

Abstract
EZH2 overexpression promotes cancer by increasing histone methylation to silence tumor suppressor genes, but how EZH2 levels become elevated in cancer is not understood. In this study, we investigated the mechanisms by which EZH2 expression is regulated in non-small cell lung carcinoma cells by oncogenic KRAS. In cells harboring KRASG12C and KRASG12D mutations, EZH2 expression was modulated by MEK-ERK and PI3K/AKT signaling, respectively. Accordingly, MEK-ERK depletion decreased EZH2 expression in cells harboring the KRASG12C mutation, whereas PI3K/AKT depletion decreased EZH2 expression, EZH2 phosphorylation, and STAT3 activity in KRASG12D mutant cell lines. Combined inhibition of EZH2 and MEK-ERK or PI3K/AKT increased the sensitivity of cells with specific KRAS mutations to MEK-ERK and PI3K/AKT targeted therapies. Our work define EZH2 as a downstream effector of KRAS signaling and offer a rationale for combining EZH2 inhibitory strategies with MEK-ERK- or PI3K/AKT-targeted therapies to treat lung cancer patients, as stratified into distinct treatment groups based on specific KRAS mutations.
Keywords: EZH2, NSCLC, KRAS, MEK-ERK pathway, PI3K/AKT pathway
INTRODUCTION
Enhancer of zeste homolog 2 (EZH2), which functions as a lysine N-methyltransferase, is the catalytic subunit of polycomb repressive complex 2 (1-3). EZH2 mediates repression of gene transcription through the trimethylation of histone H3 in lysine 27 (H3K27) (2, 4, 5). Overexpression of EZH2 has been described in a wide variety of human cancers, including non–small cell lung carcinoma (NSCLC) (3, 5-8). In lung cancer, EZH2 overexpression has been associated with poor outcome, making it an attractive therapeutic target. In an attempt to determine the importance of EZH2 in lung adenocarcinoma pathogenesis, we recently demonstrated that EZH2 depletion decreased the malignant potential of lung adenocarcinoma cell lines and increased sensitivity of those cells to both platinum-based and VEGFR-2–targeted therapies (9). However, the mechanisms driving EZH2 expression in lung cancer are not fully understood, and their identification would likely lead to new therapies targeting EZH2.
KRAS mutations are one of the most frequent alterations in cancer (10, 11), including pancreatic cancer (~70-90%) (12), colorectal cancer (50%) (13), and lung adenocarcinoma (30%) (14). KRAS mutations are generally associated with poor overall survival and resistance to therapy (11, 15, 16). KRAS point mutations occur specifically at codons 12, 13, and 61, with codon 12 being the most frequently mutated (11). The mutations at codons 12 and 13 lead to wild-type glycine (G) being replaced by cysteine (C), valine (V), aspartic acid (D), arginine (R), alanine (A), or serine (S) (17, 18). KRAS has the ability to activate multiple downstream signaling pathways, including RAF/MEK/ERK and PI3K/AKT. In the recent years, a number of agents inhibiting distinct downstream pathways activated by mutant KRAS have been developed; however, the effect of these strategies on lung cancer patients’ survival has been limited (19). It has been found that different amino acid substitutions in mutant KRAS may have a differential effect on the oncogenic potential, suggesting heterogeneity of behavior in different KRAS mutant subtypes (20, 21). Recently, it has been found that, in NSCLC refractory to targeted therapy with sorafenib, KRASG12C and KRASG12V mutants result in a worse prognosis compared with tumors containing other KRAS mutants, suggesting that not all KRAS mutants affect survival or downstream signaling pathways in a similar way (22). NSCLC cell lines with mutant KRASG12D had activated PI3K/AKT whereas those with mutant KRASG12C had decreased growth factor–dependent AKT activation (22). However, how each of these different KRAS amino acid substitutions leads EZH2 expression and which downstream effector molecules are involved are unknown.
The PI3K/AKT signaling pathways are frequently activated by oncogenic KRAS deregulating the control of metabolism, proliferation, and apoptosis (23-25). Previous studies have shown that the activation of AKT mediates phosphorylation of EZH2 at serine 21 (pS21-EZH2) (26). This modification would decrease EZH2-dependent histone modification, whereas phosphorylation of EZH2 by AKT stimulates direct methylation of non-histone protein targets, suggesting that EZH2, like other histone methyltransferases, might have histone methylation-independent activity to control tumorigenicity (27, 28). Recently, it has been shown in glioblastoma stem-like cells that pS21-EZH2 interacts with the transcription factor STAT3 (29). This interaction allows the methylation of STAT3 by EZH2, leading to enhanced STAT3 activity by increased phosphorylation of STAT3 and promoting tumor growth (29). This identifies STAT3 as a downstream effector of the AKT-EZH2 axis to control tumorigenesis. No study has investigated whether oncogenic KRAS mediates the AKT-dependent phosphorylation of EZH2 by increasing the activity of STAT3.
