

Abstract
Small cell lung cancer (SCLC) is an extremely aggressive cancer that frequently recurs Twenty-three human SCLC lines were selected representing varied Myc status. Gene expression of lung cancer, stem-like, hedgehog pathway, and notch pathway genes were determined by RT2-PCR array and Exon 1.0 ST array. Etoposide and topotecan concentration response was examined. The IC50’s for etoposide and topotecan ranged over nearly 3 logs upon 96 hrs exposure to the drugs. Myc status, TOP2A, TOP2B and TOP1 mRNA expression or topoisomerase 1 and topoisomerase 2 protein did not account for the range in the sensitivity to the drugs. γ-secretase inhibitors, RO429097 and PF-03084014, had little activity in the SCLC lines over ranges covering the clinical Cmax concentrations. MYC amplified lines tended to be more sensitive to the bromodomain inhibitor JQ1. The Smo antagonists, erismodegib and vismodegib and the Gli antagonists, HIP1 and SEN-450 had a trend toward greater sensitivity of the MYC amplified line. Recurrent SCLC is among the most recalcitrant cancers and drug development efforts in this cancer are a high priority.
Keywords: Small Cell Lung Cancer, SCLC, SCLC gene expression, hedgehog inhibitors, bromodomain inhibitors
1. INTRODUCTION
Small cell lung cancer (SCLC) is an extremely aggressive cancer that frequently recurs after conventional cytotoxic chemotherapy. SCLC cells are small with limited cytoplasm surrounding the nuclei. The cells tend to grow as floating clusters or spheroids which are often difficult to disaggregate. While SCLC is challenging to work with in culture, it tends to grow well as xenografts. SCLC is a lung malignancyof neuroendocrine origin for which there is no effective treatment. It affects >200,000 people world-wide every year with a very high mortality rate. In the US, 13–15% of lung cancer cases are SCLC. Although initially a chemotherapy and radiation-sensitive disease, SCLC recurs rapidly and <5% of patients survive five years. There has been no change in the standard of care for SCLC for the past three decades. Treatment most often involves platinum-based combination chemotherapy, hyperfractionated thoracic radiation, and prophylactic cranial irradiation [1, 2]. SCLC has unique biology and chromosomal changes, dysregulation of tumor suppressor genes, oncogenes, and signaling pathways, upregulation of receptor tyrosine kinases, growth factors and cellular markers, and activation of early development pathways [3]. From 1977 through 1992, 126 SCLC cell lines were established from patients at the NCI-Navy Medical Oncology Branch. Extensive clinical information was available on 96 patients from whom these cell lines were established. The number of SCLC lines established from previously untreated patients with both limited and extensive stage SCLC increased during the 16 years of the study. These cell lines became and remain critically important models for the study of this deadly malignancy [4].
MYC family DNA amplification was present in 16/44 (36%) SCLC lines established from previously treated patients compared to 7/52 (11%) SCLC lines established from untreated patients. MYC DNA amplification is associated with shorter patient survival [4]. The apoptosis related gene, caspase 8, is frequently silenced in SCLC by aberrant promoter methylation. In 34 SCLC lines (12 MYC amplified), caspase 8 gene and protein expression was lost in 79%. There was also a high rate of loss of expression of CASP10, DR5, FAS and FASL in SCLC. The loss of expression of proapoptotic components was higher in MYC amplified SCLC lines and these lines were completely TRAIL resistant [5]. Array comparative genomic hybridization (aCGH) karyotype analysis of 33 SCLC tumors and 13 SCLC lines showed that SCLC tumor and line karyotypes were highly aberrant with high copy number gains detected in SCLC tumors and lines in cytogenetic bands encoding JAK2, FGFR1 and MYC family members. The copy number of these genes often exceeded 100, suggesting they represent driver alterations and drug targets in SCLC. In SCLC tumors recurrent copy number alterations were observed in 203 genes. The aCGH profile of SCLC lines and clinical SCLC specimens were similar [6]. Despite the discovery of an increasing number of MYC target genes, identification of MYC target core sets corresponding to specific cellular outcomes has proved elusive.
The highly aggressive nature of SCLC suggests that this disease may have an elevated stem cell fraction. Side population cells from the NCI-H146 or NCI-H526 SCLC lines over-expressed the following genes associated with cancer stem cells and drug resistance: CD133, ABCG2, FGF1, IGF1, MYC, SOX1/2, WNT1, angiogenesis genes, and notch and hedgehog pathways [7, 8]. Cancer may be viewed as aberrant organogenesis in which progenitor/stem cells escape dependence on niche signaling through mutation in genes such as Ptch or through activation of progenitor cell pathways. Normally, the airway epithelial uses the hedgehog pathway to repopulate after injury. Activation of the hedgehog pathway has been studied in a mouse SCLC model (mSCLC) in which Rb1 and Trp53 were deleted in the lung epithelium. mSCLC expressed hedgehog pathway components in vivo and in culture. Crossing a constitutively active allele of the hedgehog pathway member, Smoothened (Smo), into Rb1-Trp53 conditional mutant mice led to an increase in the size and number of lung nodules per mouse while Smo deletion resulted in fewer and smaller nodules. Smo and Gli1 inhibitors blocked proliferation and increased death in mSCLC. In vivo, Smo inhibition following cisplatin and etoposide treatment was effective in preventing SCLC xenograft regrowth, suggesting that hedgehog pathway inhibitors may be useful therapies [9]. Hedgehog acyltransferase (Hhat)-mediated palmitoylation, a modification critical for hedgehog signaling, is a target for Shh pathway inhibition. In cells, Hhat inhibitors blocked hedgehog palmitoylation and inhibited autocrine and paracrine hedgehog signaling [10]. SCLC is characterized by high levels of SOX2, SOX4, and SOX11. The HMG box transcription factor SOX4 involved in neuronal development is amplified and overexpressed in SCLC and may be a driver oncogene [11]. CD44highCD90+ cells from primary SCLC lines had mesenchymal morphology, increased expression of mesenchymal markers N-cadherin and vimentin, increased mRNA levels of the embryonic stem cell related genes Nanog and Oct4, suggesting the CD44highCD90+ population a good candidate for the SCLC cancer stem cells [12].
