniform but instead presented as an elongated donut. In 97% of 202 cells examined, PriA was localized to the periphery (Fig. 2A2). The fluorescence signal of GFP-PriA was 17% higher at the periphery than in the center of these cells as indicated in the 3-D plot (Fig. 2A3). Similarly, localization of RecG to the periphery was observed in 97% of 202 cells examined (Fig. 2A4). The fluorescence signal of mcherry-RecG in these cells was 25% higher at the periphery than in the center (Fig. 2A6). In addition to the change in fluorescence signal appearance from uniform to elongated donut, the merged image of panels A2 and A4 shown in Figure 2A, panel 5, suggests that both PriA and RecG may be localized to the same regions of each cell as indicated by the yellow colour. Analysis of this image yields a Pearson’s correlation coefficient of 0.95 demonstrating colocalization. The values obtained from the colocalization analysis of five separate images yielded coefficients ranging from 0.88–0.95. This raises the possibility that each helicase may be bound to the same SSB tetramer resulting in their co-localization.
Figure 2. Elevated levels of SSB localize PriA and RecG to the cell periphery.


To determine localization, overnight cultures from a single colony from which expression had been verified using SDS-PAGE and bulk-phase fluorescence measurements, were diluted 1:100 into 5ml of fresh LB containing antibiotics (ampicillin, kanamycin and chloramphenicol) and grown for 4 hours in the absence of IPTG. Here, RecG and PriA were expressed from low copy number vectors while SSB was expressing from the high copy number pET28 vector. Cells were harvested by centrifugation, resuspended into 10 mM MgSO4, attached to poly-L-lysine coated coverslips and imaged as described in Experimental Procedures. Representative microscopy images are presented where SSB is in excess over the DNA helicases. The magnification in each microscope image is 400x. (A). Images of cells co-expressing, GFP-PriA, mcherry-RecG and wild-type SSB. Panel 1, DIC; 2, fluorescence image captured with the GFP filter (PriA); 3, a 3-D surface plot of a single cell in panel 2 indicated by the arrow; 4, fluorescence image captured with the mcherry filter (RecG); 5, colocalization image of panels 2 and 4; 6, a 3-D surface plot of a single cell in panel 4 indicated by the arrow. (B), 3-D surface plots of representative cells showing co-localization of PriA and RecG by wild-type SSB. Inset, the individual cells indicated by the white arrows in panel A5. Cell 3 is from a separate image. The 3-D surface plots were done using ImageJ.
In addition to the change in overall appearance in fluorescence signal location within cells, a region of enhanced fluorescence or focus was observed in 12% of all cells examined (Fig. 2A5; white arrows). Foci were often observed near cell poles but also in the approximate center of the cell (Fig. 2B). Analysis of colocalization of fluorescence signals within these cells yields Pearson’s correlation coefficient ranging from 0.92 to 0.94. Within foci, the individual fluorescence signals were 1.65- (PriA) and 1.8-fold (RecG) higher than the signal in the interior of the cell (Fig. 2B). Furthermore, the ratio of RecG to PriA in these foci, after corrected for the differences in expression levels, is 2. This ratio may indicate 2 RecG proteins targeted for each PriA. In summary, we suggest that those foci may be stalled DNA replication forks requiring rescue and to which SSB has targeted both PriA and RecG.
It is also evident that in contrast to the images in Figure 1, the overall fluorescence signal for each helicase was lower than that in cells where SSB was present at endogenous levels. This is attributed to the elevated level of expression of SSB (chromosomal and plasmid borne genes) which is now in excess of both RecG and PriA (determined by SDS-PAGE for helicases and SSB, and bulk-fluorescence measurements for helicases; data not shown). This ratio of SSB to helicase is important as SSB is typically present at a several-fold excess over both proteins in exponentially growing cells (Shlomai & Kornberg 1980; Meyer & Laine 1990; Lloyd 1991; Tanaka et al. 2003; Hruz et al. 2008; Ishihama et al. 2008). Consequently, the data in Figure 2 demonstrate that when SSB is present in excess over RecG and PriA, helicases are localized to the vicinity of the inner cell membrane in the absence of exogenous DNA damage.
PriA and RecG localization requires the SSB C-terminus
Previous in vitro studies have demonstrated that the C-terminal eight residues of SSB are essential for binding to RecG and PriA (Cadman & McGlynn 2004; Buss et al. 2008). To determine whether localization to the vicinity of the inner membrane requires the C-terminus of SSB, experiments were repeated using cells co-expressing SSBΔC8 and either GFP-PriA or mcherry-RecG from low copy-number vectors. The results show that in 43% of these cells, PriA and RecG were localized to the periphery, down from 97% observed for wild type (Fig. 3). Critically, in those cells in which peripheral localization of fluorescence was observed, the fluorescence signal was only 4–10% higher than the cell interior. This modest enhancement is likely due to the binding of RecG and PriA to the available, functional SSB C-termini present in endogenous wild-type homotetramers as well as to those in wild-type/SSBΔC8 chimeras (Liu et al. 2011). In addition, in the case of RecG, binding may also occur to other sites on the SSB tetramer (see below). Similar results were obtained when both fluorescent tagged DNA helicases were co-expressed from low copy number vectors with SSBΔC8 from a high copy number vector (Figure S3 in Supporting Information). In these cells, the fluorescence of each helicase was spread throughout the cells with no detectable localization to the vicinity of the inner membrane (Figure S3B and C). Therefore, localization to the vicinity of the inner membrane requires an excess of SSB over helicases and that the SSB C-terminus be present. By definition, these results imply that SSB must bind to each helicase to facilitate its localization.
Figure 3. Efficient localization of PriA and RecG to the cell periphery requires the SSB C-terminus and the presence of both DNA helicases.

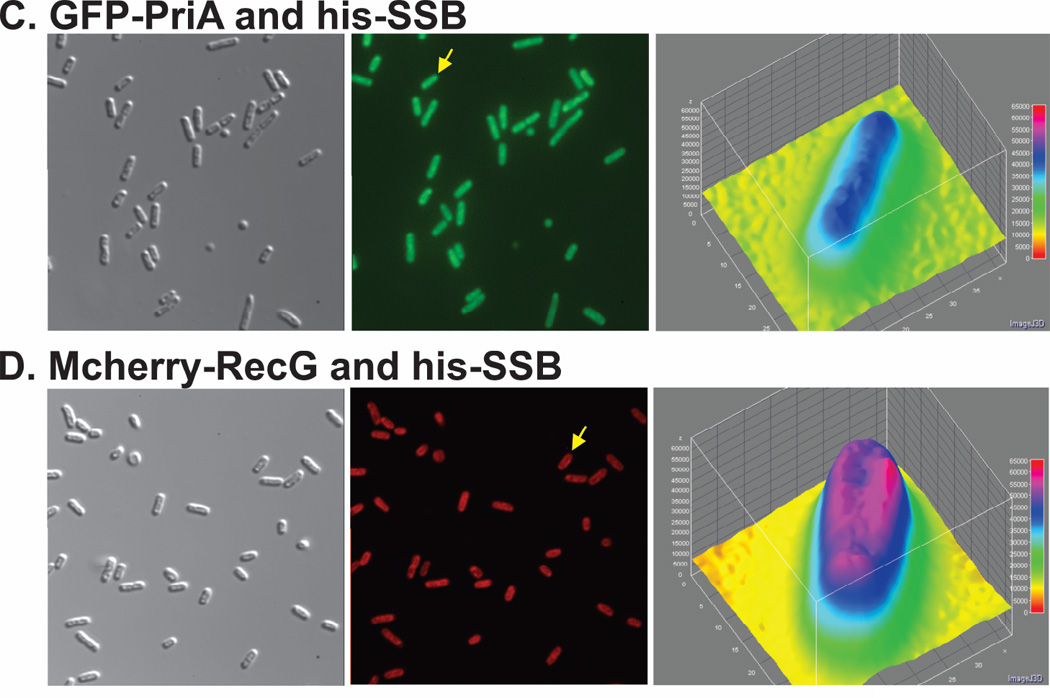
Fluorescence microscopy images and analysis of cells where fluorescent tagged helicases and SSBΔC8 were co-expressed. Mutant SSB was expressed from pET28 while the helicases were expressed from low copy number vectors. To determine localization, overnight cultures from a single colony from which expression had been verified using SDS-PAGE and bulk-phase fluorescence measurements, were diluted 1:100 into 5mL of fresh LB containing antibiotics and grown for 4 hours in the absence of IPTG. Cells were harvested by centrifugation, resuspended into 10 mM MgSO4, attached to poly-L-lysine coated coverslips and imaged as described in Experimental Procedures. Representative microscopy images are presented. The magnification in each microscope image is 400x. (A), GFP-PriA and SSBΔC8. Panel 1, DIC image; 2, GFP; 3, a 3-D surface plot of a single cell in panel 2 indicated by the yellow arrow. (B), mcherry-RecG and SSBΔC8. Panel 1, DIC image; 2, mcherry; 3, a 3-D surface plot of a single cell in panel 2 indicated by the yellow arrow. White arrows indicate cells where localization to the cell periphery can be discerned. (C), Representative images of cells expressing SSB and GFP-PriA. Panel 1, DIC image; 2, GFP; 3, a 3-D surface plot of a single cell in panel 2 indicated by the yellow arrow. (D), Representative images of cells expressing SSB and mcherry-RecG. Panel 1, DIC image; 2, mcherry; 3, a 3-D surface plot of a single cell in panel 2 indicated by the yellow arrow. The 3-D surface plots were generated using ImageJ.
