

Abstract
Cockayne syndrome (CS) is a premature aging disorder characterized by developmental defects, multisystem progressive degeneration, and sensitivity to ultraviolet light. CS is divided into two primary complementation groups, A and B, with the CSA and CSB proteins presumably functioning in DNA repair and transcription. Using laser microirradiation and confocal microscopy, we characterized the nature and regulation of the CS protein response to oxidative DNA damage, double-strand breaks (DSBs), angelicin monoadducts, and trioxsalen interstrand crosslinks (ICLs). Our data indicate that CSB recruitment is influenced by the type of DNA damage, and is most rapid and robust as follows: ICLs > DSBs > monoadducts > oxidative lesions. Transcription inhibition reduced accumulation of CSB at sites of monoadducts and ICLs, but did not affect recruitment to (although slightly affected retention at) oxidative damage. Inhibition of histone deacetylation altered the dynamics of CSB assembly, suggesting a role for chromatin status in the response to DNA damage, whereas the proteasome inhibitor MG132 had no effect. The C-terminus of CSB, and in particular its ubiquitin-binding domain, were critical to recruitment, while the N-terminus and a functional ATPase domain played a minor role at best in facilitating protein accumulation. Although the absence of CSA had no effect on CSB recruitment, CSA itself localized at sites of ICLs, DSBs and monoadducts, but not oxidative lesions. Our results reveal molecular components of the CS protein response and point to a major involvement of complex lesions in the pathology of CS.
Keywords: Cockayne syndrome, CSA/ERCC8, CSB/ERCC6, DNA Damage Response, DNA Repair
Introduction
Cockayne syndrome (CS) is a premature aging disorder that is inherited in an autosomal recessive manner 1-3. The prevalence of CS is 2-3 per one million live births. As predominant clinical features, CS patients display developmental failure, such as loss of body weight and short stature; severe multisystem progressive degeneration, including microcephaly, hearing loss and nervous system defects; and also marked photosensitivity. The various pathologies, which can vary in severity depending on the nature of the mutation and presumably other genetic and nongenetic factors, give rise to premature aging features and eventually lead to a shorter life span, which is why CS is often referred to as a segmental progeria.
CS is comprised of two major complementation groups: CSA and CSB 4, 5. Roughly two-thirds of the patients have mutations in the CSB gene 6. CS is subdivided into three primary subtypes [type I (moderate), II (severe) and III (mild)] according to the extent and onset of the disease 7. The phenotypes of CSA or CSB patients largely overlap, although the disease severity seems to be disparate. In particular, over half of the CSB (ERCC6) mutations (56%) cause the most severe forms of CS, namely type II, as well as the pediatric disorder cerebro-oculo-facio-skeletal (COFS), whereas CSA (ERCC8) mutations (75%) appear to be primarily involved in CS type I, which is a more moderate form of the disease, although the onset of symptoms still arise in early childhood.
The 396 amino acid (44 kDa) CSA protein consists almost entirely of WD40 repeat motifs, seven in total, which are typically involved in coordinating interactions among multiprotein complexes 4, 8. CSA, which itself harbors no known enzymatic activity, appears to be a component of a predicted ubiquitin E3 ligase complex comprised of CUL4, RBX1 and DDB1 9. The CSB protein of 1493 amino acids (168 kDa) is a member of the SWI2/SNF2 family of chromatin remodelers, and harbors conserved motifs for ATP binding and hydrolysis in the central portion of the polypeptide 10. Consistently, CSB is a DNA-dependent ATPase, with potential roles in chromatin reorganization. Notably, the N- and C-terminal regions of CSB are less well defined structurally and functionally, although they are thought to impart enzymatic and protein interaction specificity. Evidence indicates that the biological contributions of CSA and CSB are likely in transcription regulation and/or DNA repair.
Early work indicated that CSA and CSB participate in transcription-coupled nucleotide excision repair (TC-NER). In particular, CS patient cells display (i) hypersensitivity to ultraviolet(UV) light, which induces bulky DNA adducts, namely cyclobutane pyrimidine dimers (CPD) and pyrimidine (6-4) pyrimidone photoproducts, and (ii) failure to recover RNA synthesis following irradiation 11. In TC-NER, arrest of RNA polymerase II (RNAPII) at the site of the blocking lesion (e.g., a UV photoproduct) results in a biochemical response that involves CS protein recruitment, followed by assembly of NER factors, such as the nuclease complex ERCC1/XPF 12. Repair is subsequently carried out, before restoration of RNA transcription. Studies have documented that CSA- and CSB-deficient cells exhibit a lack of preferential repair of CPDs in actively transcribed genes, consistent with a failed TC-NER response 13, 14. TC-NER appears to also extend to bulky adducts generated by cisplatin, a compound capable of forming both intrastrand and interstrand DNA crosslinks 15.
In addition to the defects reported in the repair of helix-distorting, transcription-blocking lesions, work has revealed functions of CSA and CSB in the response to oxidative DNA damage. For example, studies have found, at least in some cases, that CS mutant cells are hypersensitive to oxidizing agents, such as γ-irradiation, paraquat and potassium bromate 16-18, and accumulate oxidative base lesions, including 8-oxoguanine and formamidopyrimidines 19, 20. Moreover, the CSB protein has been shown to communicate with the base excision repair (BER) pathway, a major system for coping with oxidative lesions, via functional interactions with the DNA glycosylases NEIL1 21 and NEIL2 22, and the apurinic/apyrimidinic endonuclease APE1 23. Whether the contribution of CSB to BER is transcription-associated or is part of a general genome response is presently under debate.
Besides functions in NER- and BER-related pathways, studies have identified roles for the CS proteins in the repair of complex lesions, such as DNA interstrand crosslinks (ICLs) and double-strand breaks (DSBs). In particular, deficiencies in CSA or CSB result in hypersensitivity to crosslinking agents, such as cisplatin, mitomycin C (MMC) or psoralen/UVA 24-27. In addition, reporter assays with MMC or cisplatin ICL substrates found that CSB-deficient cells exhibit decreased reactivation activity, indicative of failed DNA damage removal 25, 28. Moreover, our data indicate that CSB coordinates the resolution of ICLs, possibly in a transcription-associated repair mechanism involving an interaction with the 5′-3′ exonuclease SNM1A 24. Finally, it has recently been shown that CSB modulates the resolution of DSBs via homologous recombination, possibly in a transcription-associated process that engages RAD51C and RAD52, and checkpoint activation 29, 30. Studies suggest that the accumulation of DNA damage in CS cells may promote mitochondrial dysfunction via a hyperactive PARP1 response 31.
The combination of high resolution live-cell imaging and the ability to generate localized DNA damage within the nucleus has helped to reveal important insights into the involvement of proteins in the DNA damage response. For example, established confocal microscopy/microirradiation systems have uncovered that CSB accumulates at sites of UV photoproducts, ICLs and oxidative DNA damage 24, 32. Exploiting an in-house technology that integrates laser microirradiation and confocal microscopy, we aimed to define the elements that regulate CSA or CSB recruitment and retention at sites of enriched localized DNA damage, with a current emphasis on CSB.
Results
System for introducing localized DNA damage & monitoring protein responses
Over the past several years, we have developed a platform that integrates laser microirradiation and confocal microscopy to introduce targeted DNA damage and visualize protein recruitment/retention 24, 33-36. Our technology uses a 365 nm laser, which depending on the power employed and the activatable agent included in the culture medium, has the capacity to introduce mainly oxidative DNA damage or more complex lesions. In particular, we have described previously that “low” laser alone (1.7%) can activate a response by CSB, where recruitment is abrogated by the addition of the anti-oxidant N-acetylcysteine (NAC) to the medium 24. These results indicate that the major lesions created at 1.7% laser alone consist of predominantly oxidative modifications. In addition, our recent work found that CSB is recruited to sites of trioxsalen + 1.7% laser, and that this response is not affected by the addition of NAC, indicating that the predominant modification under these conditions is the trioxsalen-induced ICL species 24. Finally, prior work using angelicin + 2.2% laser - a trioxsalen analog designed to generate only monoadducts and not ICLs - was found to promote recruitment of a number of NER proteins, including the recognition factors XPA and XPC, consistent with the major DNA product being a bulky, helix-distorting substrate for the pathway 36.