The current report examines gene expression and compound response in a series of SCLC lines with a focus on their varied expression of members of the MYC family.
2. METHODS and MATERIALS
Cell Lines
The twenty-three small cell lung carcinoma (SCLC) cell lines used in the study were purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in serum free RPMI1640 (Gibco®, Grand Island, NY) containing selenium, insulin and transferrin or media supplemented with 5% fetal bovine serum (FBS, Hyclone™, Utah, USA) and glutamine. Cells were maintained in a 5% CO2-humidified incubator at 37°C. Table 1 shows the morphology and doubling time for each cell line along with the patient prior treatment and response to treatment.
Table 1.
Shown are the morphology and doubling time for each SCLC line along with the patient prior treatment, response to treatment and Myc status.
| Cell Line | Morphology, Growth Properties | Site of Origin | Prior Radiation | Prior Chemo | Chemo Regimen | Response | Myc | Doubling Time (h) |
|---|---|---|---|---|---|---|---|---|
| NCI-H82 | epithelial, suspension, multi-cell aggregates | pleural effusion | Y | Y | CMC/VAP, VP16 | CR | cMYC | 25.5 ± 4.2 |
| NCI-H211 | suspension | bone marrow | Y | Y | CMC/VAP | PR | cMYC | 55.6 ± 6.5 |
| NCI-H446 | epithelial, mixed adherent & floaters | pleural effusion | N | Y | CMC/VAP | PR | cMYC | 41.0 ± 4.6 |
| NCI-H524 | suspension | lymph node | N | Y | CMC/VAP | PR | cMYC | 38.4 ± 0.9 |
| NCI-H1650 | epithelial, adherent | pleural effusion | N | N | AD, PL | PD | cMYC | 41.3 ± 3.3 |
| NCI-H2171 | floating clusters, suspension | pleural effusion | Y | Y | VP/PL | PR | cMYC | 48.6 ± 6.7 |
| NCI-H69 | suspension, multi-cell aggregates | pleural effusion | Y | Y | CMC/VAP, VP16/iFOS | CR | N-MYC | 51.1 ± 3.1 |
| NCI-H526 | epithelial, round clusters in suspension | bone marrow | N | N | CMC/VAP | PD | N-MYC | 35.2 ± 1.7 |
| NCI-H720 | suspension | atypical carcinoid | Unknown | Unknown | Unknown | Unknown | N-MYC | 72.4 ± 26.7 |
| NCI-H378 | epithelial, suspension, multi-cell aggregates | pleural effusion | Y | Y | CMC/VAP | PR | L-MYC | 92.8 ± 12.9 |
| NCI-H510 | epithelial,, mixed adherent & floaters | brain/adrenal | Y | Y | CMC/VAP | SD | L-MYC | 152.3 ± 40.6 |
| NCI-H889 | epithelial clusters in suspension | lymph node | N | N | VP/PL | PR | L-MYC | 61.1 ± 5.1 |
| NCI-H1963 | suspension | carcinoma | N | N | VP/PL | CR | LMYC | 43.6 ± 7.6 |
| NCI-H2107 | epithelial, clusters in suspension | bone marrow | N | N | VP/PL | PR | LMYC | 63.4 ± 8.4 |
| NCI-H128 | floating aggregates | pleural effusion | Y | Y | CMC/VAP | PR | unamp | 77.3 ± 16.9 |
| NCI-H146 | epithelial, suspension, multi-cell aggregates | bone marrow | N | Y | CMC/VAP | PR | unamp | 52.9 ± 11.9 |
| NCI-H187 | suspension | pleural effusion | N | N | CAPO, MTX | PR | unamp | 67.7± 13.6 |
| NCI-H196 | suspension | pleural effusion | Y | Y | CAPO, PCI | CR | unamp | 80.1 ± 8.4 |
| NCI-H345 | epithelial, suspension, multi-cell aggregates | bone marrow | Y | Y | CMC/VAP | CR | unamp | 65.3 ± 8.6 |
| NCI-H740 | suspension | lymph node | N | N | VP/PL | PR | unamp | 79.7 ± 9.9 |
| NCI-H841 | mixed adherent & floaters | lymph node | Y | Y | CMC/VAP | NR | unamp | 31.2 ± 3.6 |
| NCI-H865 | aggregate in suspension | pleural effusion | Y | Y | CMC/VAP | CR | unamp | 80.9 ± 6.8 |
| NCI-H1688 | epithelial, adherent | derived from liver met | N | N | VP/PL | PD | unamp | 82.7 ± 15.6 |
| NCI-H1930 | aggregate in suspension | lymph node | N | N | VP/PL | CR | unamp | 147.4 ± 22.3 |
CMC/VAP: cyclophosphamide, methotrexate; AD:adriamycin; PL: cis-platinum; iFOS: ifosfamide; VP/Pl: VP16, cis-platinum; CAPO: cyclophosphamide, Adriamycin, cis-platinum, vincristine; PCI: Prophylactic cranial irradiation; Response: CR: complete remission; PR: partial remission; PD: progressive disease; SD: stable disease; NR: No response
Concentration Response Curves
NOTCH, BET-bromodomain, Hedgehog, glutamate and topoisomerase inhibitors were obtained from the Drug Synthesis and Chemistry Branch of the Developmental Therapeutics Program, National Cancer Institute (Rockville, MD). All drugs dilutions were prepared at 1000X in DMSO and stored at −80 °C. Growth inhibition assays for small cell lung carcinoma cell lines to NOTCH, BET-bromodomain, hedgehog, glutamate and topoisomerase inhibitors were performed in 96 well plates using Tecan Robotic. Cells were exposed to eight drug concentrations for 96 hrs and ATP content (CellTiter Glo®, Promega) was used as an end point. Luminescence was measured using Tecan-100 microplate reader.