Hence, we investigated the mechanisms of EZH2 expression associated with oncogenic KRAS and whether disruption of the MEK-ERK and PI3K/AKT signaling pathways would affect EZH2 expression in a panel of lung adenocarcinoma, colorectal, and pancreatic cancer KRAS mutant cell lines. Moreover, we studied the efficacy of inhibition of MEK-ERK and PI3K/AKT combined with direct EZH2 inhibition in KRAS mutant cell lines. Furthermore, we examined the ability of the oncogenic KRAS to mediate the AKT-dependent phosphorylation of EZH2 and histone methylation-independent activity of EZH2 by methylating and activating STAT3. Our study provides evidence of regulation of EZH2 by oncogenic KRAS and provides a rationale for EZH2 inhibition resulting in a significant increase in sensitivity to MEK-ERK and PI3K/AKT targeted therapy and identified EZH2 as an attractive target to decrease STAT3 activity in KRAS mutant lung tumors.
MATERIALS AND METHODS
Cell Lines and Tumor Specimens
Lung adenocarcinoma and immortalized human bronchial epithelial cells (HBEC) expressing KRAS wild-type (HBEC3-KT) and KRAS mutant with stable p53 knockdown (HBEC3-KT53KC12 and KT53KD12 and HBEC3-KTKV12)cell lines were provided by Drs. Adi Gazdar and John Minna between 2012 and 2014 (UT Southwestern Medical Center) and authenticated using DNA fingerprinting (30-32). Archived FFPE tumor specimens obtained from 279 adenocarcinomas patients who underwent surgical resection with curative intent were collected from the Lung Cancer Specialized Program of Research Excellence tissue bank at UT MD Anderson Cancer Center. Detailed clinical and pathologic information on the patients is presented in Supplementary Table 1. The study protocol was approved by the Institutional Review Board at MD Anderson Cancer Center.
Immunohistochemical Analysis
To determine the immunohistochemical expression of EZH2 in lung adenocarcinomas, we used FFPE tumor tissues placed in a tissue microarray. Tissue samples were analyzed for EZH2 expression in the nucleus of malignant cells by using antibodies against EZH2 (Novocastra, Leica Biosystems). We used a 4-value intensity score (0, 1+, 2+, and 3+) and the percentage (0% to 100%) of the extent of reactivity. The final score was obtained by multiplying the intensity and extent-of-reactivity values (range, 0–300).
Transfection of Lung Adenocarcinoma Cells with siRNAs
Lung adenocarcinoma cell lines were transfected with three-gene-specific siRNA (siRNA-3) duplexes for the KRAS, MEK1, AKT1, AKT2, AKT3, and EZH2 genes and a scrambled siRNA (siControl) (OriGene Technologies) at a final concentration of 10 nmol/L using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions. To verify the knockdown efficiency of each gene that was knocked down, mRNA and proteins were collected from transfected cells for qRT-PCR and Western blot analysis. Western blot analysis used specific antibodies against EZH2, KRAS, MEK1/2, and AKT (Cell Signaling Technology).
Treatment with MEK-ERK and PI3K/AKT Inhibitors
To determine the effect of treatment with MEK-ERK and PI3K/AKT inhibitors (i) on EZH2 expression, cell lines were treated with different concentrations of the MEK-ERKi AZD6244 (0, 0.5, and 1 μM) or the PI3K/AKTi MK2206 (0, 25, and 50 nM) for 72 hours. Protein lysates were collected from subconfluent cultures after 72 hours of growth in medium without FBS and subjected to Western blot analysis with specific antibodies against EZH2, MAPK P44/42, pMAPK P44/42, AKT, pAKT, STAT3, pSTAT3 (Cell Signaling Technology), pEZH2, and methyl lysine.
MTS Assay and Treatment of Lung Adenocarcinoma Cells with MEK-ERK and PI3K/AKT Inhibitors
MEK-ERKi AZD6244 (selumetinib) and PI3K/AKTi MK2206 2HCl (MK2206) were purchased from Selleck Chemicals. To determine the IC50 of these drugs, lung adenocarcinoma cells were seeded in octuplicate at a density of 2000 cells/well. The following day, cells were treated with the drugs at increasing concentrations, and an endpoint viability assay was performed after 72 hours of treatment using MTS assays (Promega). The dual drug studies (AZD6244 or MK2206) + EZH2 inhibitor (EZH2i) (DZNep, GSK343, GSK126 and EPZ-6438) were performed in a similar manner.