To more thoroughly define the nature of the DNA damage created under our treatment conditions (see below), we visualized YFP-tagged XRCC1, γH2AX and 53BP1 accumulation within the nucleus of laser microirradiated cells (Figure 1), thereby assessing markers for oxidative damage, complex lesions and DSBs, respectively. Our studies revealed a robust recruitment of the BER-related protein XRCC1 at 1.7 and 2.2% laser alone, consistent with the formation of oxidative DNA damage (Figure 1A). Moreover, exposure of cells to NAC prior to microirradiation greatly suppressed the XRCC1 response (Figure 1A), supporting the prior evidence that the major lesions under this treatment condition are oxidative modifications 24. γH2AX foci were observed at sites of angelicin + laser, trioxsalen + laser and high laser alone (5.5%), but not low laser alone (1.7 or 2.2%), consistent with the three former situations generating complex lesions, such as bulky monoadducts, ICLs and DSBs, respectively (Figure 1B). 53BP1 accumulation was detected at sites of high laser alone, consistent with prior studies suggesting the production of DNA DSBs in this scenario 37, and trioxsalen + laser (Figure 1B), implying the creation of DSBs or a specific role for 53BP1 in the response to ICLs 38. Thus, in light of the current findings, we expect the five different treatment conditions employed herein to produce or enrich for the following types of DNA damage: 1.7% or 2.2% laser alone = oxidative DNA damage (base lesions & single-strand breaks); 5.5% laser alone = complex lesions, such as DSBs; angelicin + 2.2% laser = bulky monoadducts; and trioxsalen + 1.7% laser = ICLs.
Figure 1.
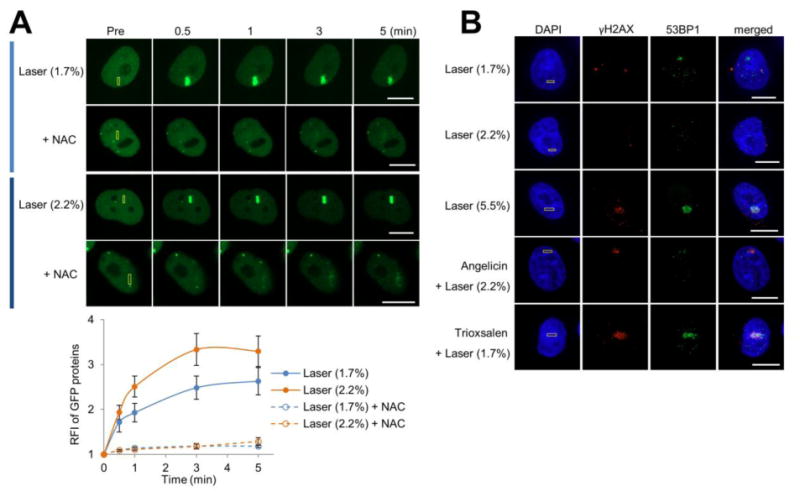
DNA damage marker profile at the different localized treatment scenarios. (A) XRCC1 recruitment to localized DNA damage, with or without NAC treatment. (Top) pXRCC1-EYFP was transfected into HeLa cells, and the indicated region (yellow box) was laser irradiated as specified: 1.7 or 2.2% laser alone. Shown are representative images of unirradiated cells (Pre), and the XRCC1 response at 0.5, 1, 3, and 5 min post-laser irradiation. Bar; 10 μm. (Bottom) Quantification of the XRCC1 response to localized DNA damage in the presence or absence of NAC. The graph reports the RFI of YFP-tagged XRCC1 at the microirradiated area relative to unirradiated (background) parts of the nucleus. Each data point is derived from a total of at least 8 independent cells, from three independent experiments. Error bars indicate SEM. (B) Accumulation of γH2AX and 53BP1 under specific DNA damage scenarios. The indicated region (yellow box) in HeLa cells was laser irradiated as specified: 1.7%, 2.2% or 5.5% laser alone, or angelicin + laser (2.2%) or trioxsalen + laser (1.7%). Cells were fixed at 10 min post-laser irradiation and stained for γH2AX, 53BP1 and DAPI. Shown are representative immunofluorescence images of γH2AX and 53BP1. Bar; 10 μm.
Response profile of CSB is determined by the nature of the DNA damage
To characterize the recruitment and retention of the CS proteins to localized DNA damage in live cells, we initially focused on the response of CSB to the different treatment scenarios outlined above, following transfection of pCSB-GFP into HeLa cells (Figure 2). Our studies reveal that CSB-GPF (confirmed to be functionally active by complementation of the UV irradiation sensitivity of CS1AN CSB mutant cells; Figure S1) responds to the lower laser doses (1.7% and 2.2%) in a similar manner, consistent with these two conditions generating a comparable spectrum of DNA alterations, likely oxidative damage. In the situation involving 5.5% laser exposure, which generates a detectable level of γH2AX or 53BP1 foci, presumed to represent DNA DSBs or other complex lesions (see above), the CSB response was comparatively more rapid and robust, particularly within the first 2 min after DNA damage induction. In conditions involving angelicin + 2.2% laser, CSB accumulated to a much greater degree relative to the 2.2% laser alone. Of all the treatment conditions, the recruitment of CSB was most rapid and pronounced to sites of trioxsalen/1.7% laser-induced ICLs, although the dispersement from the damage was also more swift. Collectively, the data indicate that the different treatment conditions generate distinct DNA damage profiles, and that the nature of the damage influences the kinetics and degree of CSB recruitment, with more complex lesions, such as ICLs, DSBs and bulky monoadducts, promoting a more robust, and often more rapid, response than simple oxidative modifications (Figure 2B).
Figure 2.
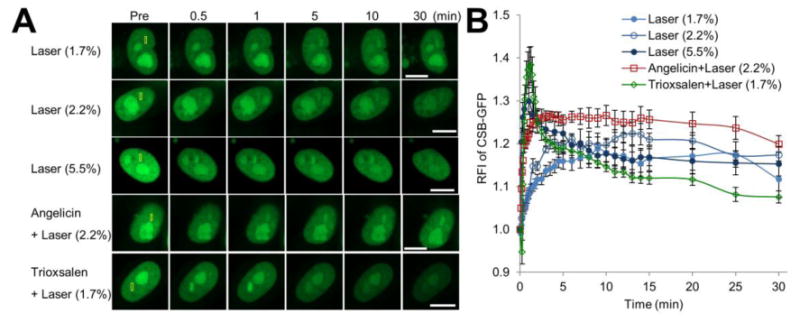
CSB accumulation at sites of DNA damage is influenced by the nature of the modification. (A) CSB recruitment and retention at localized DNA damage. pCSB-GFP was transfected into HeLa cells, and the indicated region (yellow box) was laser irradiated as specified: 1.7%, 2.2% or 5.5% laser, or angelicin + laser (2.2%) or trioxsalen + laser (1.7%). Shown are representative images of unirradiated cells (Pre), and the CSB response at 0.5, 1, 5, 10 and 30 min post-laser irradiation. Bar; 10 μm. (B) Quantification of the CSB response to localized DNA damage. The graph reports the RFI of CSB-GFP at the microirradiated area relative to unirradiated (background) parts of the nucleus. Each data point is derived from a total of at least 12 independent cells, from three independent experiments. Error bars indicate SEM.
Western blot analysis revealed that in our transient-transfection experiments using HeLa cells, GFP-CSB was expressed at roughly 48-fold higher levels than the endogenous CSB protein (Figure S2A). To reduce concerns about (i) the potential complication of the high level of the transiently-expressed protein and (ii) the potential interference of the endogenous protein on the response kinetics, we examined GFP-CSB recruitment in a stably-complemented CS1AN CSB-deficient cell line (Figure S2B, C). This cell line expressed the GFP-CSB fusion protein at levels comparable to what is seen for the endogenous protein in HeLa cells (Figure S2A). Recruitment studies using the five DNA damage scenarios revealed a response profile for CSB in the complemented cell line that is similar, although varying to differing degrees in intensity and kinetics, to that observed in the transient HeLa cell experiments (compare Figure S2B, C with Figure 2).
Transcription inhibition differentially affects CSB response to distinct forms of DNA damage
Since we had previously shown that the CSB protein response to trioxsalen ICLs is suppressed by the addition of the RNAPII inhibitor, α-amanitin, to the medium 24, we examined here the effects of RNAPII transcription inhibition on recruitment of CSB to oxidative DNA damage (or 1.7% laser alone from hereon) and angelicin monoadducts. As shown in Figure 3, treatment of HeLa cells with α-amanitin prior to microirradiation significantly reduced CSB-GFP accumulation at localized DNA monoadducts, as would be expected for a prototypical TC-NER response that is initiated by RNAPII arrest at a blocking lesion. As for oxidative DNA damage, our studies indicate that the initial phase of CSB recruitment (i.e., the first ∼5 min) is not affected by RNAPII inhibition, although the total accumulation and retention of CSB appears to be dampened (Figure 3). In particular, the relative fluorescence intensity (RFI) values comparing untreated and α-amanitin treated cells reached statistical significance (set as p < 0.05) at 30 sec for angelicin monoadducts, but not until 20 min for oxidative lesions. Collectively, our data indicate that bulky monoadducts and DNA ICLs engage the CSB pathway in a transcription-associated manner, whereas initial CSB recruitment to oxidative DNA damage does not require active RNAPII transcription. However, the organization and retention of CSB at sites of oxidative DNA damage (i.e., at the later stages after damage induction) does appear to involve active RNAPII machinery.
Figure 3.