Combination Concentration Response
Carboplatin and etoposide concentration response curves were generated for the SCLC lines. Based on the IC50 values for each compound, a suitable concentration range for combination of the two agents was determined. The selected concentrations were 3.7uM carboplatin and 0.3uM etoposide. The combination of etoposide and carboplatin was treated as a single agent for the combination experiments. The third agents were examined over a concentration range based around their clinical Cmax concentration, if known. All compound exposures were simultaneous for 96 hrs with ATP content (CellTiter Glo®, Promega) as the end point. Luminescence was measured using Tecan-100 microplate reader.
RNA Extraction and Measurement of RNA Integrity
RNA from the cell pellets was extracted using Qiagen miRNeasy Mini Kit (#217004) with on column DNase digestion according to manufacturer’s instructions. The RNA integrity in terms of RIN numbers of extracted RNA was measured on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). Samples with a RIN above 8.5 were used for gene expression profiling.
Real-time reverse transcription-Polymerase Chain Reaction (RT-PCR)
Gene expression of Hedgehog, Notch, lung cancer and stem like genes in 24 SCLC cell lines was performed using 96 well custom RT2-PCR Array (Qiagen). A total of 800 ng RNA was used for cDNA synthesis and genomic DNA elimination using the RT2 easy first strand cDNA synthesis kit (Qiagen, Germantown, MD) as recommended by the manufacturer. The qRT–PCR was performed on an Applied Biosystems 7500 system using Fast SYBR green master mix (Qiagen), following the manufacturer’s recommendations. Forty PCR cycles were run in a 96 well plate RT2-PCR array. Gene expression values were normalized using housekeeping genes. Average values of each gene for 24 cell lines was subtracted from corresponding genes for each cell line and gene expression is expressed as log 2 of the value as 2^-ΔΔCT.
Western Blots
Untreated SCLC cells cultured in log phase were collected, washed with PBS and cell pellets were stored at −70°C. The cell pellets were lysed in sodium dodecyl sulfate (SDS) lysis buffer and cell lysates were prepared as described (Ref). The protein concentrations were determined by BCA Protein Assay Kit. Total proteins were fractionated using SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto PVDF membrane (Millipore, MA) for Western blot. Membranes were blocked with 5 % nonfat milk in TBST for 1h and then incubated overnight (16h) with primary antibodies on a rocker at 4 °C. The primary antibodies used were MYC antibody (CST, #5605, 1:750); MYCN antibody (SC, #56729, 1:200); MYCL antibody (SC, #790, 1:200); ASCL-1 antibody (SC, #390794, 1:500) and β-actin (Sigma, # 1:5000); TopoI antibody (BD Pharmingen, #556597, 1:1000); TopoIIa antibody (CST, #4733, 1:1000). Primary antibody was removed; membranes were washed three times with TBST for 5 min each, and probed with corresponding secondary antibody (1:2,000 dilution) for 1h at room temperature. Finally, membranes were washed 3 times with TBST and developed using Visualizer western blot detection kit from Millipore (Billerica, MA). Images were captured using the Kodak Image Station 4000MM Pro and processed using Kodak Molecular Imaging Software (Carestream Health, New Haven, CT, USA). The protein levels were normalized by β-actin.
Exon Arrays
Total RNA was extracted from samples using Qiagen miRNeasy Mini Kit (#217004) to isolate RNA including the miRNA fraction, according to manufacturer’s instructions. Agilent RIN >8.5 indicated good quality RNA for all samples. Sense strand cDNA from 100ng total RNA was fragmented and labeled using Affymetrix WT terminal labeling kit. Samples were hybridized with Human Exon 1.0 ST Arrays (Affymetrix) at 45°C, 60 rpm for 16 hrs. Arrays were washed and stained using Affymetrix Fluidics Station 450, and scanned on Affymetrix GeneChip scanner 3000 7G.
3. RESULTS
Twenty-three human SCLC lines were selected for study to represent varied Myc status. Six lines had ampified c-Myc, three lines had amplified n-Myc, five lines had amplified l-Myc and the remaining ten lines had no Myc amplification. Fourteen lines were from patients who had prior chemotherapy and/or radiation therapy and most lines were derived from samples of pleural effusion or lymph node. The generation or doubling times of the cell lines ranged from 25.5 hrs to 152 hrs (Table 1). Two NSCLC lines, A549 and NCI-H1650, were used as comparators.
Gene expression of lung cancer, stem-like, hedgehog pathway, and notch pathway genes were determined by RT2-PCR array and Exon 1.0 ST array. There was good agreement between gene expression by RT-PCR and exon array. Most of the SCLC lines express high levels of ASCL1 (Figure 1A). Generally, the SCLC lines which were c-Myc amplified were among the lowest ASCL1 mRNA expressers; however, the five c-Myc amplified SCLC lines expressed ASCL1 protein at levels similar to the other SCLC lines studied (Figure 1B). High c-Myc mRNA levels were evident in the c-Myc amplified lines by both RT-PCR and exon array, while expression of NANOG and POU5F1 (OCT4) are uniformally lower (Figure 1A). There is a broad range of expression of SOX2 mRNA with the c-Myc lines tending to be low. The three n-Myc amplified lines have detectable n-Myc protein and l-Myc was present in the five l-Myc amplified lines.
Figure 1.


Panel A Expression of genes associated with lung cancer and stem-like properties in 23 SCLC lines as determined by the 1/ΔCT value from RT-PCR or as log2 from gene expression derived from exon arrays. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). Panel B: Western blot showing the expression of c-myc, n-myc, l-myc and α-tubulin in the 23 SCLC lines.