Efficient localization requires both PriA and RecG
The images in Figure 2 show that both DNA helicases are targeted to the vicinity of the inner membrane. Results in Figure 3A and B, show that the last eight residues of SSB are required for this localization to occur. It is conceivable that the presence of both helicases may be required for localization or alternatively, it may only occur when one is bound to SSB. To test this, strains containing a high copy number vector expressing SSB were transformed with low copy number vectors expressing GFP-PriA and separately, mcherry-RecG. The results show that efficient localization does not occur under these conditions (Figure 3C and D). There is a slight enhancement of RecG at the cell periphery, where the signal is on average, 7% higher than in the cell interior (Figure 3D, panel 3). Therefore, efficient localization to the vicinity of the inner membrane requires wild type SSB and both RecG and PriA.
SSB binds to fork rescue helicases in vivo
The microscopy data imply that SSB is binding to RecG and PriA in vivo. This follows because when the last eight residues of SSB which are required for binding in vitro are eliminated, localization to the inner membrane is essentially eliminated. To test whether SSB-helicase binding is occurring, histidine-tagged helicases (RecG or PriA) were independently expressed with wild-type SSB. In separate experiments, individual wild-type helicases were co-expressed with his-SSB in Tuner (λDE3) cells, respectively (Table I and II). One liter cultures of cells expressing proteins from high copy number vectors (Table II), were grown, lysed and the cleared cell lysates subjected to nickel column chromatography. After extensive washing, proteins were eluted using a linear, imidazole gradient as described in the Experimental Procedures. Purifications were done using both high (600 mM) and low (150 mM) KCl conditions. The rationale for using two separate salt conditions is as follows. First, in vitro binding of SSB to RecG or PriA requires 1.3 M ammonium sulfate. Thus binding could be an artifact as this salt does not occur in the cytoplasm of E. coli. Second, as the 600 mM concentration present in the standard nickel column chromatography buffers could also lead to artifacts, we lowered the salt concentration used in lysis and chromatographic steps to 150 mM, which is below that of the cytoplasm of E. coli (Neidhardt et al. 1996).
Table I.
Strains used in this study
| Strain | Relevant characteristics | Source |
|---|---|---|
| PB 27775 | Tuner(λDE3)/ pET21a(+)-SSB & pET28a(+)-his-RecG | This study |
| PB 27776 | Tuner(λDE3)/ pET21a(+)-SSBΔC8 & pET28a(+)-his-RecG | This study |
| PB 27805 | Tuner(λDE3)/ pET28a(+)-his-SSB & pEAW106-RuvA | This study |
| PB 27806 | Tuner(λDE3)/ pET28a(+)-his-SSB & pEAW106-RuvB | This study |
| PB 27807 | Tuner(λDE3)/ pET28a(+)-his-SSB & pGS775-RuvC | This study |
| PB 27829 | Tuner(λDE3)/ pET28a(+)-his-SSB & pGS772-RecG | This study |
| PB 27834 | Tuner(λDE3)/ pET28a(+)-his-SSB & pET15b–PriA | This study |
| PB 27876 | Tuner(λDE3)/ pET21a(+)-SSB & pET28a(+)-his-PriA | This study |
| PB 27879 | Tuner(λDE3)/ pET28a(+)-his-SSBΔC8 & pET15b–PriA | This study |
| PB 27880 | Tuner(λDE3)/ pET28a(+)-his-SSBΔC8 & pGS772-RecG | This study |
| PB 27903 | Tuner(λDE3)/ pET28a(+)-his-PriA & pET21a(+)-SSBΔC8 | This study |
| PB 28031 | Tuner(λDE3)/ pDuet-his-mcherry-linker-RecG & pWKS30-his-GFP-PriA & pET28a(+)-SSB |
This study |
| PB 28032 | Tuner(λDE3)/ pDuet-his-mcherry-linker-RecG & pWKS30-his-GFP-PriA & pET28a(+)- SSBΔC8 |
This study |
| PB 28038 | Tuner(λDE3)/ pDuet-his-mcherry-linker-RecG & pWKS30-his-GFP-PriA |
This study |
| PB 28048 | Tuner(λDE3)/ pWKS30-his-GFP-PriA | This study |
| PB 28051 | Tuner(λDE3)/ pDuet-his-mcherry-linker-RecG | This study |
| PB 28074 | Tuner(λDE3)/ pET28a(+)-PriA-his & pET21a(+)-SSB | This study |
| PB 28079 | Tuner(λDE3)/ pET28a(+)-RecG-his & pET21a(+)-SSB | This study |
| PB 28137 | Tuner(λDE3)/ pET28a(+)-RecG-his & pET15b–PriA & pET21a(+)-SSB |
This study |
| PB 28146 | Tuner(λDE3)/ pET28a(+)-PriA-biotin & pET15b–his-SSB & pET21a(+)-BirA |
This study |
| PB 28219 | Tuner(λDE3)/ pET28a(+)-RecG-his & pET15b–PriA | This study |
| PB 28261 | Tuner(λDE3)/ pET28a(+)-RecG-his & pPAL7-profinity-SSB | This study |
Table II.
Plasmids used in this study
| Plasmids | Relevant characteristic | Source |
|---|---|---|
| pWKS30 | Low copy number vector; AmpR | (Wang & Kushner 1991) |
| pET21a(+)-SSB | Expression of wild-type SSB; AmpR | (Shereda et al. 2007) |
| pET21a(+)-SSB | Expression of wild-type SSB; CamR | This study |
| pET28a(+)-SSB | Expression of wild-type SSB; KanR | This study |
| pET28a(+)-his-SSB | Expression of N-terminal his-SSB; KanR | (Liu et al. 2011) |
| pET21a(+)-SSBΔC8 | Expression of SSBΔC8; AmpR | (Shereda et al. 2007) |
| pET28a(+)-SSBΔC8 | Expression of SSBΔC8; KanR | This study |
| pET28a(+)-his-SSBΔC8 | Expression of N-terminal his-SSBΔC8; KanR | This study |
| pPAL7-profinity-SSB | Expression of N-terminal profinity-SSB; AmpR | This study |
| pGS772-RecG | Expression of wild-type RecG; AmpR | (Lloyd & Sharples 1993; Cadman & McGlynn 2004) |
| pET28a(+)-his-RecG | Expression of N-terminal his-linker-RecG; KanR | This study |
| pET28a(+)-RecG-his | Expression of C-terminal RecG-his; KanR | This study |
| pDuet-his-mcherry-linker-RecG | Expression of N-terminal his-mcherry-linker-RecG; CamR | (Liu et al. 2011; Ryzhikov et al. 2011) |
| pET15b–PriA | Expression of wild-type PriA; AmpR | K. Marians |
| pET28a(+)-his-PriA | Expression of N-terminal his-PriA; KanR | This study |
| pET28a(+)-PriA-his | Expression of C-terminal PriA-his; KanR | This study |
| pET28a(+)-PriA-biotin | Expression of C-terminal PriA-biotin; KanR | This study |
| pWKS30-his-GFP-PriA | Expression of N-terminal his-GFP-PriA; AmpR | This study |
| pEAW106-RuvA | Expression of wild-type RuvA; AmpR | (Iype et al. 1994) |
| pEAW106-RuvB | Expression of wild-type RuvB; AmpR | (Iype et al. 1994; Purnapatre et al. 1999; Buss et al. 2008) |
| pGS775-RuvC | Expression of wild-type RuvC; AmpR | (Sharples & Lloyd 1991) |
| pET21a(+)-BirA | Expression of wild-type BirA; CamR | This study |
Regardless of which salt condition was used, the results show that his-SSB and wild-type RecG co-eluted from the column (Fig. 4A and B). In fact, more complex eluted in the lower salt condition. Co-elution was observed whether the histidine tag was present on either RecG or SSB (Fig. 4 and data not shown). In addition, there was no difference in the amount of complex eluted when the tag was on either the N- or C-terminus of RecG (data not shown). When the tag was on SSB, proteins eluted as a broad peak between 166 and 434 mM imidazole with the apex at 280 mM (Figure S4 in Supporting Information). The broad peak observed contained a large amount of protein (>150 mg) with the RecG and SSB present in each fraction at an almost 1:1 stoichiometry (Fig. 4A). This is consistent with each SSB tetramer being able to bind as many as 2 RecG monomers as demonstrated previously (Buss et al. 2008). When the tag was placed on either the N- or C-terminus of RecG, SSB and the helicase eluted as a narrow peak with the apex at 240 mM imidazole (Figure S4 in Supporting Information). Gel analysis of this purification revealed that free wild-type SSB was found in the column flow through, indicating that while all of the histidine-tagged RecG was in a complex with SSB, not all of the SSB was complexed with helicase (data not shown). Co-elution was not due to untagged RecG or SSB binding to the column resin as separate purifications of wild type versions of each protein showed that no SSB eluted from the column, and a small but detectable amount of RecG was observed (data not shown). Instead, the majority of each protein was present in the flow through and wash fractions (data not shown). Therefore, RecG and SSB form a complex that elutes from the nickel column when either protein is histidine-tagged. Similar results were obtained when cells were grown in the absence of IPTG, although the overall levels of protein obtained was significantly lower (Figure S5, left panel in Supporting Information).