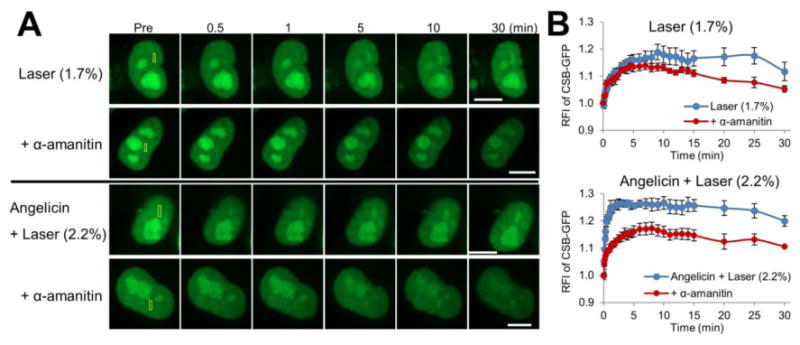
CSB recruitment to complex lesions, but not oxidative damage, is dependent on transcription. (A) CSB recruitment and retention at localized DNA damage following α-amanitin treatment. Scenarios included 1.7% laser alone or angelicin + laser, and the yellow box indicates the region of microirradiation. Shown are representative images of unirradiated cells (Pre), and the CSB response at 0.5, 1, 5, 10 and 30 min post-laser irradiation. Bar; 10 μm. (B) Quantification of the CSB response to site-specific DNA damage. See Figure 2B legend for further details, with each data point being derived from 8 independent cells from at least two experiments. Error bars indicate SEM. Data were analyzed using repeated measures two-way ANOVA followed by post hoc Bonferroni' test. 1.7% laser alone, p = 0.014 at 20 min. Angelicin + laser, p < 0.05 between 30 sec and 30 min.
Histone acetylation status, but not the proteasome mediated-degradation pathway, modulates the CSB response
Since CSB has been shown to associate with chromatin and participate in chromatin remodeling 39-42, we examined whether the status of histone acetylation would affect the recruitment or persistence of CSB at sites of DNA damage. Towards this end, following pCSB-GFP plasmid transfection into HeLa cells, but prior to laser microirradiation, cells were pre-treated with a histone deacetylase (HDAC) inhibitor, either sodium butyrate (NaB) or trichostatin A (TSA); these inhibitors prevent histone deacetylation, resulting in chromosome relaxation. We observed that CSB-GFP exhibited a trend towards increased accumulation at localized oxidative DNA damage in cells treated with either HDAC inhibitor, as compared to the untreated control cells (Figure 4). As for ICLs, we observed an interesting phenomenon where in the presence of either NaB or TSA, the initial burst of CSB-GFP recruitment (30 sec to 1 min), which is seen in the absence of the HDAC inhibitor, is lost; however, the overall accumulation and retention of CSB (5 min and later) is greater in the presence of NaB or TSA relative to the untreated control (Figure 4). These studies reveal that open chromatin by-and-large results in an apparent increase in accumulation of CSB at sites of localized DNA damage (potentially due to the increased accessibility of DNA), although an open chromatin state may reduce the need for rapid recruitment of CSB in situations involving complex lesions, such as DNA ICLs, possibly reflecting a specialized role for CSB in chromatin remodeling. We also found that pretreatment of HeLa cells with the proteasome inhibitor MG132 did not observably alter the CSB response to either oxidative DNA damage or DNA ICLs (Figure S3), suggesting that CSB recruitment and retention are not obviously influenced by proteasome-mediated degradation.
Figure 4.
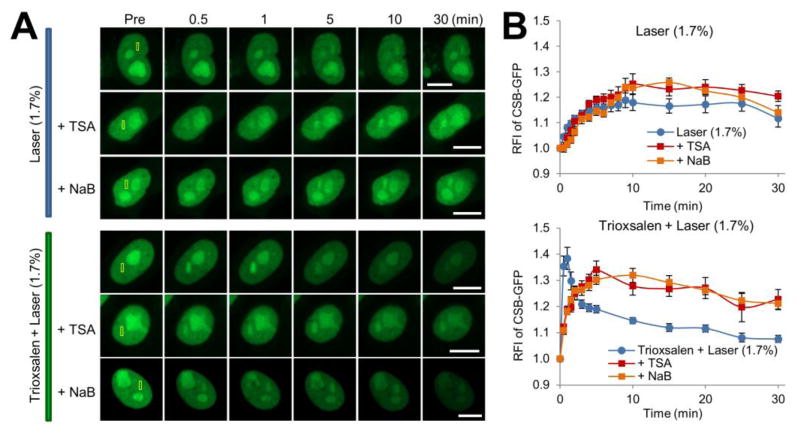
The status of histone acetylation affects the CSB protein DNA damage response. (A) CSB recruitment and retention at localized DNA damage following treatment with TSA or NaB. Scenarios included 1.7% laser alone or trioxsalen + laser, and the yellow box indicates the region of microirradiation. Shown are representative images of unirradiated cells (Pre), and the CSB response at 0.5, 1, 5, 10 and 30 min post-laser irradiation. Bar; 10 μm. (B) Quantification of the CSB response to localized DNA damage with HDAC inhibitors. Each data point (RFI) is derived from 10 independent cells, from at least three independent experiments. Error bars indicate SEM. Data of 1.7% laser alone was analyzed relative to TSA or NaB supplementation using repeated measures two-way ANOVA: with or without TSA, p=0.070, or NaB, p = 0.6015. Data of trioxsalen + laser relative to conditions that include TSA or NaB was analyzed using repeated measures two-way ANOVA followed by post hoc Bonferroni' test: for TSA, p < 0.0001 at 30 sec-1min and p < 0.05 after 5 min; for NaB, p < 0.05 between 30 sec-1 min.
The terminal regions of CSB direct its response to DNA damage
As noted earlier, the central portion of the CSB protein harbors a conserved SWI2/SNF2 helicase-like ATPase domain (Figure 5A, yellow bar). The N- and C-terminal portions of CSB, however, are less well defined, both in terms of their structural and functional properties; although there are reports that they may regulate chromatin association and the remodeling activities of the protein 40. To determine the contribution(s) of the different regions of CSB to the DNA damage response, we created three different constructs that express GFP-tagged CSB fragments: N-CSB (amino acids 1-507), M-CSB (455-1009) and C-CSB (1010-1493) (Figure 5A).
Figure 5.
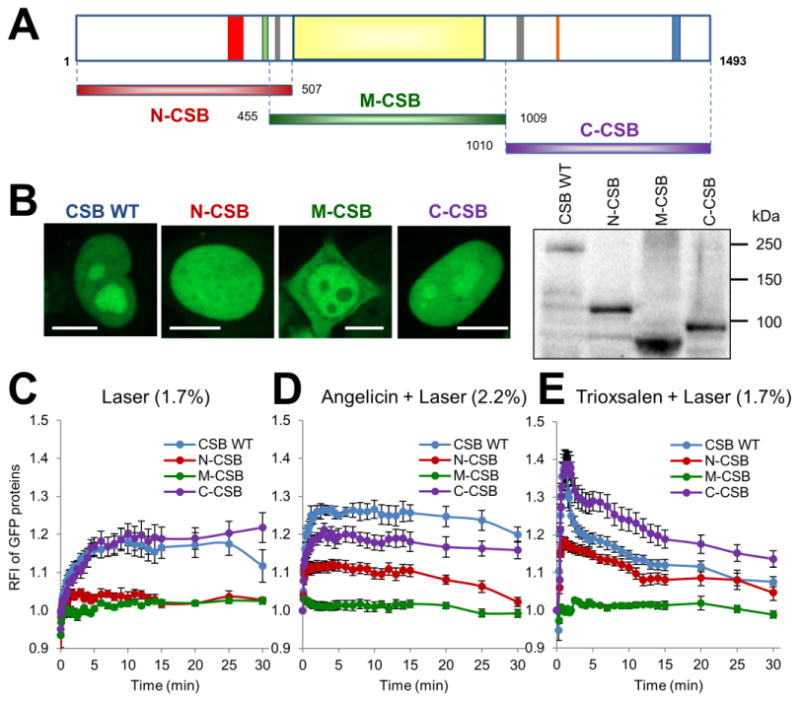
C-terminus of CSB, and to lesser extent its N-terminus, regulate recruitment to DNA damage. (A) Schematic of the domain structure of CSB, and N-terminally GFP-tagged CSB fragments used in this study. Fragments include N-CSB (red, amino acids 1-507), M-CSB (green, 455-1009) and C-CSB (purple, 1010-1493). Regions of interest in CSB are: red: acidic region (356-394), green: glycine rich region (442-450), grey: two putative nuclear localization signals (467-481, 1038-1055), orange: nucleotide binding fold (1134-1138), blue: UBD (1400-1428). The ATPase domain is identified as the light yellow central portion of the protein. (B) Localization pattern and protein expression of N-CSB, M-CSB or C-CSB in HeLa cells. (Left) Shown are representative images of GFP-tagged CSB WT (full-length CSB), N-CSB, M-CSB or C-CSB in HeLa cells. Bar; 10 μm. (Right) Shown is expression of CSB proteins as determined by western blot analysis of whole cell extracts prepared from HeLa cells transfected with pCSB-GFP, pGFP-N-CSB, pGFP-M-CSB or pGFP-C-CSB. (C-E) Quantification of the response of CSB fragments to (C) laser alone, (D) angelicin + laser or (E) trioxsalen + laser. The graphs shown report the RFI of GFP-tagged proteins at the microirradiated area relative to unirradiated background. Each data point for WT CSB, N-CSB, M-CSB and C-CSB is derived from at least 10 independent cells, from at least three independent experiments. Error bars indicate SEM.