The topoisomerase 2 inhibitor, etoposide, and the topoisomerase 1 inhibitor, topotecan, are approved for treatment of SCLC. Etoposide and topotecan concentration response was examined in the 23 SCLC lines (Figure 2A). The IC50’s for etoposide ranged from 0.003 to 10 uM and the IC50’s for topotecan ranged from 0.0015 to 1.8 uM in the 23 SCLC lines upon 96hrs exposure to the drugs. The expression of TOP1, TOP2A mRNA and TOP2B mRNA as well as topoisomerase 1 and toposiomerase 1A protein were examined in the 23 SCLC lines as well as 3 normal cell types and 2 NSCLC lines (Figure 2B). The SCLC lines expressed higher levels of TOP2A than did the normal cells and the NSCLC lines. The expression of TOP1 mRNA was relatively high in all of the cell types tested. SLFN11 expression has been associated with topoisomerase inhibitors. SLFN11 mRNA was heterogeneously expressed in the SCLC lines. However, Myc status, and TOP2A, TOP2B and TOP1 mRNA expression or topoisomerase 1 and topoisomerase 2 protein did not account for the nearly 3 log range in the sensitivity of the SCLC lines to etoposide or topotecan; however, there was a strong correlation between sensitivity to etoposide and senstivity to topotecan (Figure 2C).
Figure 2.



Panel A TOP2A, TOP2B gene expression in the 23 SCLC lines, 3 normal cell types and 2 non-small cell lung cancer lines as determined by log2 from gene expression derived from exon arrays. The data were sorted on TOP2A expression from highest to lowest expressers, and TOP1 gene expression as determined by log2 from gene expression derived from exon arrays, sorted from highest to lowest TOP1 expression. PANEL B: Concentration response curves for the 23 SCLC lines exposed to 0.001 – 10 uM of the topoisomerase 2 inhibitor, etoposide, or the topoisomerase 1 inhibitor, topotecan, for 96 h. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). The dotted line is the clinical Cmax for topotecan. The experiments were repeated 3–4 times. PANEL C: Relationship between sensitivity to etoposide and topotecan showing a strong correlation (R2 = 0.8606).
Gene expression for a broad spectrum of notch pathway and related genes were present in the 23 SCLC lines (Figure 3A). Proteolytic cleavage of notch receptors by the presenilin/γ-secretase complex is required for the activation of the notch pathway. γ-secretase inhibitors block notch activity [13, 14]. Concentration response experiments were carried out with two γ-secretase inhibitors, RO429097 and PF-03084014, covering concentrations including the clinical Cmax concentration for each compound (Figure 3B). At concentrations up to 10 uM RO429097 had little effect on the growth of the 23 SCLC with cell growth between 90–100% of the control at the clinical Cmax. Similarly, PRF-03084014 exposure resulted in 90–100% of control cell growth at the clinical Cmax concentration. At the highest concentration tested, 2 of 5 l-Myc amplified SCLC lines reached an IC50. When exposure to a concentration range of PF-03084014 was combined simultaneously with exposure to etoposide (0.3 uM) and carboplatin (3.7 uM) in 4 representative SCLC lines additive to sub-additive SCLC killing was obtained as assessed by simple Bliss additivity (Figure 3C).
Figure 3.


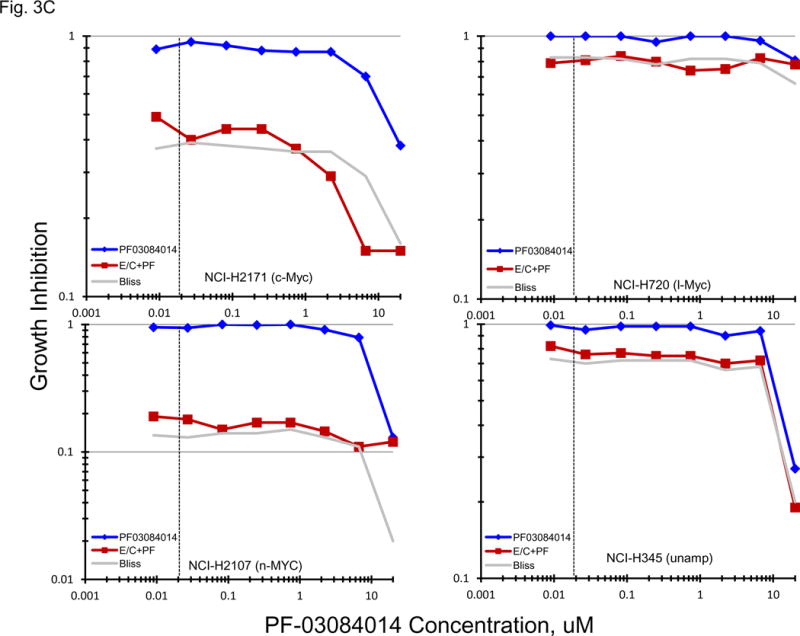
PANEL A Expression of genes associated with the Notch pathway in 23 SCLC lines as determined by the 1/ΔCT value from RT-PCR or as log2 from gene expression derived from exon arrays. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). PANEL B: Concentration response curves for the 23 SCLC lines exposed to 0.001 – 10 uM of the γ-secretase inhibitors, RO429097 and PF-03084014, for 96 h. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). The dotted lines are the clinical Cmax for each compound. The experiments were repeated 3–4 times. PANEL C: Concentration response curves for 4 representative SCLC lines exposed to 0.01 – 20 uM of PF-03084014 alone (blue line) or in simultaneous combination with 3.7uM carboplatin and 0.3uM etoposide (red line). The gray line is calculated simple Bliss additivity for the combination regimen.