Figure 4. PriA and RecG bind to SSB in vivo.

1L cultures of cells expressing PriA and SSB, and separately, RecG and SSB were grown to early log phase, IPTG added to 200 µM and growth continued until early stationary phase. Cells were harvested by centrifugation, lysed and the cleared cell lysate applied to a 5ml nickel column as described in the Experimental Procedures. Proteins were then eluted using an imidazole gradient following extensive washing to remove unbound proteins. Aliquots from various fractions throughout the purification were subjected to electrophoresis. The resulting 12% SDS-PAGE gels of the purification of helicase-SSB complexes are presented. Panels (A) - (C), co-purification of RecG and SSB; panels (D) - (F), co-purification of PriA and SSB. In (A) and (D), purification was done with buffer containing 600mM NaCl. In panels (B) and (E), the [KCl] was 150 mM. Panel (C), purification of the his-RecG/profinity-SSB complex; left panel, protein gel stained with Coomassie Brilliant Blue; right panel, protein gel stained with silver. Panel (F), purification of the his-SSB/PriA-biotin complex. The numbers at the top of lanes indicates fraction numbers from the peak in the corresponding elution profiles (Figure S4 in Supporting Information). M, molecular weight marker; CCL, clear cell lysate; FT, flow through. Ni, pooled fractions from the nickel column; Av, pooled fractions from the avidin column; Pro, complex eluted from the profinity eXact column.
To further verify that the SSB-RecG complex exists in vivo and is not occurring once cells are lysed, separate 1 L cultures of his-SSB and RecG were grown, harvested and lysed as before and then the two cleared cell lysates were mixed. The mixture was then divided into two equal fractions. Fraction 1 was immediately applied to a nickel column, while Fraction 2 was stirred for 30 minutes to allow for complex formation prior to loading on a separate nickel column. Following elution using an imidazole gradient, fractions were analyzed by SDS-PAGE. In both cases only his-SSB eluted from the column at a level comparable to that shown in Figure 4 (Figure S6 in Supporting Information). Therefore, we conclude that the SSB-RecG complex exists prior to cell lysis. Collectively, the data presented above demonstrate that RecG binds to SSB in vivo, consistent with a genomics study (Butland et al. 2005).
To further study complex formation, we subjected the SSB-RecG complexes to further purification steps using Q-sepharose and ssDNA cellulose chromatography. The two proteins co-elute from Q-sepharose in two peaks (data not shown). The first peak eluted at 530mM and the second at 690mM NaCl. SDS-PAGE analysis revealed that the first peak contained a slight excess of SSB relative to RecG while the second contained approximately equal ratios of the two proteins. When purified alone, RecG elutes from Q-sepharose at 340mM NaCl. Similarly, the salt concentration for complex elution from ssDNA cellulose is 700mM whereas when purified separately, RecG elutes at 450mM NaCl and SSB elutes at salt concentrations above 1M. These results imply that the SSB-RecG complex is very stable.
Previous work demonstrated that over-expression of RecG was detrimental to plasmid copy number (Vincent et al. 1996; Fukuoh et al. 1997). This is attributed to RecG displacing R-loops that are required for replication initiation of the ColE1 plasmids used for expression (Itoh & Tomizawa 1979). Consistent, in our hands and when over-expressed on its own, low levels of RecG are produced with a typical RecG purification yielding ∼2 mg of protein per liter of cells (Slocum et al. 2007). Similar yields are obtained with PriA (data not shown). In contrast, our data show that in the presence of SSB, the expression levels of the DNA helicases are significantly higher (Figure 4A and D). In fact, more than 200 mg of helicase-SSB complex can be purified from a single liter of cells with the helicases representing at least one third of the total protein, approximately 70mg. This represents a 35-fold increase in the levels of RecG. Similar results were observed for PriA (see below). We attribute this increase to SSB binding to all of the available RecG and PriA thereby sequestering the helicase to the inner membrane. This may also stabilize these helicases making them less sensitive to degradation and/or turnover.
As for RecG, co-elution of wild-type PriA and his-SSB from the nickel affinity column was observed at either the high or low salt condition (Fig. 4D and E). When the histidine tag was on SSB, proteins eluted in a broad peak with the apex at 298 mM imidazole (Figure S4 in Supporting Information). When PriA was tagged, proteins eluted in a narrow peak centered at 250 mM imidazole and free wild-type SSB was observed in the flow through suggesting that all of the PriA was bound to SSB and that there was an excess of SSB present (data not shown). Significantly more protein was recovered when the tag was on SSB than when it was on PriA and, the ratio of PriA to his-SSB in the peak fraction was 1.5, whereas the ratio of RecG to his-SSB was 1. This suggests that more PriA binds to each SSB tetramer than does RecG. Finally, co-elution was not due to untagged PriA binding to the column resin as purification of wild-type PriA showed that the protein was present in the flow through and wash fractions only (data not shown). Therefore, PriA and SSB form a complex in vivo that can be eluted from the nickel column when either protein is histidine-tagged.
Even though untagged SSB and helicases did not bind to the nickel column when expressed on their own (data not shown), we wanted to verify the results from the nickel columns. To do this, helicases and SSB were differentially tagged, co-expressed and complexes purified using two different columns. First, cells expressing RecG-his were transformed with N-terminal, Profinity-tagged SSB. As before, proteins were co-expressed, cells lysed and the cleared cell lysates applied to a nickel column. The peak fractions, which contained both RecG-his and Profinity-SSB, were pooled and applied to a profinity column. The profinity tag was cleaved using sodium fluoride releasing both SSB and RecG-his (Fig. 4C). Analysis of the final lane from the profinity columns reveals that proteins are present at a 1:1 ratio. This confirms that SSB and RecG exist as a complex.
Next, cells containing his-SSB were transformed with PriA-biotin. The resulting cleared cell lysate was applied to a nickel column and proteins were eluted with an imidazole gradient. As expected, both his-SSB and PriA-biotin were eluted from the column (Fig. 4F). Peak fractions were pooled, applied to an avidin column and eluted with buffer containing biotin. Both proteins eluted from the column, confirming that PriA and SSB exist as a complex. Further, and as for RecG, analysis of the gel demonstrates that the stoichiometry of the SSB-PriA complex is 1. Therefore, these data demonstrate that RecG and PriA bind to SSB in vivo, consistent with the fluorescence microscopy data showing colocalization.
Complex formation requires the SSB C-terminus
Our imaging data demonstrate that SSBΔC8 does not efficiently target RecG and PriA to the vicinity of the inner membrane (Fig. 3A and B). This result is consistent with a model wherein SSB binds to these enzymes via its C-terminus to facilitate targeting, consistent with the above-mentioned complex purifications and also in vitro studies which show that SSBΔC8 does not form complexes with either RecG or PriA (Cadman & McGlynn 2004; Buss et al. 2008). However, as the fluorescence microscopy and purification conditions in the preceding sections used very low and extreme over-expression conditions respectively, it is conceivable that mass action is driving protein-protein interactions that may not otherwise occur. We thought this unlikely, but decided to test this possibility nonetheless. To do this, we over-expressed SSBΔC8 with each of the helicases separately. If mass action is driving complex formation, we expected to purify SSBΔC8-helicase complexes. If however it is not, and complex formation requires the SSB C-terminus as expected, SSBΔC8 will not form complexes with either PriA or RecG in vivo.
To test complex formation, we again made dual-expression strains, but replaced wild-type SSB with SSBΔC8, and his-SSB with his-SSBΔC8. The results show that for both histidine tagged DNA helicases, SSBΔC8 was detected only in the flow through but not in the peak fractions, even when lanes were significantly over-loaded (Fig. 5A and B; data not shown). When tagging was reversed, some RecG was detected in the peak fractions whereas PriA was undetectable (Fig. 5C and D). In these experiments, RecG eluted earlier than his-SSBΔC8, with a small amount of overlap between the two protein peaks. The amount of RecG present is greater than that observed in purifications using only wild type RecG (data not shown). Thus, we propose that there may be residues of SSB separate from its C-terminus that are involved in RecG binding. This finding is consistent with the microscopy data demonstrating that a small amount of RecG is targeted to the inner membrane when this mutant form of SSB is present. As co-elution of either PriA or RecG was not observed with SSBΔC8, complex formation requires the C-terminus of SSB. Consequently, mass action resulting from over-expression of these proteins is not the driving force behind complex formation.
Figure 5. Complex formation requires the SSB C-terminus.