Xenograft Studies
Female athymic nude mice, 6 to 8 weeks old, were injected subcutaneously in the flank with 1.5 × 106 of H23 and HCC461 NSCLC cell lines. Tumors were allowed to grow until the tumors reached an average volume of about 0.05 cm3, and the mice were then randomly divided into control and treatment groups (n = 10 animals/group). The following treatments were administered (10 mice/treatment) as follows: vehicle alone, DZNep PBS 1 mg/kg (50 μl, intraperitoneal injection), AZD6244, and MK2206 were suspended in 1% (w/v) aqueous polysorbate 80, 50 mg/kg each (50 μl, oral gavage), or the combination at the indicated doses (DZNep 1 mg/kg + AZD6244 50 mg/kg or DZNep 1 mg/kg + MK2206 50 mg/kg). DZNep was administered three times per week for 3 weeks, and AZD6244 and MK2206 were administered daily. Tumors were measured twice a week with calipers. Tumor volumes were calculated according to the formula (L × 2W)/2. All animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at MD Anderson.
Statistical Analysis
Data obtained from cell culture assays were summarized using descriptive and inferential statistics accompanied by graphs from Prism software program (GraphPad). Western blot analyses were performed multiple times and normalized to β-actin protein. The patients’ demographic and clinical information was compared using the chi-square, Fisher exact, Wilcoxon rank-sum, and Kruskal-Wallis tests.
RESULTS
EZH2 Expression is Associated with Oncogenic KRAS Mutation Subtypes
To study the association of EZH2 activation and oncogenic KRAS mutations in lung cancer, we first examined the levels of EZH2 immunohistochemical expression in lung adenocarcinoma tumor samples with known KRAS mutation status. Of the 265 surgically resected lung adenocarcinoma tumors analyzed, 82 tumors (30.9%) had a KRAS mutation: 72 (87.8%) tumors had a mutation at codon 12, 10 (12.2%) had a mutation at codon 13, and no mutation was detected in codon 61 (Supplementary Table 1). Although EZH2 expression did not correlate with the presence of KRAS mutation, we found that EZH2 expression levels were significantly higher in tumors with a KRASG12C mutation (P = 0.0036) compared with the other types of KRAS amino acid substitutions (Fig. 1A and B). Because mutations affecting codon 12 of KRAS were the most frequent in NSCLC and other tumors, we characterized EZH2 expression by Western blot in a panel of eight lung adenocarcinoma cell lines harboring different amino acid substitutions in KRAS codon 12 and the wild-type gene (KRASWT). We found that all lung adenocarcinoma cell lines had detectable levels of EZH2 protein expression, and we observed higher levels of EZH2 expression in cell lines with the KRASG12C mutation and moderate levels in cell lines having KRASG12D, KRASG12R, KRASG12S, and KRASG12V, compared with cell lines with KRASWT (Fig. 1C, left panel). To confirm these findings, we studied the effect of KRAS mutations on EZH2 expression in isogenic cell lines. We used HBECs previously characterized (31, 32), having KRASWT and transfected with the most common subtypes of KRAS mutant detected in lung adenocarcinoma (KRASG12C, KRASG12D, and KRASG12V). Western blot analysis indicated that HBEC lines having KRAS mutations had elevated levels of EZH2 expression compared with the HBEC line having KRASWT (Fig. 1C, right panel). Together, these findings suggest that specific subtypes of oncogenic KRAS regulate EZH2 expression in lung cancer. To confirm the role of oncogenic KRAS in regulating EZH2 expression, we knocked down KRAS expression using treatment with a siKRAS in a panel of five lung adenocarcinoma cell lines with KRASWT and KRAS mutants. Knockdown of KRAS downregulated EZH2 expression in cell lines harboring KRAS mutation compared with non-transfected cells or cells transfected with siControl (Fig. 1D); interestingly, this downregulation was more pronounced in cell lines having KRASG12C and KRASG12D mutations than in cell lines with KRASG12S and KRASG12V, and no change in EZH2 expression was detected in KRASWT cells (Fig. 1D). These findings suggest that different amino acid substitutions in oncogenic KRAS differentially modulate EZH2 expression in lung adenocarcinoma cells.
Figure 1.