Bromodomain and extra-terminal (BET) domain proteins including BRD2, BRD3, BRD4 and BRDT, have been associated with acetylated chromatin and facilitate transcriptional activation [15–17]. Several reports have associated c-Myc transcription with BET bromodomain protein activity [15, 16, 18]. BRD4 has been most clearly associated with c-Myc activity. The gene expression for BRD1, BRD2, BRD3, BRD4 and BRDT by the 23 SCLC lines, 3 normal cell types and 2 NSLC lines indicate that the expression of the mRNA for these genes is not highly varied in the SCLC lines (Figure 4A). When the data were organized by BRD4 expression, c-MYC amplified SCLC lines distributed over the range of expression. Concentration response experiments were performed with JQ1, a known BET bromodomain inhibitor (Figure 4B). The mean JQ1 IC50 for the c-MYC amplified lines was 0.47 uM, for the n-MYC amplified lines was 0.22 uM, for the l-MYC amplified lines was 0.68uM and for the unamplified lines was 1.9 uM. Overall, there was a trend in the MYC amplified lines for greater sensitivity to JQ1 than the unamplified lines. In a larger series of SCLC lines, the mean JQ1 IC50 in 10 lines derived from treatment naïve patients was 5.37 uM, and the mean JQ1 IC50 in 29 lines derived from previously treated patients was 6.19 uM; this difference was not significant. The combination of JQ1 with etoposide and carboplatin was explored in four SCLC lines with different MYC status (Figure 4C). With each of the four SCLC lines, the addition of JQ1 to treatment with etoposide and carboplatin resulted in greater-than-additive cytotoxicity the magnitude of which increased with increasing JQ1 concentration. The least effect was observed with the NCI-H720 line and the greatest effect was observed with the NCI-2107 line. These lines were similarly responsive to JQ1 as a single agent.
Figure 4.


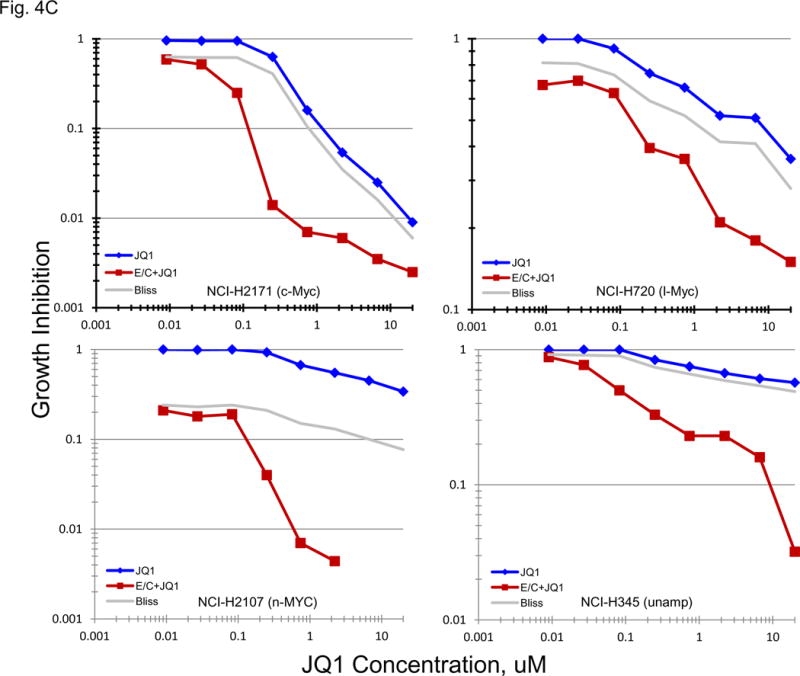
PANEL A BRD1, BRD2, BRD3, BRD4 and BRDT gene expression in the 23 SCLC lines, 3 normal cell types and 2 non-small cell lung cancer lines as determined by log2 from gene expression derived from exon arrays. The data were sorted on BRD4 expression from highest to lowest expressers. PANEL B: Concentration response curves for the 23 SCLC lines exposed to 0.001 – 10 uM of the BET bromodomain inhibitor, JQ1, and the glutamate inhibitor, riluzole, for 96 h. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). The dotted line is the clinical Cmax for riluzole. The experiments were repeated 3–4 times. PANEL C: Concentration response curves for 4 representative SCLC lines exposed to 0.01 – 20 uM of JQ1 alone (blue line) or in simultaneous combination with 3.7uM carboplatin and 0.3uM etoposide (red line). The gray line is calculated simple Bliss additivity for the combination regimen.
Normally, an active hedgehog pathway is required for embryo development. Activation of the hedgehog pathway has been implicated in many cancers and appears to be potentially causal in basal cell carcinoma. mRNA for a broad spectrum of hedgehog pathway and related genes were present in the 23 SCLC lines (Figure 5A). The Smo antagonists, erismodegib and vismodegib as well as the Gli antagonists, HIP1 and SEN-450, were assessed in the 23 SCLC lines (Figure 5B). There was a trend for the MYC amplified lines to be more responsive to HIP1 than the MYC unamplified lines; however, the effect was small. The same trend was observed with SEN-450. Clinical Cmax concentrations for erismodegib and vimodegib occurred at concentrations near the IC50s for both compounds. There was a trend toward the MYC amplified lines to be more responsive to both erismodegib and visomodegib than the MYC unamplified lines; however, the effect was small. The combination of erismodegib or vismodegib with etoposide and carboplatin was explored in four SCLC lines with different MYC status (Figure 5C). With each of the four SCLC lines, the combinations were generally additive to sub-additive.
Figure 5.