1L cultures of cells expressing PriA and SSBΔC8, and separately, RecG and SSBΔC8 were grown to early log phase, IPTG added to 200 µM and growth continued until early stationary phase. Cells were harvested by centrifugation, lysed and the cleared cell lysate applied to a 5ml nickel column as described in the Experimental Procedures. Proteins were then eluted using an imidazole gradient following extensive washing to remove unbound proteins. Aliquots from various fractions throughout the purification were subjected to electrophoresis. The resulting 12% SDS-PAGE gels are shown. Panels (A) and (B), helicases are N-terminal histidine tagged. Panels (C) and (D) SSBΔC8 is N-terminal histidine tagged. M, molecular weight marker; CCL, cleared cell lysate; FT, flow through. Numbers at the top of lanes indicate fraction numbers from the peak.
To further test the mass action model, separate strains over-expressing his-SSB and the individual proteins of the RuvA/B/C resolvasome were made. Previous work has shown that SSB does not bind to either RuvA, RuvB or RuvAB in vitro (Buss et al. 2008). Therefore, we did not anticipate their binding to SSB, unless mass action resulting from the over-expression is driving these interactions. As before, cleared cell lysates were subjected to nickel column chromatography. The results show that in each case, only his-SSB eluted from the column with RuvA, RuvB, and RuvC eluting in the flow through and wash fractions (Figure S7 in Supporting Information). Thus, SSB does not bind to the components of the resolvasome in vivo. Therefore, mass action resulting from the over-expression cannot be driving interactions between SSB and either RecG or PriA, that otherwise normally would not occur.
Purified helicase-SSB complexes do not contain DNA
Our microscopy results show that SSB binds to RecG and PriA and localizes them to the vicinity of the inner membrane in the absence of DNA damage. It is possible that this may be the “storage form” of these helicases and consequently, they should not be bound to DNA. Our in vitro data and that of others demonstrating SSB-helicase binding occurs in the absence of DNA is consistent with this interpretation (Cadman & McGlynn 2004; Shereda et al. 2007; Buss et al. 2008; Suski & Marians 2008). If our model is correct, then the purified helicase-SSB complexes should not contain DNA. In contrast, if they do, then close to stoichiometric ratios of DNA to protein should be observed in the peak fractions and consequently, the presence of DNA will be required for complex formation.
To determine whether DNA is required for SSB-helicase complex formation, 19 and 29 mg of the SSB-PriA and SSB-RecG complexes respectively, were treated with benzonase as described in Experimental Procedures. Thereafter, the protein mix was subjected to nickel column chromatography. As before, SSB and helicases co-eluted (Fig. 6). It is possible that the broad-specificity benzonase nuclease may have not been able to displace proteins have so that complete DNA could occur. Therefore, the pooled proteins from the above-mentioned chromatography step were dialyzed into a compatible buffer, treated with a high concentration of DNaseI and re-chromatographed. Again, SSB co-eluted with the DNA helicases (data not shown). As the protein complexes remained intact following the treatment with nucleases, these results suggest that DNA is not required for their formation.
Figure 6. Complex formation with SSB is DNA-independent.

1L cultures of cells expressing RecG and his-SSB, and separately, PriA and his-SSB were grown to early log phase, IPTG added to 200 µM and growth continued until early stationary phase. Cells were harvested by centrifugation, lysed and the cleared cell lysate applied to a 5ml nickel column as described in the Experimental Procedures. Proteins were then eluted using an imidazole gradient following extensive washing to remove unbound proteins. The fractions containing both helicase and SSB were pooled, dialyzed to remove NaCl, treated with benzonase, applied to the nickel column and eluted with an imidazole gradient. The resulting 12% SDS-PAGE gels showing protein after the first column; following benzonase treatment and fractions from the second nickel column are presented. (A), The SSB-RecG complex does not require DNA for its formation or stability; (B), The SSB-PriA complex does not require DNA for formation or stability. Pre, pooled protein fractions eluted from the nickel column prior to benzonase treatment; post, protein sample following nuclease treatment; FT, flow through, W, wash; Lanes 6 – 14, fractions from the elution step of the second passage through the nickel column.
However, there may still be small fragments of DNA present to which the complexes bind with high affinity thereby making the nucleic acid inaccessible to nuclease digestion. Therefore, to determine whether DNA may be present, 226 – 352 pmoles of complex from the benzonase/column-treated pool of proteins were denatured, treated with phosphatase, labeled with 32P-ATP and subjected to denaturing acrylamide gel electrophoresis. The results show that only 3 – 4 pmoles of nucleic acid could be detected and which migrated with an apparent size of 15 – 17 nt (data not shown). As 226 to 352 pmoles of complex were treated, this result means that 99% of the complexes present do not contain DNA.
It is possible that the failure to label DNA that might be present could be due to the presence of an inhibitor of alkaline phosphatase or T4 polynucleotide kinase. To test this, the same amounts of protein were mixed with a control, 31-mer oligonucleotide prior to the first boiling step. Then, the protein-DNA mix was processed as before. The results show that in each fraction, the 31-mer oligonucleotide labeled with an efficiency identical to that in the control lane containing the same amount of DNA (data not shown). Thus, the failure to label DNA is not due to the presence of an inhibitor of the phosphatase/ kinase enzymes but must be due to the absence of DNA. Consequently, and consistent with in vitro data, complex formation between SSB and RecG and between SSB and PriA, does not require DNA.
PriA and RecG bind to the same SSB tetramer
Previous work has shown that the combined actions of PriA and RecG are critical to the rescue of stalled replication forks (Gregg et al. 2002). Furthermore, an in vitro study has shown both helicases can co-exist on a fork and their interactions are complex (Tanaka & Masai 2006). Thus far we have shown that in vivo, RecG binds to SSB and separately, PriA binds to SSB. The microscopy analysis shows that for efficient localization to occur, both RecG and PriA must be present. In addition, SSB colocalizes both RecG and PriA to foci, raising the possibility that both helicases may be bound to the same tetramer.
To determine whether this may be occurring, we co-expressed RecG-his with Profinity-SSB and PriA-biotin. The resulting cleared cell lysate should contain at least four complexes (Fig. 7A). These are the RecG-his/PriA-biotin, RecG-his/profinity-SSB, PriA-biotin/profinity-SSB and the RecG-his/profinity-SSB/PriA-biotin complexes. When this lysate is applied to the nickel column, the SSB/PriA complex, which does not contain a histidine tag, is found in the flow through (data not shown). As expected, three potential complexes elute from the column: RecG-his/profinity-SSB, RecG-his/PriA-biotin and the three protein complex, RecG-his/profinity-SSB/PriA-biotin (Fig. 7A, left side). The SDS-PAGE gel in Figure 7B demonstrates the presence of three proteins with the expected molecular weights for SSB, RecG and PriA. The fractions from the nickel column were pooled and applied to the Profinity column. Now the RecG/PriA complex which does not contain a profinity tag, is found in the flow through (Fig. 7A, left). Importantly, two complexes could be eluted from the profinity column: the SSB-RecG and the SSB/RecG/PriA complexes. Analysis of the SDS-PAGE gels demonstrates that SSB, RecG-his and PriA-biotin are all present. The only way PriA-biotin could elute from both the nickel and Profinity columns is if it were in a complex with both SSB and RecG simultaneously.
Figure 7. PriA and RecG bind to SSB simultaneously.



1L cultures of cells expressing RecG-his, PriA-biotin and profinity-SSB were grown to early log phase in LB+ antibiotics and biotin, IPTG added to 200 µM and growth continued until early stationary phase. Cells were harvested by centrifugation, lysed and the cleared cell lysate applied to a 5ml nickel column as described in the Experimental Procedures. Proteins were then eluted using an imidazole gradient following extensive washing to remove unbound proteins. Pooled fractions were applied to the profinity column, washed and cleavage allowed to occur overnight at 4°C. The next day, proteins were eluted using elution buffer as described by the manufacturer (Biorad). (A), scheme of the triple-tagged complex purification. (B), SDS-PAGE of relevant fractions from the purification. (C), blots from PAGE gels of the same fractions probed with anti-6x–his tag antibody (left) or streptavidin (right). Key: M, marker; Ni, nickel pool, PF, profinity flow through and PE, proteins eluted from the profinity column. (D), SDS-PAGE criterion gel lanes of fractions eluted from a nickel and gel filtration column. (E), blots of relevant lanes of the RecG-his/PriA-biotin purification. Key: M, marker; CCL, cleared cell lysate; FT, flow through; Ni, nickel pool and GF, peak fraction from the gel filtration column.
As RecG and PriA are similar in size we wanted to determine whether both proteins were present in the fractions eluted from the Profinity column. To determine identity, the same fractions were again subjected to SDS-PAGE, transferred to membranes and probed with either anti-his tag antibody to detect for the presence of RecG and streptavidin coupled to alkaline phosphatase to detect for the presence of PriA. The resulting blots show that both RecG and PriA eluted from the Profinity column (Fig. 7C). Therefore, both helicases can bind to the same SSB tetramer simultaneously.