Following transfection of the different GFP-tagged constructs into HeLa cells, we initially observed a distinct intracellular localization pattern for each protein fragment that is worthy of comment (Figure 5B). In particular, unlike full-length CSB (CSB WT), which is specifically localized to the nucleus, with nucleolar enrichment; N-CSB-GFP showed strictly pan-nuclear staining, without nucleolar enhancement; M-CSB-GFP showed a broad distribution throughout the cell, including in the cytoplasm, with noticeable nucleolar exclusion; and C-CSB-GFP exhibited a pattern more similar to CSB WT, although with perhaps a slightly reduced nucleolar concentration. These data suggest that the N- and C-terminal ends of CSB regulate localization to the nucleus, with the C-terminal portion perhaps harboring the strongest signals for nucleolar targeting, though confirmation of these conclusions is currently underway.
Regarding the response of the various CSB fragments to either oxidative DNA damage (1.7% laser alone), angelicin monoadducts or trioxsalen ICLs, we found that in all situations the C-CSB-GFP protein was most similar to CSB WT, although with slight variation in total accumulation, particularly in the latter two conditions (Figure 5C-E; Figure S4). The N-CSB-GFP protein also showed a broad response to DNA damage induction, yet in all scenarios, the response was significantly reduced in comparison to CSB WT or C-CSB-GFP. Strikingly, the M-CSB-GFP protein did not respond to any DNA damage condition, indicating that the ATPase domain on its own is not involved in substrate recognition or repair protein complex assembly, molecular events that are apparently mediated by the terminal portions of the CSB, particularly its C-terminus.
Ubiquitin-binding domain (UBD), but not ATPase function, is critical to CSB response to DNA damage
As shown above, M-CSB, which contains the ATPase domain, does not respond to DNA damage. To explicitly evaluate the contribution of the ATPase function of CSB in recruitment, we created an ATPase mutant construct, where we site-specifically introduced a Walker A motif mutation (CSBK538A) known to inactivate the ATPase function 43. Interestingly, we note that full-length CSBK538A was mainly localized in the nucleoplasm, with less accumulation in the nucleolus as compared to CSB WT (compare Figure 2A and 6A), perhaps implicating the ATPase activity in regulating the intracellular distribution of the protein. Nevertheless, recruitment of CSBK538A to oxidative lesions, angelicin monoadducts or trioxsalen ICLs was effectively identical to CSB WT (Figure 6A), indicating that the ATPase function of CSB is not essential for protein assembly at sites of DNA damage.
Figure 6.
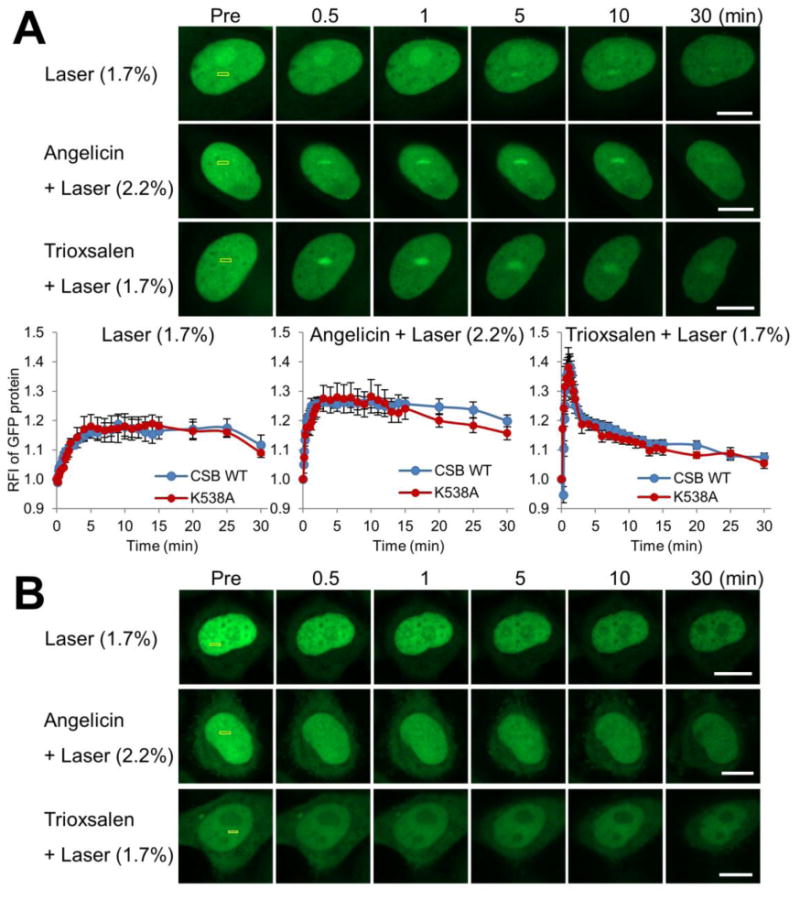
UBD of CSB, but not its ATPase activity, facilitates recruitment to localized DNA damage. (A) CSB recruitment and retention at localized DNA damage. pCSB-GFPK538A was transfected into HeLa cells, and the indicated region (yellow box) was laser irradiated under the conditions specified. (Upper) Shown are representative images of unirradiated cells (Pre), and the CSB response at 0.5, 1, 5, 10 and 30 min post-laser irradiation. (Bottom) Quantification of CSBK538A response to localized DNA damage for laser (1.7%), angelicin + laser or trioxsalen + laser. Each data point (RFI) is derived from at least 8 independent cells, from three independent experiments. Error bars indicate SEM. (B) CSB UBD mutant does not respond to site-specific DNA damage. Shown are representative images of unirradiated cells (Pre), and the response of GFP-CSB UBDmut at 0.5, 1, 5, 10 and 30 min post-laser irradiation. Bar; 10 μm.
Prior amino acid sequence analysis of CSB identified a CUE or ubiquitin-binding associated domain, termed the UBD, in the C-terminus of CSB (residues 1400 to 1428), which has the ability to bind mono- or poly-ubiquitin substrates; in addition, mutation analysis revealed its importance in TC-NER following UV irradiation 44. To determine if the UBD influences CSB recruitment to DNA damage, we created an UBD mutant expression construct (CSB UBDmut), where L1427L1428→G1427G1428 mutations were introduced into the full-length protein. First, it is noteworthy that the CSB UBDmut displays a more diffuse intracellular distribution in comparison to CSB WT, including localization in the cytoplasm, as well as reduced accumulation in the nucleolus (Figure 6B). Second, and quite strikingly, we did not detect a response of CSB UBDmut to any of the DNA damage scenarios, i.e., oxidative DNA damage, angelicin monoadducts or trioxsalen ICLs (Figure 6B). These observations imply that ubiquitin binding by CSB may not only regulate intracellular localization, but may be a critical molecular component for assembly of the protein at sites of DNA damage and repair.
CSA does not regulate CSB recruitment to DNA damage
To explore for a potential link between the CSA and CSB proteins, we determined whether CSA status influences the recruitment of CSB to localized DNA damage. In particular, CSB-GFP was expressed in CSA-deficient (CSA(-)) or CSA-corrected (CSA(+)) CS3BE cells (Figure 7A), and then monitored for accumulation at oxidative damage, angelicin monoadducts or trioxsalen ICLs. These studies revealed that the absence of CSA has no discernable effect on CSB recruitment to any of the DNA damage scenarios (Figure 7B-D), in line with prior studies that demonstrated that CSB operates upstream of CSA in the UV DNA damage response 45, 46.
Figure 7.