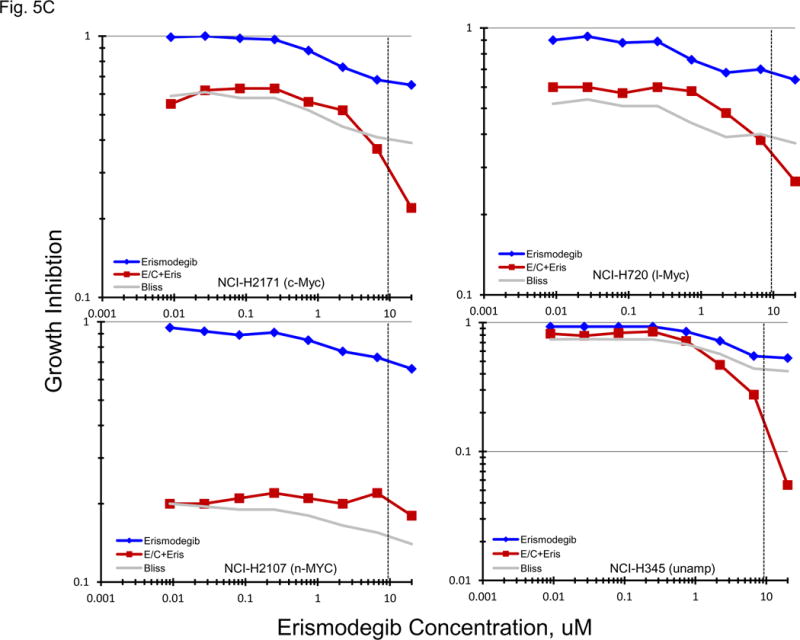

PANEL A Expression of genes associated with the Hedgehog pathway in 23 SCLC lines as determined by the 1/ΔCT value from RT-PCR or as log2 from gene expression derived from exon arrays. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). PANEL B: Concentration response curves for the 23 SCLC lines exposed to 0.01 – 100 uM of the Gli inhibitors, HPI1 and SEN-450, and the Smo inhibitors, erismodegib and vismodegib, for 96 h. The blue symbols represent c-MYC amplified SCLC lines (n = 5), green symbols represent n-MYC amplified SCLC lines (n = 3), red symbols represent l-MYC amplified SCLC lines (n = 5) and gray symbols represents MYC unamplified SCLC lines (n = 10). The dotted lines are the clinical Cmax for erismodegib and vismodegib. The experiments were repeated 3–4 times. PANEL C: Concentration response curves for 4 representative SCLC lines exposed to 0.01 – 20 uM of erismodegib or vismodegib alone (blue line) or in simultaneous combination with 3.7uM carboplatin and 0.3uM etoposide (red line). The gray line is calculated simple Bliss additivity for the combination regimen.
4. DISCUSSION
Drug discovery is exceptionally challenging for SCLC cancer. Genetic and epigenetic changes including gene mutations, deletions, amplifications, translocations and methylation induced gene silencing are frequent in SCLC cell lines and tumors; however, questions regarding the genomic stability, lack of differentiation and absence of stromal–vascular–inflammatory cell compartments are significant issues [19]. Since biopsies are rare in SCLC diagnosis, SCLC cell lines remain the main tool for SCLC biological characterization and translational research. The lack of SCLC progenitor cell type knowledge prevents identification of SCLC gene expression changes compared to the most appropriate normal cell [20].
Targets such as Myc have been known in SCLC for some time, but remain difficult to approach therapeutically. Other targets have only recently become of interest. In the current study, the response of 23 human SCLC lines of varied Myc status was assessed to 10 targeted anticancer agents. MYC status was confirmed by gene expression and protein expression. In addition, gene expression determined by exon array and by RT-PCR for lung cancer related genes, stem-like genes, notch and hedgehog pathway genes.
c-Myc is a transcription factor which promotes cancer growth by upregulation of a transcriptional program influencing metabolic adaptation, maintenance of stem cells, cell division and survival [21–24]. MYC is a transcriptional amplifier, increasing the transcription of genes that are switched on in tumor cells, lymphocytes and stem cells. MYC effects are broad and cell-type-specific because MYC amplifies existing genetic outputs. The c-Myc protein is implicated in physiological and pathological growth, proliferation, apoptosis, metabolism, and differentiation via regulation of numerous target genes [25]. In tumor cells expressing high c-Myc, Myc binds in promoter regions of active genes and causes transcriptional amplification, producing increased transcripts from active genes [26, 27]. Myc overexpression stabilizes HIF1a under normoxic conditions and enhances HIF1α accumulation under hypoxic conditions [28]. c-MYC overexpression in cancer stem cells leads to increased expression of CHK1 and CHK2 and subsequent activation of the DNA-damage-checkpoint response resulting in radio-resistance. CHK1 and CHK2 expression loss reverses radioresistance in cancer stem cells [29].
Direct therapeutic targeting of MYC protein remains difficult. The Myc promoter contains a guanine-rich sequence (PU27) capable of forming quadruplex (four-stranded) DNA, which may negatively regulate myc transcription. Exposure of Myc over-expressing cells to an oligonucleotide encoding the genomic PU27 sequence induced cell cycle arrest and death [30]. Pharmacologically interfering with the bromodomain and extraterminal (BET) proteins depletes MYCN in cells resulting in cytotoxicity [31]. The bromodomain and extraterminal (BET) protein Brd4, described as a general transcriptional regulator, recruits transcriptional regulatory complexes to acetylated chromatin [32, 33]. The therapeutic effects of bromodomain inhibitors have been attributed to a specific set of downstream target genes whose expression is disproportionately sensitive to pharmacological targeting of BET proteins. Brd4 engages in direct regulatory interactions with several DNA-binding transcription factors to influence their function [34]. BET inhibitors engage the bromodomain pocket in a competitive manner with acetylated peptide binding, thereby causing the displacement of BET pro-proteins from chromatin in cells exposed to these compounds. The bromodomain reader protein family has a role in translating histone modifications with transcriptional consequences; thus, bromodomain proteins are potential therapeutic targets [35–37]. The BET bromodomain inhibitor JQ1 was more effective in the MYC amplified lines, and, interestingly, JQ1 in combination with etoposide and carboplatin resulted in greater-than-additive cytotoxicity even in the MYC unamplified cell line tested (Figure 4B and C). There is developing evidence that factors in addition to MYC expression may be important in the response to the BET bromodomain class of compounds in SCLC and other cancers in culture and in xenografts [38, 39]. Several BET bromodomain inhibitors are currently in early clinical trial in hematologic and solid malignancies.