To further evaluate complex formation, a cleared cell lysate of his-SSB/wtRecG/WtPriA was subjected to nickel column chromatography. Fractions containing all three proteins were pooled, concentrated and applied to a Superose-6 column. The elution profile shows that the proteins eluted in two overlapping peaks with calculated molecular weights of 401 and 214 kDa (Figure S8 in Supporting Information). These molecular weights correspond to complexes consisting of 4 helicases and a single SSB tetramer and 2 helicases and a single SSB tetramer, respectively. The identity of the proteins present was confirmed by SDS-PAGE in Criterion gels (data not shown). To ensure that all 3 proteins were in fact present, a cleared cell lysate of RecG-his/PriA-biotin/profinity-SSB was subjected to nickel and Profinity column chromatography as before. The fractions eluted from the latter column were concentrated and subjected to gel filtration as described in the Experimental Procedures. The theoretical MW of this three protein complex is 238 KDa. The calculated MW of the single peak containing RecG-his+PriA-biotin+SSB was found to be 202 kDa (data not shown). Fractions from this peak were analyzed by SDS-PAGE and were found to contain all three proteins at a stoichiometry identical to that in the load, consistent with the existence of a three protein complex containing 1 molecule of each. The size is smaller than expected but larger than a dimeric complex consisting of either RecG and PriA or RecG/PriA and SSB (154 KDa each). Therefore, we conclude that SSB can exist in a tripartite complex with both DNA helicases.
It is conceivable that PriA and RecG could interact with one another on the surface of SSB, possibly leading to complex stabilization. The binding of the two helicases to one another would be consistent with genetic and biochemical data (Al-Deib et al. 1996; Tanaka & Masai 2006). To test whether this is occurring, RecG-his and PriA-biotin were co-expressed and the resulting lysate was applied to a nickel column and proteins eluted using an imidazole gradient (Figure S9 in Supporting Information). Both helicases were detected in peak fractions at approximately equal amounts, indicating that these proteins also bind to one another in vivo (Fig. 7D).
To verify that these are in fact RecG and PriA, peak fractions were subjected to electrophoresis, transferred to membranes and probed as before. The results show that both RecG and PriA are present as bands of the expected sizes were detected in the western and biotin blots, respectively (Fig. 7E). Finally, the peak fraction from the nickel column gradient was applied to a gel filtration column to determine whether the two helicases were present as an aggregate or as a dimer. The predicted molecular weight of the RecG/PriA complex is 162 kDa. We observed the proteins eluting as a sharp peak with a molecular weight of 145 kDa as calculated from the Kav value (data not shown). SDS-PAGE analysis shows that both helicases were present in this peak with their identities confirmed by blots (Fig. 7D and E; GF lanes). The calculated MW of the complex in the peak is slightly smaller than expected and may be due to the proteins interacting with the superose resin or possibly to the fact that RecG is more compact in solution than expected (Sun et al. 2015).
DISCUSSION
The primary conclusion of this study is that SSB forms complexes in vivo with both PriA and RecG, DNA helicases critical to the rescue and reactivation of stalled DNA replication forks. In the absence of exogenous DNA damage, SSB protein localizes these DNA helicases to the periphery of cells, possibly to the inner membrane. For this to occur, SSB binds via its C-terminus to RecG or PriA, in the absence of DNA. In addition to binding SSB separately, our results show that PriA and RecG can bind to the same SSB tetramer and also to one another. In vivo binding of SSB to RecG is consistent with genomics studies, but this is the first time that binding of SSB or RecG to PriA has been shown.
The fluorescence microscopy image analysis revealed that in the absence of DNA damage, SSB localized both PriA and RecG to the cell periphery. For this to occur, three key elements were required. First, the SSB C-terminus must be present as SSBΔC8 eliminated localization. Second, both helicases must be present since efficient localization was only observed when GFP-PriA and mcherry-RecG were coexpressed along with SSB. Third, SSB must be in excess over each helicase, a situation that mimics that of exponentially growing cells where SSB is present at 1 to 2,000 tetramers per cell, RecG is present at 7 molecules per cell and PriA is present at 30–50 enzymes per cell (Shlomai & Kornberg 1980; Meyer & Laine 1990; Lloyd 1991; Tanaka et al. 2003; Ishihama et al. 2008; Taniguchi et al. 2010). When helicases were expressed in excess over the endogenous levels of SSB, localization was not observed. Instead, when SSB is present in excess over RecG and PriA, there are now a sufficient number of SSB C-termini are available for binding, leading to helicase localization to the vicinity of the inner membrane. Membrane localization of repair enzymes is not without precedent. A recent study has demonstrated that following its synthesis, UmuC is sequestered at the inner cell membrane. Its release into the cytosol requires the RecA* nucleoprotein filament-mediated cleavage of UmuD to UmuD′ (Robinson et al. 2015). Support for the implication of membranes in SSB function comes from a previous study identifying the ssb-3 allele (Schmellik-Sandage & Tessman 1990). This mutation (G15D) renders the cell extremely sensitive to UV irradiation, with a survival curve similar to that for a ΔrecA mutation. Ssb-3 also leads to an alteration in membrane permeability. It has been proposed that as this residue lies at the juncture between the first hydrophobic and a hydrophilic domains, that hydrophobic interactions may be perturbed by this mutation (Meyer & Laine 1990). It is conceivable that this may be the membrane binding site of SSB. Work is currently in progress to test this possibility.
Localization of RecG and PriA to the vicinity of the inner membrane by SSB in the absence of exogenous DNA damage implies two things. First, the SSB-helicase complex(es) could be the storage form of these enzymes. If this model is correct then when ssDNA regions become exposed due to the replication machinery stalling and/or dissociating from the DNA, SSB can rapidly target helicases to these exposed regions. Consistent, in those cells where SSB was over-expressed relative to helicases, foci were observed in approximately 12% of cells examined. This number of foci would be consistent with SSB targeting helicases to sites of fork stalling and their number are consistent with the predicted number of forks requiring rescue in an exponentially growing cell culture (Cox et al. 2000). Second, if these are the storage forms of the helicases, then their complexes with SSB should not contain DNA. Our data from the treatment of purified SSB-helicase complexes with nucleases demonstrates that DNA was not present and consequently is not required for complex formation. Furthermore, for binding to occur in the cytoplasm or at the membrane, the SSB C-termini cannot be sequestered but must instead be available. Support for the availability of the SSB C-termini in solution comes from a recent study from the Dixon laboratory (Su et al. 2014). They demonstrated that in solution in the absence of DNA the SSB C-terminus is highly mobile and that the last 8 residues are in fast exchange between free and bound states. However, the fraction of C-termini in the bound state was found to be low. Therefore, the exposed C-termini of SSB are available for binding to interactome partners in the absence of DNA.
The fluorescence microscopy data demonstrate that SSB targets both PriA and RecG to the vicinity of the inner membrane and that they can be observed colocalized. In addition, when these proteins are required for the rescue of a stalled fork, our data suggest that SSB is able to target both proteins to the DNA and this was visualized as focus formation. The triple protein-complex purification data SSB demonstrate that both helicases can bind to the same SSB tetramer. Further, purification of the RecG-PriA complex suggests that helicases could also bind to one another, further stabilizing the resulting three-protein complex and be a contributing factor to the combined helicase actions at stalled replication forks (Gregg et al. 2002). Support for the simultaneous presence of PriA and RecG on a fork comes from in vitro studies showing that these helicases function in a coordinated manner to generate a structure in which an arrested fork is stabilized for further repair and/or replication restart (Tanaka & Masai 2006). Once RecG is loaded, the fork is regressed into a substrate for RuvAB as demonstrated recently (Manosas et al. 2013). Alternatively, once fork processing has taken place and it is time for the replisome to be reloaded, it is the already loaded PriA (or an SSB-PriA complex) that would lead to reloading of DnaB (Xu & Marians 2003; Gabbai & Marians 2010). Alternatively, the simultaneous loading of PriA and RecG may facilitate distinct repair pathways, depending on the nature of the DNA present at the fork (Al-Deib et al. 1996; Marians 1999; Gregg et al. 2002; Xu & Marians 2003; Gabbai & Marians 2010; Rudolph et al. 2010).