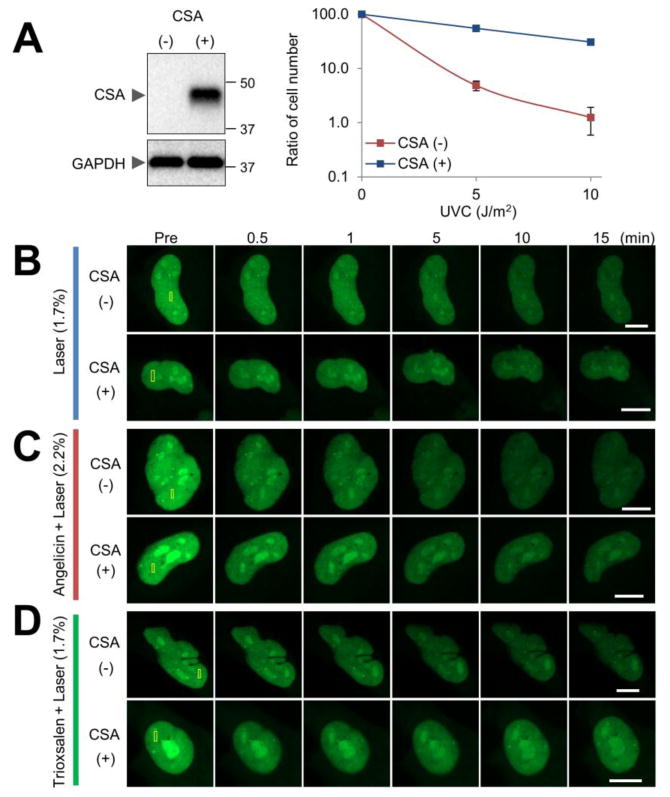
CSA does not regulate CSB recruitment to DNA damage. (A) (Left) Expression of CSA protein in CSA-corrected (CSA(+)) or CSA-deficient (CSA(-)) cells. Western blot images of the indicated whole cell extracts are shown for CSA and GAPDH (loading control). (Right) UV sensitivity of CSA(+) and CSA(-) cells. Cell viability 6 days after UVC irradiation (0, 5, or 10 J/m2) was determined by using a hemocytometer. Reported is the ratio of the cell count determined at the indicated UVC dose in comparison to the untreated control sample. Each data point represents the combined results from three independent experiments. Error bars indicate SD. (B-D) CSA-independent recruitment and retention of CSB at localized DNA damage. CSA(+) or CSA(-) cells were transfected with pCSB-GFP, and the indicated region (yellow box) was laser irradiated: (B) laser alone, (C) angelicin + laser, (D) trioxsalen + laser. Shown are representative images of unirradiated cells (Pre), and the CSB response at 0.5, 1, 5, 10 and 15 min post-laser irradiation. Bar; 10 μm.
CSA detectably recruits to complex lesions, not oxidative DNA damage
Since there are mixed reports about the coordination between CSA and CSB 47, 48, we used the same microirradiation/confocal microscopy strategy to begin to define the potential participation of GFP-tagged CSA in the various DNA damage responses. As described above for the CSB-GFP construct, we first determined the functionality of the GFP-tagged CSA fusion protein. In particular, we created two CSA constructs that expressed the protein either as an N- or C-terminal GFP-tag fusion, and monitored intracellular localization in HeLa cells. The N-terminal GFP-CSA did localize to the nucleoplasm (but not the nucleolus), but was more prominently distributed to the cyctoplasm (Figure S5A). Conversely, the C-terminal GFP-CSA was primarily detected in the nucleoplasm, as would be expected for CSA based on prior reports 46, 49-51, with little nucleolar localization (Figure S5A). Consistent with C-terminal GFP-CSA retaining normal folding and activity, we found that the fusion protein corrected the UV sensitivity of CS3BE CSA mutant cells (Figure S5B).
Using the C-terminal CSA-GFP expression system, we examined, following transfection of the construct into HeLa cells, the recruitment of CSA to the four major DNA damage scenarios: 1.7% laser alone, 5.5% laser alone, angelicin + laser, and trioxsalen + laser. We found quantitatively measureable responses of CSA to the three latter conditions involving the more complex lesions (albeit to a lesser degree and more diffuse than seen for CSB), but no quantitative response to the low dose 1.7% laser exposure (Figure 8A). As with CSB (see earlier), we examined CSA-GFP recruitment in a stably-complemented CS3BE CSA-deficient cell line (Figure S5C), and observed similar results, although with some variability in signal intensity and kinetics, to what was seen in the HeLa cell experiments (Figure 8B). Collectively, our studies imply a prominent role for CSA in the repair response to bulky monoadducts, DSBs and ICLs, but not simple oxidative DNA lesions.
Figure 8.
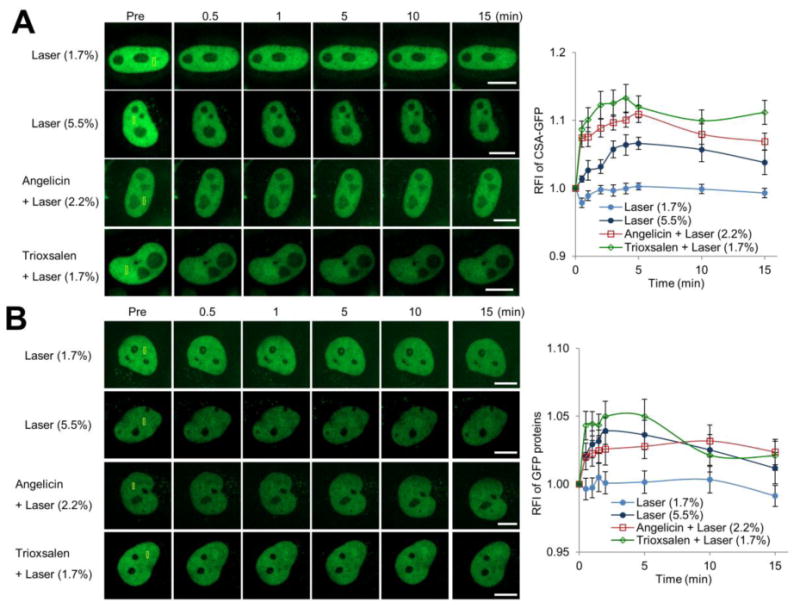
CSA accumulation is seen at more complex DNA damage, but not oxidative lesions. CSA recruitment and retention at localized DNA damage in (A) HeLa cells following transient transfection of pCSA-GFP or (B) a CSA-GFP stably expressing CSA-deficient CS3BE cell line. (Left) Shown are representative images of unirradiated cells (Pre), and the CSA-GFP response at 0.5, 1, 5, 10 and 15 min post-laser irradiation. The indicated region (yellow box) was laser irradiated under the specified conditions: 1.7% laser, 5.5% laser, angelicin + laser or trioxsalen + laser. Bar; 10 μm. (Right) Quantification of the CSA response to localized DNA damage. The graph reports the RFI of CSA-GFP at the microirradiated area relative to unirradiated background. Each data point is derived from at least 10 independent cells, from at least two independent experiments. Error bars indicate SEM.
Discussion
There has been some debate over the years regarding the involvement of the CS proteins in DNA repair. The early evidence of UV irradiation sensitivity, failed recovery of RNA synthesis following UV light exposure, and reduced excision of UV photoproducts from actively transcribed regions of the genome, established the paradigm that the CS proteins participate in a process termed TC-NER. However, it is recognized that UV light is not responsible for the major clinical phenotypes of CS patients, particularly the developmental abnormalities and neurological defects. As such, the search for potential causative DNA lesions in CS was put into motion. One possible candidate was determined to be the cyclopurines, which are bulky oxidative base modifications that can be removed via a TC-NER mechanism, and have been shown to accumulate in CSB-deficient cells 52, 53. In addition, work has suggested roles for the CS proteins in general oxidative stress resistance and damage processing, arguing that CSA and CSB function more broadly in coping with common endogenous DNA lesions. To better define the major DNA substrates of the CS proteins, and the elements that regulate their functional responses, we exploited an established laser microirradiation/confocal microscopy set-up that is capable of monitoring protein recruitment to and retention at enriched sites of DNA damage. Our studies found that: (i) CSB accumulates most rapidly and robustly at sites of ICLs, although its persistence is less sustained compared with other lesions; (ii) DSBs elicit a CSB response similar to that of ICLs, yet the initial recruitment phase is less pronounced and dispersement is more gradual; and (iii) the CSB reaction to monoadducts or oxidative DNA damage is similar in its kinetic profile, though quantitatively greater for the former. We also report on CSA assembly in live cells at more complex DNA lesions, such as ICLs, DSBs and monoadducts, but not at oxidative DNA modifications.
In TC-NER, transcription is required for efficient assembly of participating proteins at the site of DNA damage, since the response is initiated by stalling of an elongating RNAPII at a genomic obstruction 54. Consistent with ICLs being recognized, at least in part, via a transcription-associated repair mechanism, we found that α-amanitin, an RNAPII inhibitor, significantly reduced CSB accumulation at trioxsalen DNA crosslinks 24. Recent studies have also indicated that DSBs may be processed in a CSB-facilitated mechanism that involves active transcription 29. We report herein that RNAPII inhibition similarly impairs CSB recruitment to angelicin monoadducts, a phenomenon that might be predicted for these bulky modifications, and is generally consistent with prior experiments demonstrating that α-amanitin reduces TC-NER protein (e.g., CSB and SMARCA5) accumulation at UV photoproducts 55. Notably, we did not observe an effect of α-amanitin treatment on the early recruitment phase of CSB to sites of oxidative DNA damage, although there was a slight, albeit significant, reduction in total protein accumulation/retention after 20 min. Thus, our results suggest that oxidative lesions are not initially identified as part of a transcription-associated repair response involving CSB, although CSB may function in some fashion later during the process.