The mainstays of SCLC standard of care are the toposiomerase II inhibitor etoposide and the topoisomerase I inhibitor topotecan. The response of the 23 SCLC lines to these drugs was highly heterogeneous and unrelated to the Myc status of the lines (Figure 2A). The expression of the TOP2A and 2B genes as well as the TOP1 gene expression were determined in the 23 SCLC lines and 3 normal cell types and 2 non-small cell lung lines. The expression of TOP2A was lower in the normal cells and non-small cell lung cancer lines than in the SCLC lines while the expression of TOP2B and TOP1 was similar in all of the cells (Figure 2B). Neither the gene expression nor the protein expression of the topoisomerases reflected the great heterogeneity of the response of the SCLC lines to etoposide and topotecan. However, there was a very strong correlation between the response of the SCLC lines to etoposide and topotecan which could not be accounted for in the gene expression data (Figure 2C).
The notch pathway is an intercellular signaling mechanism required for embryonic development. Key factors in the pathway are notch transmembrane receptors and delta (or delta-like) and jagged ligands. Mutations in notch pathway members are rare in SCLC and studies indicate that a hyperactive notch pathway may supress SCLC and down-regulate ASCL1 expression [40–42]. The SCLC lines were not responsive to γ-secretase inhibitors alone or in combination with etoposide and carboplatin (Figure 3B and C). The Hedgehog signaling pathway is one of the key regulators of embryo development. The hedgehog pathway regulates the survival and proliferation of several tissue progenitor and stem populations promoting the expression of several well-known stem cell and proliferative genes, including genes encoding MYC, cyclin D1, insulin-like growth factor 2 (IGF2) and BMI. In some cancers, dysregulation of hedgehog pathway signaling in a stem cell population can explain tumor formation [43]. The highly aggressive nature of SCLC suggests that this disease may have an elevated stem cell fraction. Neither the Gli inhibitors nor the Smo inhibitors had marked effects on the SCLC as single agents or in combination with etoposide and carboplatin (Figure 5B and C). Trends associated with MYC status were quite weak for both the notch pathway inhibitors and the hedgehog pathway inhibitors.
Recurrent SCLC is among the most recalcitrant cancers. Additional drug development efforts focused on achieving clinical benefit in this disease are a high priority.
Highlights.
In 23 SCLC lines, etoposide and topotecan response ranges over 3 logs and response between the two drugs is highly correlated but is not correlated with top1 or top2 levels.
Bromodomain inhibitor JQ1 has a trend to be more active in MYC amplified lines and is synergistic with etoposide and carboplatin.
γ-Secretase, Smo and Gli inhibitors are not effective in SCLC lines alone or in combination with etoposide and caroplatin.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute.
Footnotes
Conflict of Interest Statement
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paumier A, Le Pechoux C. Radiotherapy in small cell lung cancer: where should it go. Lung Cancer. 2010;69:133–40. doi: 10.1016/j.lungcan.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez E, Lilenbaum RC. Small cell lung cancer: past, present and future. Curr Oncol Rep. 2010;12:327–34. doi: 10.1007/s11912-010-0120-5. [DOI] [PubMed] [Google Scholar]
- 3.D’Angelo SP, Pietanza MC. The molecular pathogenesis of small cell lung cancer. Cancer Biol Ther. 2010;10:1–10. doi: 10.4161/cbt.10.1.12045. [DOI] [PubMed] [Google Scholar]
- 4.Johnson BE, Russell E, Simmons AM, Phelps R, Steinberg SM, Ihde DC, et al. MYC family DNA amplification in 126 tumor cell lines from patients with small cell lung cancer. J Cell Biochem Suppl. 1996;24:210–7. doi: 10.1002/jcb.240630516. [DOI] [PubMed] [Google Scholar]
- 5.Shivapurkar N, Reddy J, Matta H, Sathyanarayana UG, Huang CX, Toyooka S, et al. Loss expression of death-inducing signaling complex (DISC) components in lung cancer cell lines and the influence of MYC amplification. Oncogene. 2002;21:8510–4. doi: 10.1038/sj.onc.1205941. [DOI] [PubMed] [Google Scholar]
- 6.Voortman J, Lee HH, Killian JK, Suuriniemi M, Wang Y, Lucchi M, et al. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc Natl Acad Sci USA. 2010;107:13040–45. doi: 10.1073/pnas.1008132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salcido CD, Larochelle A, Taylor BJ, Dunbar CE, Varticovski L. Molecular characterization of side population cells with cancer stem cell-like characteristics in small cell lung cancer. Br J Cancer. 2010;102:1636–44. doi: 10.1038/sj.bjc.6605668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang B, Yang H, Huang YZ, Yan RH, Liu FJ, Zhang JN. Biologic characteristics of the side population of human small cell lung cancer cell line H446. Chin J Cancer. 2010;29:254–60. doi: 10.5732/cjc.009.10330. [DOI] [PubMed] [Google Scholar]
- 9.Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ, et al. Characterization of the cell of origin for small cell lung cancer. Cell Cycle. 2011;10:2806–15. doi: 10.4161/cc.10.16.17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrova E, Rios-Esteves J, Ouergelli O, Glickman JF, Resh MD. Inhibitors of hedgehog acyltransferase block sonic hedgehog signaling. Nat Chem Biol. 2013;9:247–9. doi: 10.1038/nchembio.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo SD, Matheu A, Mariani N, Carretero J, Lopez-Rios F, Lovell-Badge R, et al. Novel transcriptional targets of the SRY-HMG box transcription factor SOX4 link its expression to the development of small cell lung cancer. Cancer Res. 2011;72:176–86. doi: 10.1158/0008-5472.CAN-11-3506. [DOI] [PubMed] [Google Scholar]