Collectively the data presented herein are consistent with our model of the temporal sequence of events that transpire at a stalled DNA replication fork (Buss et al. 2008). It can now be expanded to include a role for SSB in delivery of both PriA and RecG to the fork prior to RuvAB. The high affinity of SSB for ssDNA enables RecG (and PriA) to outcompete other proteins thereby increasing the likelihood of them playing a dominant role in stalled replication fork rescue. In fact a recent study has demonstrated that SSB loads RecG onto a model fork and during the process remodels RecG (Sun et al. 2015). However, it is unclear whether the same SSB tetramer is responsible for both helicase storage and fork loading. We note that the existence of these SSB-helicase complexes at the inner membrane does not preclude the possibility that helicases may be released from their storage location resulting in sequential binding of other SSB tetramers followed by the helicases to the DNA. This may be a daunting but not impossible task as there is a 142 to 286-fold excess of SSB tetramers relative to RecG present in a single cell (Shlomai & Kornberg 1980; Meyer & Laine 1990; Lloyd 1991; Tanaka et al. 2003; Ishihama et al. 2008; Taniguchi et al. 2010). A similar argument can be made for PriA. Alternatively, storage SSB could release from the inner membrane and act as a potent helicase loader for RecG and PriA, thereby enabling these helicases to effectively outcompete other proteins vying for the ssDNA/fork regions (Dillingham et al. 2008; Kunzelmann et al. 2010). At present, studies are in progress to determine how PriA and RecG are transferred from the inner membrane to stalled replications forks.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were reagent grade, made up in Nanopure water and passed through 0.2µm pore size filters. Yeast Extract and tryptone were from Becton Dickinson and Company (MD, USA). NaCl, sucrose, tris-base, KCl, Na2HPO4, NaH2PO4, EDTA, acetic acid, methanol and nickel sulfate were from J.T. Baker (NJ, USA). Ampicillin was from Fisher (NJ, USA). IPTG was from OmniPur (NJ, USA). Kanamycin, chloramphenicol, lysozyme and sodium deoxycholate were from Sigma (MO, USA). Benzonase was from Novagen (NJ, USA). Imidazole was from EMD (NJ, USA). Coomassie brilliant blue R-250 was from Bio-Rad Laboratories (CA, USA). Glucose was from Mallinckrodt (KY, USA). Nonidet P40 was from USB (OH, USA). Cross-linked agarose for the 1 and 5 mL HisTrap FF columns were from GE Healthcare Life Sciences (NJ, USA). Cross-linked agarose for the 25 mL nickel column was from Life Technologies (CA, USA). In-Fusion® HD Cloning Plus was from Clontech (CA, USA). D-biotin was from AMRESCO (OH, USA). Pierce® Monomeric Avidin UltraLink® Resin was from Thermo Scientific (IL, USA). 4–20% Criterion™ TGM™ Gel, 12+2 wells, 45 µl #567–1093 was from Bio-Rad Laboratories (CA, USA). Tuner (λDE3) cells were from Novagen (WI, USA). pET-15b, pET-21a(+), pET-28a(+) and pDuet vectors were from Novagen (WI, USA). pPAL7 supercoiled vector was from Bio-Rad Laboratories (CA, USA). Profinity eXact resin-placed 5 mL Bio-Scale cartridge (CA, USA). Restriction endonucleases were from New England BioLabs Inc. (MA, USA). The molecular weight calibration kit was from GE Healthcare biosciences. The anti-his tag antibody was from Qiagen and streptavidin conjugated to alkaline phosphatase was from Bio-Rad.
RecG cloning
A plasmid containing N-terminal linker-recG clone was a gift from GS Briggs and RG Lloyd (details to be published elsewhere). To create N- and C-terminal his6-linker-recG, the linker-recG gene from this vector was amplified by PCR and inserted between the NdeI and BamHI sites, and separately, the NcoI and HindIII sites, of pET-28a(+) vector using Infusion cloning. Positive clones were identified by restriction enzyme mapping.
PriA cloning
A pET-15b plasmid overexpressing PriA was obtained from Dr. Ken Marians (Sloan Kettering). To create N-terminal histidine tagged PriA construct, The ApaI and EcoRI fragment containing PriA was sub-cloned from pET-15b into pET-28a(+) vector. C-terminal histidine tagging of PriA was constructed by PCR amplification and infusion cloning into pET-28a(+) vector between NcoI and HindIII sites. C-terminal biotinylation of PriA was achieved by inserting a biotinylation sequence between HindIII and XhoI sites into pET-28a(+) vector (Schatz 1993). To create low copy number his-GFP-PriA, his-GFP-PriA genes from the pET-15b vector were PCR amplified and inserted between BamHI and BglII sites of the pWKS30 vector. Positive clones were identified by restriction enzyme mapping.
SSB cloning
A pET-21a(+) plasmid overexpressing a mutant form of SSB with the last 8 C-terminal residues removed was a gift from Dr. James Keck (UW-Madison). To create N-terminal his6-ssbΔC8, the ssbΔC8 gene from this vector was amplified by PCR and inserted between the NdeI and BamHI sites of pET-28a(+) vector using Infusion cloning. N-terminal profinity tagging of ssb was achieved by Infusion cloning of the ssb gene into the pPAL7 vector. The drug maker exchange from ampicillin to chloramphenicol was achieved by inserting the PCR amplified chloramphenicol gene into the ampicillin gene at ScaI site. Positive clones were identified by restriction enzyme mapping.
Dual and triple-plasmid expression
Tuner (λDE3) competent cells were separately co-transformed with SSB and plasmids expressing RecG, PriA, RuvA, RuvB, or RuvC (Tables I and II). Expression was verified using 5 mL cultures grown at 37°C for 2 hours, IPTG was added to 100 µM or 300 µM (final), followed by an additional three hours of growth. Induced levels were evaluated using 12% SDS-PAGE. Cultures expressing SSB and the relevant test proteins were grown overnight with antibiotics and 0.2% glucose and these overnights were used to inoculate 0.1, 1 or 10 liter cultures the following day as described below.
Cell growth
A fresh overnight culture was added to LB media at a ratio of 1 to 100. Antibiotics were used at the following concentration for cell growth: 100 µg/mL of carbenicillin, 250 µg/mL of ampicillin, 25 µg/mL of kanamycin and 25 µg/mL of chloramphenicol. For 100 mL or 1 L cultures, cells were grown in flasks at 37°C with vigorous shaking (280 rpm). For 5 or 10 L cultures, cells were grown at 37°C in a 16 L fermentor (New Brunswick, NJ). When the OD600 was 0.4 to 0.6, IPTG was added to 100 µM or 300 µM (final), and grown until the culture reached the stationary phase. Cells were harvested by centrifugation at 4°C and cell pellets were resuspended in lysis buffer (50 mM Tris-HCl (pH 8.0), 20% sucrose) using 4 mL/g of cell paste. If the volume of the resuspension mixture was less than 10 mL, additional lysis buffer was added to adjust the volume to a total of 10 mL. For biotinylated proteins, final concentration of 0.5 mM D-biotin was added into LB media prior to autoclaving. The resulting mixture was stored at −20°C or stirred at 4°C overnight for lysis the following day.
Protein Purification
To lyse cells, lysozyme (1 mg/mL final), and benzonase were added (3 µl, 1 L and 10 µL for 5 L of culture, separately), and the mixture stirred for 30 minutes at 4°C. Deoxycholate was added to 0.05% final, and the mixture stirred for an additional 30 minutes. Imidazole and KCl or NaCl were added to a final concentration of 30 and 600 (high salt) or 150 (low salt) mM, respectively. The whole cell lysate was centrifuged at 37,000×G at 4°C for 1 hour. The cleared cell lysate of 1 L culture was loaded onto a 5 mL HisTrap FF column equilibrated in Binding Buffer (30 mM Imidazole; 15.4 mM Na2HPO4; 4.5 mM NaH2PO4; 600 mM KCl; (pH 7.4)). The cleared cell lysates of 5 or 10 L cell cultures were applied to a 25 mL nickel column which has similar chromatography condition to the 5mL HisTrap FF column but scaled accordingly. The nickel column was subjected to three washes sequentially: Binding Buffer (50 column volume (CV)), Binding Buffer with 0.2% NP40 (40 CV), Binding Buffer (30 CV). Proteins were eluted using a linear, imidazole gradient (30 mM to 500 mM in the same buffer). Proteins were identified by 12% SDS-PAGE. Following electrophoresis, gels were stained with Coomassie Brilliant Blue, destained and photographed. Photographed gels were quantitated using Image Quant Software (v5.2, GE Biosciences).
Purification of biotinylated protein following HisTrap FF column chromatography
Phosphate-Buffer Saline (PBS), Biotin Elution Buffer (BEB), Regeneration Buffer were prepared based on the manual (Pierce® Monomeric Avidin UltraLink® Resin Instructions). The avidin resin (5 mL) was washed, blocked and regenerated (Pierce Monomeric Avidin UltraLink Resin, Thermo Scientific, 53146). Proteins collected from the 5 mL HisTrap FF column were combined and subjected to avidin column chromatography. After loading, 10 mL of additional PBS was added to force the proteins into the resin bed. The resin was incubated at 4°C for 1 hour to enhance binding. The column was then washed with 50 mL PBS until UV trace had achieved baseline. To elute the target proteins, 50 mL BEB was added to the resin. Proteins were collected, combined and then precipitated using 50% ammonium sulfate. The protein solution was centrifuged at 40,000 rpm for 90 minutes and the resulting protein pellet was resuspended in nickel column Binding Buffer to one tenth its original volume.
Purification of Profinity-tagged protein following HisTrap FF column chromatography
Proteins eluted from the HisTrap FF column were applied directly to a 5 mL Profinity eXact column. Cell lysis and chromatography conditions were identical to that described above except sodium acetate was used instead of sodium chloride to prevent premature cleavage of the profinity tag. Buffers (Binding/Wash Buffer, Elution Buffer, Storage Buffer and Cleaning Solution) were prepared according to Profinity eXact™ Protein Purification System Instruction Manual. The column was loaded, washed and proteins were cleaved as described in the instruction manual.