In our analysis, we have examined arbitrarily at least 8 cells per each recruitment/retention response, and have looked at over 400 live cell profiles (299 for CSB, 102 for CSA). In none of the individual cases have we seen any gross deviation in the response kinetics of CSA or CSB to a specific DNA damage form. As such, there is no evidence that either CSA or CSB react in a cell cycle-specific manner, implying that transcription-associated repair occurs via a preserved mechanism during the different cell cycle phases. Additionally, we found that CSB recruitment was generally increased to localized DNA damage in the presence of HDAC inhibitors, which produce an open chromatin state by preventing histone deacetylation. This finding is in itself perhaps not surprising, as it may simply imply that in situations of open chromatin there is more DNA accessible for CSB binding and assembly. What was noteworthy in our studies is the observation that the initial burst of CSB accumulation at ICLs was suppressed upon HDAC inhibition. This finding may suggest that CSB is needed to facilitate chromatin remodeling at sites of complex lesions 56, such as ICLs; whereas in situations of pre-existing open chromatin, such as with HDAC inhibitors, there is no longer a requirement for CSB remodeling activity. However, we cannot entirely exclude potential indirect effects of these small molecules on the CSB response due to broader consequences on gene transcription. Another notable result from our analysis was that the proteasome inhibitor MG132 had no effect on CSB recruitment to oxidative lesions or DNA ICLs. Prior studies have suggested that CSB is ubiquintinated, possibly by BRCA1 or the CSA-DDB1-CUL4-RBX1 complex 57, 58, and subsequently targeted for degradation by the proteasome; yet our studies do not support a model involving proteasome-mediated degradation of CSB as part of the DNA damage response.
Our CSB domain analysis clearly indicates that the N- and C-terminal portions of CSB, particularly the latter, mediate its recruitment to sites of DNA damage. Indeed, the M-CSB fragment, which spans the central SWI2/SNF2 DNA-dependent ATPase domain, did not respond to any of the DNA damage scenarios. Thus, the generally non-conserved ends of CSB either facilitate DNA binding, or more likely, participate in molecular interactions that are integral to the DNA damage response and complex assembly. We also found that mutation of an essential residue for the ATPase activity of CSB (K538A) did not alter the recruitment of the protein to oxidative DNA lesions, angelicin monoadducts or trioxsalen ICLs, indicating that targeted accumulation of CSB does not require enzymatic activity. Our finding is generally consistent with the observation that a CSB ATPase mutant protein detectably localized to sites of DSBs 29 and associated with chromatin normally following treatment of cells with the genotoxin etoposide 30. Moreover, prior reconstitution studies found that assembly of the TC-NER complex, involving proteins such as TFIIH, XPA, RPA, XPG, XPF and CSB, was independent of ATP 59. However, Lake et al. reported that a functional ATPase domain was needed for efficient chromatin association of CSB following UV irradiation 40, an observation that was substantiated by Batenburg et al. 30. Thus, it appears that the precise nature of the DNA damage or the associated chromatin modifications may dictate the need for a functional ATPase domain during the recruitment phase.
While our studies indicate that the ATPase function of CSB is not critical for its initial assembly at sites of DNA damage, it is important to emphasize that our experimental approach does not evaluate the contribution of this activity to the actual downstream repair events (i.e., complex assembly and substrate excision). Indeed, a CSB ATPase mutant protein poorly complements the UV light sensitivity of CS cells, indicating that this functionality plays a pivotal role in DNA damage removal 43, 60. Furthermore, the ATPase activity of CSB has been reported to be vital for the assembly of NER or homologous recombination proteins needed to execute resolution of DNA photoproducts or DSBs 29, 45. That said, additional work has suggested that, while the ATPase activity of CSB may be important to TC-NER and DSB repair, this enzymatic function does not influence the processing of the oxidative base lesion 8-oxoguanine 61. Future work will need to more thoroughly assess the biological significance of both the ATPase activity and the reported ATP-independent functions of CSB, including its ability to stimulate BER protein activities in vitro 22.
Significantly, we found that mutation of the UBD within the C-terminal portion of CSB essentially abolished protein recruitment to oxidative DNA lesions, angelicin monoadducts or trioxsalen ICLs. This observation points to ubiquitination at the DNA damage site as being key to the CSB protein response. Consistent with this domain operating in TC-NER, prior analysis found that mutation of the UBD in CSB impaired its complementation efficiency for UV light sensitivity and RNA synthesis recovery following UV irradiation 44. This previous work, which primarily examined intranuclear mobility following UV irradiation, and not “site-specific” accumulation, however, suggested that a UBD mutant of CSB assembles normally at UV photoproducts, but becomes trapped (or generally immobile) at the site of DNA damage, thereby interfering with the TC-NER response 44. Moreover, a recent report by Wei et al. found that a C-terminal deletion mutant of CSB, which lacks the UBD, detectably assembled at DSBs, although quantitation of the efficiency of recruitment was not provided and visibly appears less than full-length CSB 29. While the reason behind the apparent discrepancies may stem from the nature of the DNA lesion or the experimental approach, the compilation of the results identify binding of an ubiquitinated substrate by CSB as an important component of the repair response.
Importantly, previous work has supported a prominent role for ubiquitination during transcription and the TC-NER response 62. Indeed, Schwertman et al. found several NER-related proteins, including CSB, DDB2, XPC, RNAPII and UVSSA (a protein defective in UV sensitive syndrome group A), to be major components of the UV light-induced ubiquitination network 63. Besides CSB being a target of ubiquitin-ligases, and itself being able to bind mono- and poly-ubiquitin chains, reports have indicated that the ubiquitination status of RNAPII changes upon DNA damage induction and presumably transcription stalling 64. Thus, it is reasonable to speculate that it is this molecular alteration of RNAPII that contributes to CSB recruitment. However, future work will need to be conducted to identify the precise ubiquitin-ligase and ubiquitination target that mediate CSB protein assembly at sites of DNA damage. Importantly, the E3 ubiquitin-protein ligase does not appear to be the CSA-DDB1-CUL4-RBX1 complex, as we found the CSB response to be normal in CSA-deficient cells.
We report that CSA accumulates at sites of localized DNA damage in live cells, although the overall accumulation of CSA was quantitatively less than seen for CSB. Prior experiments also described a low level of recruitment of GFP-tagged CSA to sites of microirradiation in living cell nuclei 65. In our more comprehensive profiling, CSA was found to recruit only to the more complex lesions, i.e., ICLs, DSBs and DNA monoadducts, and not to oxidative DNA damage, supporting a role for the protein in the first three DNA repair responses. Considering the results in toto, and in light of the fact that CSA and CSB mutations give rise to similar clinical phenotypes, it is tempting to conclude that more complex DNA lesions, which perhaps have a greater propensity to interfere with transcription, are the major pathogenic substrates of the CS protein response, as opposed to simple oxidative damage.
Materials and methods
Synthetic oligonucleotides
All primer oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and are described in Table S1.
Plasmid constructs
Total RNA was prepared from SH-SY5Y cells using TRIzol (Life Technologies, Grand Island, NY), and first-strand cDNA was generated using high-capacity cDNA reverse transcription kit (Life Technologies). To create untagged, or N-terminal or C-terminal GFP-tagged, CSA expression plasmids, the CSA coding region was amplified from the above cDNA material by PrimeSTAR Max DNA polymerase according to the manufacturer's instruction (TaKaRa, Shiga, Japan) using a primer set of: CSA pcDNA Fw/CSA pcDNA Rv, CSA N-GFP Fw/CSA N-GFP Rv, or CSA C-GFP Fw/CSA C-GFP Rv. The PCR products were digested accordingly and subcloned into the XhoI and KpnI restriction sites of pcDNA3.1(-) (Life Technologies), pEGFP-C1 with linker 1 24, or pAcGFP-N1 (Clontech, Mountain View, CA) to create pcDNA-CSA, pEGFP-CSA, or pCSA-AcGFP, respectively. To create GFP-tagged N-CSB, M-CSB or C-CSB plasmids, the appropriate truncated CSB coding region was amplified from pCSB-GFP 24 by PrimeSTAR Max DNA polymerase and a primer set of N-CSB Fw/N-CSB Rv, M-CSB Fw/M-CSB Rv, or C-CSB Fw/C-CSB Rv, respectively. The PCR products were digested and subcloned into the XhoI and BamHI restriction sites of pEGFP with linker 1 to create pGFP-N-CSB, pGFP-M-CSB and pGFP-C-CSB. The nucleotide sequences of independent clones were confirmed at Eurofins Genomics (Huntsville, AL). The pXRCC1-EYFP expression construct has been described previously 35, 66.
Site-directed mutagenesis
The specific nucleotide changes corresponding to CSBK538A or UBD mutant L1427L1428→G1427G1428 (UBDmut) were introduced into pCSB-GFP using PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies, , Santa Clara, CA) and the primer set of CSB K538A Fw/CSB K538A Rv or CSB LL1427,8GG Fw/CSB LL1427,8GG Rv, respectively. After digestion with DpnI (TaKaRa) to remove the template plasmid, the PCR-amplified DNA was transformed into NEB 5-alpha High Efficiency Competent E. coli (New England Biolabs, Ipswich, MA), and the sequences of selected clones were confirmed as above.