- 12.Wang P, Gao Q, Suo Z, Munthe E, Solberg S, Ma L, et al. Identification and characterization of cells with cancer stem cell properties in human primary lung cancer cell lines. PLoS ONE. 2013;8(3):e57020. doi: 10.1371/journal.pone.0057020. http://dx.doi.org/10.1371/journal.pone.0057020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Aster JC, Blacklow SC. Targeting the notch pathway: twists and turns on the road to rational therapeutics. J Clin Oncol. 2012;30:2418–20. doi: 10.1200/JCO.2012.42.0992. [DOI] [PubMed] [Google Scholar]
- 14.Palomero T, Ferrando A. Therapeutic targeting of notch1 signaling in T-cell lymphoblastic leukemia. Clin Lymphoma Myeloma. 2009;9(suppl 3):S205–10. doi: 10.3816/CLM.2009.s.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108:16669–74. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol. 1979;122:549–4. [PubMed] [Google Scholar]
- 18.Pulvertaft JV. Cytology of Burkitt’s Tumour (African Lymphoma) Lancet. 1964;1:238–40. doi: 10.1016/s0140-6736(64)92345-1. [DOI] [PubMed] [Google Scholar]
- 19.Gazdar AF, Gao B, Minna JD. Lung cancer cell lines: useless artifacts or invaluable tools for medical science? Lung Cancer. 2010;68:309–18. doi: 10.1016/j.lungcan.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coe BP, Lee EHL, Chi B, Girard L, Minna JD, Gazdar AF, et al. Gain of a region on 7p22. 3, containing MADILI, is the most frequent event in small cell lung cancer cell lines. Gene Chrom Cancer. 2006;45:11–9. doi: 10.1002/gcc.20260. [DOI] [PubMed] [Google Scholar]
- 21.Guo J, Price DH. RNA Polymerase II Transcription Elongation Control. Chem Rev. 2013;113:8583–603. doi: 10.1021/cr400105n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyoshi S, Ooike S, Iwata K, Hikawa H, Sugaraha K. WO/2009/084693. International Patent No. PCT/JP2008/073864. 2009
- 23.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stine Z, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5:1024–39. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscrptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2011;72:1–9. doi: 10.1158/0008-5472.CAN-11-2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WJ, Wu SP, Liu JB, Shi YS, Huang X, Zhang QB, et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2012;73:1219–31. doi: 10.1158/0008-5472.CAN-12-1408. [DOI] [PubMed] [Google Scholar]
- 30.Sedoris KC, Thomas SD, Clarkson CR, Muench D, Islam A, Singh R, et al. Genomic c-Myc quadruplex DNA selectively kills leukemia. Mol Cancer Ther. 2012;11:66–76. doi: 10.1158/1535-7163.MCT-11-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnepp RW, Maris JM. Targeting MYCN: a good BET for improving neuroblastoma therapy? Cancer Discov. 2013;3:255–7. doi: 10.1158/2159-8290.CD-13-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014;1839:676–85. doi: 10.1016/j.bbagrm.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature Rev Drug Discov. 2014;13:337–56. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 34.Trabucco SE, Gerstein RM, Evens AM, Bradner JE, Shultz LD, Greiner DL, Zhang H. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin Cancer Res. 2015;21:113–22. doi: 10.1158/1078-0432.CCR-13-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharm Sci. 2012;33:146–53. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Melichar B, Adenis A, havel L, Lockhart AC, Bennouna J, Schusterbauer C, et al. Phase (Ph) I/II study of investigational Aurora A kinase (AAK) inhibitor MLN8237 (alisertib): updated ph II results in patients (pts) with small cell lung cancer (SCLC), non-SCLC (NSCLC), breast cancer (BrC), head and neck squamous cell carcinoma (HNSCC), and gastroesophageal cancer (GE) J Clin Oncol. 2013;31(Suppl):Abstr 605. [Google Scholar]
- 37.Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, Peterson R, Schweizer L, Purandare A, Ross-Macdonald P, Fairchild C, Wong T, Wee S. Sensitivity of small cell lung cancer to BET inhibition is mediated by regulation of ASCL1 gene expression. Molec Cancer Therap. 2015;14:1–8. doi: 10.1158/1535-7163.MCT-15-0037. [DOI] [PubMed] [Google Scholar]
- 38.Riveiro ME, Kwee I, Astrogues-Xerri L, Bekradda M, Vazquez R, Rinaldi A, Cvitkovic E, Bertoni F. Gene expression profile of OTX015, a BET bromodomain inhibitor, in preclinical models of non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) models. Proc AACR. 2015:Abstr # 3530. [Google Scholar]
- 39.Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, Rinaldi A, Testoni M, Cascione L, Ponzoni M, Mensah AA, Stathis A, Stussi G, Riveiro ME, Herait P, Inghirami G, Cvitkovic E, Zucca E, Bertoni F. The BET bromodomain inhibitor OTX015 affects pathogenic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res. 2015;21:1628–38. doi: 10.1158/1078-0432.CCR-14-1561. [DOI] [PubMed] [Google Scholar]
- 40.Siruranpong V, Borges MW, Ravi RK, Arnold DR, Nelkin BD, Baylin SB, Ball DW. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200–5. [PubMed] [Google Scholar]
- 41.Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. Integrative genome analyses identify key somatic driver mutations of small cell lung cancer. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rudin CM, Durinick S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small cell lung cancer. Nat Genet. 2012;44:1111–16. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briscoe J, Therond PP. The mechanisms of hedgehog signaling and its role in development and disease. Nature Rev Molec Cell Biol. 2013;14:416–29. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]