Small Scale, Step-elution of proteins
Cell lysis was identical to that of the 1 L culture. The OD280 of the cleared cell lysates were measured and the same number of OD units were loaded onto each 1 mL HisTrap FF column. The columns were subjected to sequential washes: Binding Buffer (40 CV), Binding Buffer plus 0.02% NP40 (30 CV) and then Binding Buffer (20 CV). Proteins were eluted using 5, 1 mL applications of Elution Buffer containing 500 mM imidazole. The majority of the protein was eluted in the second fraction, as determined by 12% SDS-PAGE.
Nuclease treatment and radioactive Labeling of Column Fractions
Peak fractions from the nickel column were pooled and dialyzed into a Tris buffer. 1µL of PMSF treated benzonase was added to the protein sample. After incubation at 4°C for 30 minutes, the sample was applied to a 1mL nickel column. For treatment with DnaseI, fractions from the above-mentioned nickel column were pooled and dialyzed into a Tris buffer. MgCl2 and CaCl2 in the protein sample were added to a final concentration of 2.5mM and 0.1mM respectively. 2 µL DnaseI (50 U/µL, Thermo Scientific) was added to the protein sample. After incubation at 37°C for 20 minutes, the sample was subjected 1mL nickel column chromatography.
To determine if nucleic acids were present, 500 µl aliquots of fractions were dialyzed against buffer (20 mM Tris-HCl (pH 8.0); 1mM EDTA; 500 mM NaCl) to remove imidazole. Then samples of dialyzed peak fractions were labeled with γ32P-ATP as follows: 10 µl of the peak fraction was diluted to a final volume of 30 µl in 1×polynucleotide kinase (PNK) buffer, boiled for 5 minutes at 100°C and placed on ice. 2 units of shrimp alkaline phosphatase were added and the mixture incubated at 37°C for 60 minutes followed by 20 minutes at 70°C for inactivation. Inactivated samples were placed on ice, the volume adjusted to 40 µl with PNK buffer, and 10 units of PNK and 1 µl of 32P-γ-ATP added. Samples were transferred to 37°C and incubated for 60 minutes to allow labeling to occur. Next, 10 µl of stop mix containing gel loading dye, 1% SDS and 20 mM EDTA was added, samples were boiled at 100°C for 5 minutes and subjected to electrophoresis in 12% urea gels. The control DNA was a 37-mer oligonucleotide that was 5’-end labeled. Following electrophoresis, gels were placed on phosphorimager screens, exposed and scanned using a Typhoon Scanner. Quantitation of all gels was done using Image Quant Software (v5.2, GE Biosciences).
Fluorescence titrations
5nM mcherry-RecG or GFP -PriA was incubated with the maximum concentration of SSB-GFP ¼ (for mcherry-RecG) or SSB-mcherry ¼ (for GFP-PriA) possible based on the stock concentrations in a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% glycerol, 1 mM DTT, and 1.3 M (NH4)2SO4 at 3°C in a SLM Amnico-Bowman fluorescence spectrometer. Samples were diluted with a solution of 5 nM mcherry-RecG or GFP-PriA in buffer lacking SSB until FRET was not observed. GFP was excited at 458 ± 2 nm and mcherry emission was monitored at 610 ± 4 nm.
Western and Biotin blots
To determine whether proteins had been biotinylated in vivo, samples from cell extracts were subjected to electrophoresis using SDS-PAGE, and transferred to nitrocellulose membranes using standard Western blotting techniques. Instead of probing the membranes with antibodies, Streptavidin conjugated to alkaline phosphatase (Bio-Rad, cat #1703554) was used as a probe for the presence of biotin; detection was with alkaline phosphatase colour reagents used according to the instructions supplied with the kit. For histidine-tagged proteins, separate membranes were probed with the anti-his tag antibody using the instructions from the manufacturer.
Protein co-precipitation
was done as described previously (Buss et al. 2008). Reactions were done on ice in a volume of 20 µl and contained 20mM Tris-HCl (pH 7.5), 150mM NaCl, 5% glycerol, 1mM DTT, 24.9µM SSB monomers. Where present, RecG was added to a final concentration of 20µM. Following mixing, tubes were incubated on ice for 15 min, mixed with an equal volume of ammonium sulfate precipitation buffer [reaction buffer plus 2.6M NH4(SO4)] and incubated for an additional 15 min. The mixtures were subjected to centrifugation at 15,000xg at 4C° for 1 min. The supernatant was transferred to a fresh tube and the pellet was then washed three times followed by an identical centrifugation run. The final pellet was resuspended into 40µl of 2× SDS-PAGE loading, and also 10µl of the same loading buffer was added into the supernatant. Thereafter, both pellet and supernatant were boiled for 2min and 20µl of each sample subjected to electrophoresis, gels were stained with Coomassie brilliant blue, destained and photographed. Quantitation of all gels was done using Image Quant Software.
Gel filtration
Chromatography runs were done exactly as described previously except that the buffer contained 300 mM NaCl (Liu et al. 2011). 250 µl of individual Molecular Weight Markers and protein samples were applied separately to the Superose 6 10/300 GL equilibrated with buffer containing 20 mM Tris-HCl, pH8.0, 1 mM EDTA and 300 mM NaCl. The column was run using Bio-Rad Biologic Duoflow Chromatography system. A calibration curve was generated using Kav of molecular weight markers that were chromatographed in the same buffer: Ferritin (440,000 Da), Catalase (232,000 Da), Aldolase (158,000 Da), Alcohol dehydrogenase (150,000 Da), Conalbumin (75,000 Da), Ovalbumin (44,000 Da), Carbonic anhydrase (29,000 Da) and Ribonuclease A (13,700 Da). Elution volume was determined by measuring the volume of the eluent from the point of injection to the center of elution peak. The void volume (7.2 mL) of this column was estimated to be 30% of the column volume (24 mL). The Kav value of each molecular weight standards was calculated and a calibration curve was graphed with a plot of Kav vs log of molecular weight. The molecular weight of each protein sample was determined by extrapolating the Kav from the calibration curve.
Fluorescence microscopy
Overnight cultures of cells containing the relevant plasmids were used to inoculate 5mL LB cultures containing antibiotics and grown for 3 hours. Cultures were subjected to centrifugation and resuspended in 10 mM MgSO4. 10 µl of cells were deposited on poly-L-lysine treated coverslips and allowed to bind for 10 minutes. Unbound cells were removed by aspiration, followed by 2 washes with 10 mM MgSO4. Dried coverslips were placed onto a 20 µl water drop on a microscope slide, sealed with nail polish and imaged using epi-fluorescence microscopy.
Imaging was done as described previously using four second exposures (Liu et al. 2011). Captured images were analyzed using Image-Pro Plus v6.1 (Media Cybernetics) and Fiji (Schneider et al. 2012).
Supplementary Material
ACKNOWLEDGEMENTS
Work in the Bianco laboratory is supported by National Institutes of Health Grant GM100156 (to P. R. B.).