Laser microirradiation and confocal imaging
Recruitment/retention studies were performed on a set-up involving a Nikon Eclipse TE2000-E microscope integrated with an UltraVIEW VoX 3D imaging system (PerkinElmer, Waltham, MA), and a NL100 nitrogen laser (Stanford Research Systems, Sunny Vale, CA) adjusted via MicroPoint ablation technology (Photonics Instruments, St. Charles, IL) to generate a wavelength of 365 nm. The basic methodology (i.e., sample preparation) for assessing protein responses has been described previously24, 33-36. Where indicated, cells were grown in the presence of 40 μM angelicin or 6 μM trioxsalen for 30 min prior to microirradiation. The other specific pretreatment conditions are specified in each figure legend, but in brief, were as follows: 50 mM NAC for 30 min, 5 μM TSA for 6 hr, 10 mM NaB for 6 hr, 10 μM MG132 for 2 hr, or 10 μM α-amanitin for 8 hr. Data were analyzed and reported as RFI of the fluorescent-tagged protein at the microirradiated area relative to unirradiated (background) parts of the nucleus, with the specific cell numbers being provided in the relevant figure legend.
Cell line construction and maintenance
HeLa cells were grown in normal culture media: high glucose Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum and 1% penicillin/streptomycin. To generate cell lines stably harboring either a vector control or a CSA expression system, the SV40-transformed CSA-deficient CS3BE cell line (Coriell Institute, Camden, NJ)67 was transfected with pcDNA-CSA or pCSA-AcGFP (or the corresponding empty vector) using JetPrime reagent (Polyplus-transfection, Illkirch, France) for 24 hr. Cell lines harboring either a vector control or a CSB expression system were similarly created by stably integrating pGFP or pCSB-GFP into the SV40-transformed CSB-deficient CS1AN cell line (Coriell Institute)5. In brief, transfected cells were cultured for two days in normal media, before being placed in media containing 400 μg/mL geneticin for a week. Individual geneticin-resistant colonies were isolated by limited dilution, and were periodically checked by western blotting to measure CSA or CSB protein expression. Stable cell lines were maintained in normal culture media with 200 pg/mL geneticin. All cell lines were grown in a cell culture incubator maintained at 5% CO2 and 37°C.
Immunostaining
After microirradiation, cells were incubated at 37°C for 10 min and fixed immediately with 10% formaldehyde in phosphate-buffered saline (PBS) at room temperature for 15 min. Fixed cells were permeabilized with 0.3% Triton X-100, 1% bovine serum albumin, 100 mM glycine and 0.2 mg/mL ethylenediaminetetraacetic acid in PBS on ice for 10 min. The cells were subsequently digested with RNase A at 37°C. Cells were blocked with Block ACE (DS Pharma. Biomedical Co., Osaka, Japan) in PBS for 1 hr at room temperature. For immunofluorescence staining, cells were incubated simultaneously for 90 min at room temperature with primary antibodies rabbit γH2AX (sc-101696, Santa Cruz Biotechnology, Dallas, TX) and mouse 53BP1 (612523, BD Transduction Laboratories, San Jose, CA), washed with PBS-T (PBS + 0.05% Tween20) three times, and incubated for 60 min at room temperature with Alexa Fluor 647 donkey anti-rabbit and Alexa Fluor 488 donkey anti-mouse IgG (A-31573 or A-21202, Thermo Fisher Scientific Inc., Waltham, MA). After washing twice with PBS-T and then PBS, coverslips were mounted onto glass slides using Vectashield mounting medium containing 4′-diamino-2-phenylindole (Vector Laboratories, Burlingame, CA), and subsequently visualized to detect the different DNA damage markers.
Western blotting
Western blotting was performed using standard procedures and the following primary antibodies: anti-GFP (ab6556), anti-CSA (ab137033) and anti-CSB (ab66598) from Abcam (Cambridge, MA), and anti-GAPDH (MAB374; Millipore, Billerica, MA). Secondary HRP-conjugated antibodies and ECL prime (GE Healthcare Bio-Sciences, Pittsburgh, PA) or SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific Inc.) were used to visualize proteins on a ChemiDoc XRS+ system (Bio-Rad Laboratories, Hercules, CA). Digitized images were obtained, quantified and processed with ImageLab version 5.1 (Bio-Rad Laboratories).
Statistical analysis
Statistical analysis was performed using Prism software (GraphPad Software, Inc., La Jolla, CA), and each method of statistical analysis is specified in the relevant figure legend. P < 0.05 was considered statistically significant.
Supplementary Material
Research Highlights.
The dynamics of CSB recruitment to sites of DNA damage are determined by the nature of the modification, with more pronounced accumulation being observed at complex lesions.
CSB recruitment to complex lesions, but not oxidative DNA damage, is largely dependent on active RNAPII transcription.
Histone acetylation, and thus chromatin status, but not the proteasome pathway, affects the CSB protein DNA damage response.
The C-terminus of CSB, particularly its ubiquitin-binding domain, but not its ATPase activity, facilitates recruitment to DNA damage.
While CSA does not regulate the CSB response, CSA accumulates at complex lesions, but not oxidative damage, pointing to complex DNA modifications as pathogenic in Cockayne syndrome.
Acknowledgments
We thank Drs. Michael Seidman and Anne Tseng (NIA) for their critical reading of the manuscript, Dr. Manikandan Paramasivam (NIA) for his helpful advice on the confocal microscopy and laser microirradiation experiments, Dr. Raghavendra Shamanna (NIA) for his helpful advice on the immunostaining studies, and Dr. Vilhelm Bohr (NIA) for his continued support of the ongoing CS investigations. This work was supported by the Intramural Research Program at the NIH, National Institute on Aging.
Abbreviations
- BER
the base excision repair
- CS
Cockayne syndrome
- CPD
cyclobutane pyrimidine dimers
- DSB
double-strand breaks
- GFP
green fluorescent protein
- HDAC
histone deacetylase
- ICLs
interstrand crosslinks
- PBS
phosphate-buffered saline
- RFI
relative fluorescence intensity
- RNAPII
RNA polymerase II
- TC-NER
transcription-coupled DNA nucleotide excision repair
- UBD
ubiquitin-binding domain
- UV
ultraviolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev. 2013;134:161–170. doi: 10.1016/j.mad.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Lanzafame M, Vaz B, Nardo T, Botta E, Orioli D, Stefanini M. From laboratory tests to functional characterisation of Cockayne syndrome. Mech Ageing Dev. 2013;134:171–179. doi: 10.1016/j.mad.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 4.Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995;82:555–564. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 5.Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 6.Laugel V, Dalloz C, Durand M, Sauvanaud F, Kristensen U, Vincent MC, et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat. 2010;31:113–126. doi: 10.1002/humu.21154. [DOI] [PubMed] [Google Scholar]
- 7.Natale V. A comprehensive description of the severity groups in Cockayne syndrome. Am J Med Genet A. 2011;155A:1081–1095. doi: 10.1002/ajmg.a.33933. [DOI] [PubMed] [Google Scholar]
- 8.Zhou HX, Wang G. Predicted structures of two proteins involved in human diseases. Cell Biochem Biophys. 2001;35:35–47. doi: 10.1385/CBB:35:1:35. [DOI] [PubMed] [Google Scholar]
- 9.Saijo M. The role of Cockayne syndrome group A (CSA) protein in transcription-coupled nucleotide excision repair. Mech Ageing Dev. 2013;134:196–201. doi: 10.1016/j.mad.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Lake RJ, Fan HY. Structure, function and regulation of CSB: a multi-talented gymnast. Mech Ageing Dev. 2013;134:202–211. doi: 10.1016/j.mad.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayne LV, Lehmann AR. Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne's syndrome and xeroderma pigmentosum. Cancer Res. 1982;42:1473–1478. [PubMed] [Google Scholar]
- 12.Spivak G, Ganesan AK. The complex choreography of transcription-coupled repair. DNA Repair (Amst) 2014;19:64–70. doi: 10.1016/j.dnarep.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 13.van der Horst GT, Meira L, Gorgels TG, de Wit J, Velasco-Miguel S, Richardson JA, et al. UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amst) 2002;1:143–157. doi: 10.1016/s1568-7864(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 14.van Hoffen A, Natarajan AT, Mayne LV, van Zeeland AA, Mullenders LH, Venema J. Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells. Nucleic Acids Res. 1993;21:5890–5895. doi: 10.1093/nar/21.25.5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones JC, Zhen WP, Reed E, Parker RJ, Sancar A, Bohr VA. Gene-specific formation and repair of cisplatin intrastrand adducts and interstrand cross-links in Chinese hamster ovary cells. J Biol Chem. 1991;266:7101–7107. [PubMed] [Google Scholar]
- 16.D'Errico M, Parlanti E, Teson M, Degan P, Lemma T, Calcagnile A, et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene. 2007;26:4336–4343. doi: 10.1038/sj.onc.1210232. [DOI] [PubMed] [Google Scholar]
- 17.Tuo J, Muftuoglu M, Chen C, Jaruga P, Selzer RR, Brosh RM, Jr, et al. The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J Biol Chem. 2001;276:45772–45779. doi: 10.1074/jbc.M107888200. [DOI] [PubMed] [Google Scholar]
- 18.de Waard H, de Wit J, Andressoo JO, van Oostrom CT, Riis B, Weimann A, et al. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol. 2004;24:7941–7948. doi: 10.1128/MCB.24.18.7941-7948.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003;17:668–674. doi: 10.1096/fj.02-0851com. [DOI] [PubMed] [Google Scholar]