REFERENCES
- Abd Wahab S, Choi M, Bianco PR. Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J Biol Chem. 2013;288:26397–26409. doi: 10.1074/jbc.M113.500223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Deib AA, Mahdi AA, Lloyd RG. Modulation of recombination and DNA repair by the RecG and PriA helicases of Escherichia coli K-12. J Bacteriol. 1996;178:6782–6789. doi: 10.1128/jb.178.23.6782-6789.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss JA, Kimura Y, Bianco PR. RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res. 2008;36:7029–7042. doi: 10.1093/nar/gkn795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- Cadman CJ, McGlynn P. PriA helicase and SSB interact physically and functionally. Nucleic Acids Research. 2004;32:6378–6387. doi: 10.1093/nar/gkh980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, Polard P. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010;6:e1001238. doi: 10.1371/journal.pgen.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annual Review of Genetics. 2001;35:53–82. doi: 10.1146/annurev.genet.35.102401.090016. [DOI] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- Curth U, Genschel J, Urbanke C, Greipel J. In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res. 1996;24:2706–2711. doi: 10.1093/nar/24.14.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, Tibbles KL, Hunter JL, Bell JC, Kowalczykowski SC, Webb MR. Fluorescent single-stranded DNA binding protein as a probe for sensitive, real-time assays of helicase activity. Biophys J. 2008;95:3330–3339. doi: 10.1529/biophysj.108.133512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoh A, Iwasaki H, Ishioka K, Shinagawa H. ATP-dependent resolution of R-loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction-specific helicase. EMBO J. 1997;16:203–209. doi: 10.1093/emboj/16.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbai CB, Marians KJ. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg A, McGlynn P, Jaktaji R, Lloyd R. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol Cell. 2002;9:241–251. doi: 10.1016/s1097-2765(02)00455-0. [DOI] [PubMed] [Google Scholar]
- Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W, Zimmermann P. Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Adv Bioinformatics. 2008;2008:420747. doi: 10.1155/2008/420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, Schmidt T, Rappsilber J, Mann M, Hartl FU, Kerner MJ, Frishman D. Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics. 2008;9:102. doi: 10.1186/1471-2164-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Tomizawa J. Initiation of replication of plasmid ColE1 DNA by RNA polymerase, ribonuclease H, and DNA polymerase I. Cold Spring Harb Symp Quant Biol. 1979;1(43 Pt):409–417. doi: 10.1101/sqb.1979.043.01.047. [DOI] [PubMed] [Google Scholar]
- Iype L, Wood E, Inman R, Cox M. RuvA and RuvB proteins facilitate the bypass of heterologous DNA insertions during RecA protein-mediated DNA strand exchange. J Biol Chem. 1994;269:24967–24978. [PubMed] [Google Scholar]
- Jones JM, Nakai H. Duplex opening by primosome protein PriA for replisome assembly on a recombination intermediate. Journal of Molecular Biology. 1999;289:503–516. doi: 10.1006/jmbi.1999.2783. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC. Initiation of genetic recombination and recombination-dependent replication. Trends in Biochemical Sciences. 2000;25:156–165. doi: 10.1016/s0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- Kozlov AG, Cox MM, Lohman TM. Regulation of single-stranded DNA binding by the C termini of Escherichia coli single-stranded DNA-binding (SSB) protein. J Biol Chem. 2010a;285:17246–17252. doi: 10.1074/jbc.M110.118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AG, Jezewska MJ, Bujalowski W, Lohman TM. Binding specificity of Escherichia coli single-stranded DNA binding protein for the chi subunit of DNA pol III holoenzyme and PriA helicase. Biochemistry. 2010b;49:3555–3566. doi: 10.1021/bi100069s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann S, Morris C, Chavda AP, Eccleston JF, Webb MR. Mechanism of interaction between single-stranded DNA binding protein and DNA. Biochemistry. 2010;49:843–852. doi: 10.1021/bi901743k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO Journal. 2007;26:4239–4251. doi: 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Choi M, Stanenas AG, Byrd AK, Raney KD, Cohan C, Bianco PR. Novel, fluorescent, SSB protein chimeras with broad utility. Protein Sci. 2011;20:1005–1020. doi: 10.1002/pro.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG. Conjugational recombination in resolvase-deficient ruvC mutants of Escherichia coli K-12 depends on recG. J Bacteriol. 1991;173:5414–5418. doi: 10.1128/jb.173.17.5414-5418.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RG, Sharples GJ. Dissociation of synthetic Holliday junctions by E. coli RecG protein. EMBO J. 1993;12:17–22. doi: 10.1002/j.1460-2075.1993.tb05627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annual Review of Biochemistry. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- Manosas M, Perumal SK, Bianco P, Ritort F, Benkovic SJ, Croquette V. RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun. 2013;4:2368. doi: 10.1038/ncomms3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ. PriA: at the crossroads of DNA replication and recombination. Progress in Nucleic Acid Research & Molecular Biology. 1999;63:39–67. doi: 10.1016/s0079-6603(08)60719-9. [DOI] [PubMed] [Google Scholar]
- Marians KJ. PriA-directed replication fork restart in Escherichia coli. Trends in Biochemical Sciences. 2000a;25:185–189. doi: 10.1016/s0968-0004(00)01565-6. [DOI] [PubMed] [Google Scholar]
- Marians KJ. Replication and recombination intersect. Current Opinion in Genetics & Development. 2000b;10:151–156. doi: 10.1016/s0959-437x(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Marians KJ. Mechanisms of replication fork restart in Escherichia coli. Philos Trans R Soc Lond B Biol Sci. 2004;359:71–77. doi: 10.1098/rstb.2003.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Arai K. DnaA- and PriA-dependent primosomes: two distinct replication complexes for replication of Escherichia coli chromosome. Frontiers in Bioscience. 1996;1:d48–d58. doi: 10.2741/a115. [DOI] [PubMed] [Google Scholar]
- McGlynn P, Lloyd R. RecG helicase activity at three- and four-strand DNA structures. Nucleic Acids Res. 1999;27:3049–3056. doi: 10.1093/nar/27.15.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Lloyd R. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002a;18:413–419. doi: 10.1016/s0168-9525(02)02720-8. [DOI] [PubMed] [Google Scholar]
- McGlynn P, Lloyd R. Replicating past lesions in DNA. Mol Cell. 2002b;10:700–701. doi: 10.1016/s1097-2765(02)00687-1. [DOI] [PubMed] [Google Scholar]
- McGlynn P, Mahdi A, Lloyd R. Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res. 2000;28:2324–2332. doi: 10.1093/nar/28.12.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RR, Laine PS. The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev. 1990;54:342–380. doi: 10.1128/mr.54.4.342-380.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC III, Curtiss R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, SCHAECHTER M, Umbarger HE, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd. Washington, D.C.: ASM Press; 1996. [Google Scholar]
- Nurse P, Liu J, Marians KJ. Two modes of PriA binding to DNA. Journal of Biological Chemistry. 1999;274:25026–25032. doi: 10.1074/jbc.274.35.25026. [DOI] [PubMed] [Google Scholar]
- Purnapatre K, Handa P, Venkatesh J, Varshney U. Differential effects of single-stranded DNA binding proteins (SSBs) on uracil DNA glycosylases (UDGs) from Escherichia coli and mycobacteria. Nucleic Acids Research. 1999;27:3487–3492. doi: 10.1093/nar/27.17.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, McDonald JP, Caldas VE, Patel M, Wood EA, Punter CM, Ghodke H, Cox MM, Woodgate R, Goodman MF, van Oijen AM. Regulation of Mutagenic DNA Polymerase V Activation in Space and Time. PLoS Genet. 2015;11:e1005482. doi: 10.1371/journal.pgen.1005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph CJ, Upton AL, Briggs GS, Lloyd RG. Is RecG a general guardian of the bacterial genome? DNA Repair (Amst) 2010;9:210–223. doi: 10.1016/j.dnarep.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Ryzhikov M, Koroleva O, Postnov D, Tran A, Korolev S. Mechanism of RecO recruitment to DNA by single-stranded DNA binding protein. Nucleic Acids Res. 2011;39:6305–6314. doi: 10.1093/nar/gkr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandigursky M, Mendez F, Bases RE, Matsumoto T, Franklin WA. Protein-protein interactions between the Escherichia coli single-stranded DNA-binding protein and exonuclease I. Radiat Res. 1996;145:619–623. [PubMed] [Google Scholar]
- Schatz PJ. Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Bio/Technology. 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- Schmellik-Sandage CS, Tessman ES. Signal strains that can detect certain DNA replication and membrane mutants of Escherichia coli: isolation of a new ssb allele, ssb-3 . J Bacteriol. 1990;172:4378–4385. doi: 10.1128/jb.172.8.4378-4385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich S, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- Sharples G, Lloyd R. Resolution of Holliday junctions in Escherichia coli: identification of the ruvC gene product as a 19-kilodalton protein. J Bacteriol. 1991;173:7711–7715. doi: 10.1128/jb.173.23.7711-7715.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shereda RD, Bernstein DA, Keck JL. A central role for SSB in Escherichia coli RecQ DNA helicase function. Journal of Biological Chemistry. 2007;282:19247–19258. doi: 10.1074/jbc.M608011200. [DOI] [PubMed] [Google Scholar]
- Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol. 2008;43:289–318. doi: 10.1080/10409230802341296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomai J, Kornberg A. A prepriming DNA replication enzyme of Escherichia coli. I. Purification of protein n’: a sequence-specific, DNA-dependent ATPase. Journal of Biological Chemistry. 1980;255:6789–6793. [PubMed] [Google Scholar]
- Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Slocum SL, Buss JA, Kimura Y, Bianco PR. Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J Mol Biol. 2007;367:647–664. doi: 10.1016/j.jmb.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XC, Wang Y, Yagi H, Shishmarev D, Mason CE, Smith PJ, Vandevenne M, Dixon NE, Otting G. Bound or free: interaction of the C-terminal domain of Escherichia coli single-stranded DNA-binding protein (SSB) with the tetrameric core of SSB. Biochemistry. 2014;53:1925–1934. doi: 10.1021/bi5001867. [DOI] [PubMed] [Google Scholar]
- Sun Z, Tan HY, Bianco PR, Lyubchenko YL. Remodeling of RecG Helicase at the DNA Replication Fork by SSB Protein. Sci Rep. 2015;5:9625. doi: 10.1038/srep09625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suski C, Marians KJ. Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell. 2008;30:779–789. doi: 10.1016/j.molcel.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Masai H. Stabilization of a stalled replication fork by concerted actions of two helicases. Journal of Biological Chemistry. 2006;281:3484–3493. doi: 10.1074/jbc.M510979200. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Taniyama C, Arai K, Masai H. ATPase/helicase motif mutants of Escherichia coli PriA protein essential for recombination-dependent DNA replication. Genes to Cells. 2003;8:251–261. doi: 10.1046/j.1365-2443.2003.00630.x. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S, Mahdi A, Lloyd R. The RecG branch migration protein of Escherichia coli dissociates R-loops. J Mol Biol. 1996;264:713–721. doi: 10.1006/jmbi.1996.0671. [DOI] [PubMed] [Google Scholar]
- Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- Xu L, Marians KJ. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell. 2003;11:817–826. doi: 10.1016/s1097-2765(03)00061-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.