- 20.Osterod M, Larsen E, Le Page F, Hengstler JG, Van Der Horst GT, Boiteux S, et al. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene. 2002;21:8232–8239. doi: 10.1038/sj.onc.1206027. [DOI] [PubMed] [Google Scholar]
- 21.Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner T, Rybanska I, et al. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J Biol Chem. 2009;284:9270–9279. doi: 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aamann MD, Hvitby C, Popuri V, Muftuoglu M, Lemminger L, Skeby CK, et al. Cockayne Syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech Ageing Dev. 2014;135:1–14. doi: 10.1016/j.mad.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM., 3rd Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35:4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyama T, Lee SY, Berquist BR, Gileadi O, Bohr VA, Seidman MM, et al. CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 2015;43:247–258. doi: 10.1093/nar/gku1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enoiu M, Jiricny J, Scharer OD. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012;40:8953–8964. doi: 10.1093/nar/gks670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002;62:4899–4902. [PubMed] [Google Scholar]
- 27.McKay BC, Becerril C, Ljungman M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene. 2001;20:6805–6808. doi: 10.1038/sj.onc.1204901. [DOI] [PubMed] [Google Scholar]
- 28.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, et al. Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand crosslinks. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei L, Nakajima S, Bohm S, Bernstein KA, Shen Z, Tsang M, et al. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1507105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batenburg NL, Thompson EL, Hendrickson EA, Zhu XD. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015;34:1399–1416. doi: 10.15252/embj.201490041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014;20:840–855. doi: 10.1016/j.cmet.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menoni H, Hoeijmakers JH, Vermeulen W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J Cell Biol. 2012;199:1037–1046. doi: 10.1083/jcb.201205149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson JY, et al. Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol. 2013;33:2212–2227. doi: 10.1128/MCB.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNeill DR, Paramasivam M, Baldwin J, Huang J, Vyjayanti VN, Seidman MM, et al. NEIL1 responds and binds to psoralen-induced DNA interstrand crosslinks. J Biol Chem. 2013;288:12426–12436. doi: 10.1074/jbc.M113.456087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berquist BR, Singh DK, Fan J, Kim D, Gillenwater E, Kulkarni A, et al. Functional capacity of XRCC1 protein variants identified in DNA repair-deficient Chinese hamster ovary cell lines and the human population. Nucleic Acids Res. 2010;38:5023–5035. doi: 10.1093/nar/gkq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM. Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J Biol Chem. 2009;284:27908–27917. doi: 10.1074/jbc.M109.029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh DK, Karmakar P, Aamann M, Schurman SH, May A, Croteau DL, et al. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–371. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vare D, Groth P, Carlsson R, Johansson F, Erixon K, Jenssen D. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA Repair (Amst) 2012;11:976–985. doi: 10.1016/j.dnarep.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, et al. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lake RJ, Geyko A, Hemashettar G, Zhao Y, Fan HY. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell. 2010;37:235–246. doi: 10.1016/j.molcel.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103:9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho I, Tsai PF, Lake RJ, Basheer A, Fan HY. ATP-dependent chromatin remodeling by Cockayne syndrome protein B and NAP1-like histone chaperones is required for efficient transcription-coupled DNA repair. PLoS Genet. 2013;9:e1003407. doi: 10.1371/journal.pgen.1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Citterio E, Rademakers S, van der Horst GT, van Gool AJ, Hoeijmakers JH, Vermeulen W. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J Biol Chem. 1998;273:11844–11851. doi: 10.1074/jbc.273.19.11844. [DOI] [PubMed] [Google Scholar]
- 44.Anindya R, Mari PO, Kristensen U, Kool H, Giglia-Mari G, Mullenders LH, et al. A ubiquitin-binding domain in Cockayne syndrome B required for transcription-coupled nucleotide excision repair. Mol Cell. 2010;38:637–648. doi: 10.1016/j.molcel.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 46.Kamiuchi S, Saijo M, Citterio E, de Jager M, Hoeijmakers JH, Tanaka K. Translocation of Cockayne syndrome group A protein to the nuclear matrix: possible relevance to transcription-coupled DNA repair. Proc Natl Acad Sci U S A. 2002;99:201–206. doi: 10.1073/pnas.012473199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 48.van Gool AJ, Citterio E, Rademakers S, van Os R, Vermeulen W, Constantinou A, et al. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997;16:5955–5965. doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Solovjeva L, Svetlova M, Sasina L, Tanaka K, Saijo M, Nazarov I, et al. High mobility of flap endonuclease 1 and DNA polymerase eta associated with replication foci in mammalian S-phase nucleus. Mol Biol Cell. 2005;16:2518–2528. doi: 10.1091/mbc.E04-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nardo T, Oneda R, Spivak G, Vaz B, Mortier L, Thomas P, et al. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci U S A. 2009;106:6209–6214. doi: 10.1073/pnas.0902113106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bradsher J, Auriol J, Proietti de Santis L, Iben S, Vonesch JL, Grummt I, et al. CSB is a component of RNA pol I transcription. Mol Cell. 2002;10:819–829. doi: 10.1016/s1097-2765(02)00678-0. [DOI] [PubMed] [Google Scholar]
- 52.Brooks PJ. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirkali G, de Souza-Pinto NC, Jaruga P, Bohr VA, Dizdaroglu M. Accumulation of (5′S)-8,5′-cyclo-2′-deoxyadenosine in organs of Cockayne syndrome complementation group B gene knockout mice. DNA Repair (Amst) 2009;8:274–278. doi: 10.1016/j.dnarep.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christians FC, Hanawalt PC. Inhibition of transcription and strand-specific DNA repair by alpha-amanitin in Chinese hamster ovary cells. Mutat Res. 1992;274:93–101. doi: 10.1016/0921-8777(92)90056-9. [DOI] [PubMed] [Google Scholar]
- 55.Aydin OZ, Marteijn JA, Ribeiro-Silva C, Rodriguez Lopez A, Wijgers N, Smeenk G, et al. Human ISWI complexes are targeted by SMARCA5 ATPase and SLIDE domains to help resolve lesion-stalled transcription. Nucleic Acids Res. 2014;42:8473–8485. doi: 10.1093/nar/gku565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 57.Wei L, Lan L, Yasui A, Tanaka K, Saijo M, Matsuzawa A, et al. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci. 2011;102:1840–1847. doi: 10.1111/j.1349-7006.2011.02037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, et al. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006;20:1429–1434. doi: 10.1101/gad.378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laine JP, Egly JM. Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J. 2006;25:387–397. doi: 10.1038/sj.emboj.7600933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brosh RM, Jr, Balajee AS, Selzer RR, Sunesen M, Proietti De, Santis L, Bohr VA. The ATPase domain but not the acidic region of Cockayne syndrome group B gene product is essential for DNA repair. Mol Biol Cell. 1999;10:3583–3594. doi: 10.1091/mbc.10.11.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Selzer RR, Nyaga S, Tuo J, May A, Muftuoglu M, Christiansen M, et al. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res. 2002;30:782–793. doi: 10.1093/nar/30.3.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson MD, Harreman M, Svejstrup JQ. Ubiquitylation and degradation of elongating RNA polymerase II: the last resort. Biochim Biophys Acta. 2013;1829:151–157. doi: 10.1016/j.bbagrm.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek AC, Laffeber C, et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat Genet. 2012;44:598–602. doi: 10.1038/ng.2230. [DOI] [PubMed] [Google Scholar]
- 64.Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci U S A. 1996;93:11586–11590. doi: 10.1073/pnas.93.21.11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mourgues S, Gautier V, Lagarou A, Bordier C, Mourcet A, Slingerland J, et al. ELL, a novel TFIIH partner, is involved in transcription restart after DNA repair. Proc Natl Acad Sci U S A. 2013;110:17927–17932. doi: 10.1073/pnas.1305009110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fan J, Otterlei M, Wong HK, Tomkinson AE, Wilson DM., 3rd XRCC1 co-localizes and physically interacts with PCNA. Nucleic Acids Res. 2004;32:2193–2201. doi: 10.1093/nar/gkh556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ridley AJ, Colley J, Wynford-Thomas D, Jones CJ. Characterisation of novel mutations in Cockayne syndrome type A and xeroderma pigmentosum group C subjects. J Hum Genet. 2005;50:151–154. doi: 10.1007/s10038-004-0228-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.