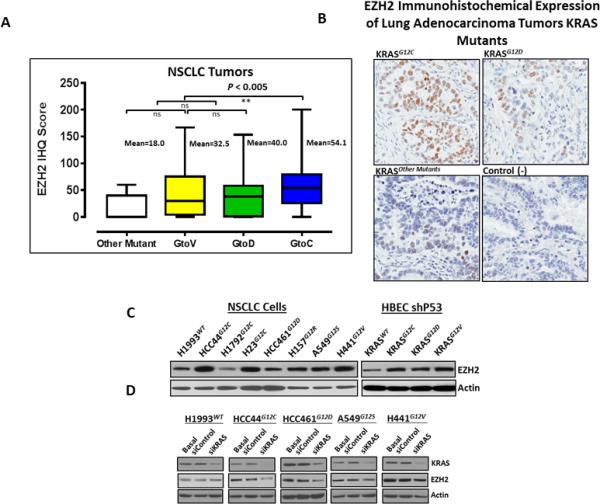
EZH2 was higher in tumors with a KRASG12C mutation compared with other KRAS mutant, and KRAS knockdown downregulates EZH2 expression in cell lines with KRAS mutant. A, Box plot of EZH2 expression of lung adenocarcinoma tumors with KRAS mutant (**P < 0.004). B, Representative photomicrographs of EZH2 immunohistochemical expression in NSCLC with different amino acids substituted on the KRAS mutant. C, EZH2 expression in NSCLC (left) and HBEC cell lines (right). D, KRAS and EZH2 expression in NSCLC cell lines upon knockdown of KRAS by treatment with siKRAS.
To further understand our finding that different amino acid substitutions in KRAS mutant differentially modulate EZH2 expression in NSCLC, we characterized by Western blot the two major downstream signaling pathways activated by KRAS, RAF/MEK/ERK and PI3K/AKT, in NSCLC and HBEC cell lines. We found that NSCLC cell lines expressing KRASWT, mutant KRASG12C, and KRASG12D had elevated levels of phosphorylated MEK1/2 (pMEK1/2) compared with other mutants (Supplementary Fig. 1). Similarly, HBEC cell lines expressing KRASG12C and KRASG12D had elevated levels of pMEK1/2 and HBEC cells expressing KRASG12V had a moderate level of pMEK1/2 compared with HBEC cells expressing KRASWT (Supplementary Fig. 1), whereas the levels of phosphorylated AKT (pAKT) were elevated in NSCLC cell lines expressing mutant KRASG12D, KRASG12R, and KRASG12V and were moderate in cell lines expressing KRASWT. We observed a decrease of pAKT in cell lines expressing KRASG12C. HBEC cells expressing mutant KRASG12C and KRASG12D showed elevated levels of pAKT compared with HBEC cells expressing KRASWT and KRASG12V (Supplementary Fig. 1). This finding supported our hypothesis that different amino acid substitutions in oncogenic KRAS may activate different downstream signaling pathways to regulate its downstream effector to controlling tumorigenesis.
Pharmacological Inhibition of the MEK-ERK Signaling Pathway Affects EZH2 Expression in KRASG12C and KRASG12S Mutant Lung Adenocarcinoma Cell Lines
To determine the mechanism involved in the regulation of EZH2 expression by oncogenic KRAS, we blocked MEK-ERK using the MEK1i AZD6244. Pharmacological disruption of signaling in the MEK-ERK pathway by AZD6244 treatment decreased the expression of EZH2 in a dose-dependent manner, and this effect correlated with the type of mutant KRAS amino acid substituted, with a higher reduction occurring in cell lines harboring KRASG12C mutation compared with the cell line harboring KRASG12S mutation; no change was observed in the KRASWT cell line or in cell lines with the other types of KRAS mutations (KRASG12D and KRASG12V) (Fig. 2A). Similar results were observed in HBEC lines, AZD6244 treatment decreased EZH2 expression in HBEC lines having KRASG12C, and no change was detected in HBEC lines with KRASWT or with KRASG12D or KRASG12V (Fig. 2B). We validated the findings using AZD6244 by knocking down MEK1 expression in three lung adenocarcinoma cell lines with KRASWT, KRASG12C, and KRASG12D using treatment with siRNA. Similar to our results with AZD6244, siMEK1 treatment decreased EZH2 expression only in lung adenocarcinoma cell lines having KRASG12C, and we did not observe any effect in other cell lines with KRASWT or KRASG12D (Fig. 2C). These findings suggest that in lung adenocarcinoma cells mutant KRASG12C preferentially regulates EZH2 expression through MEK-ERK signaling.
Figure 2.
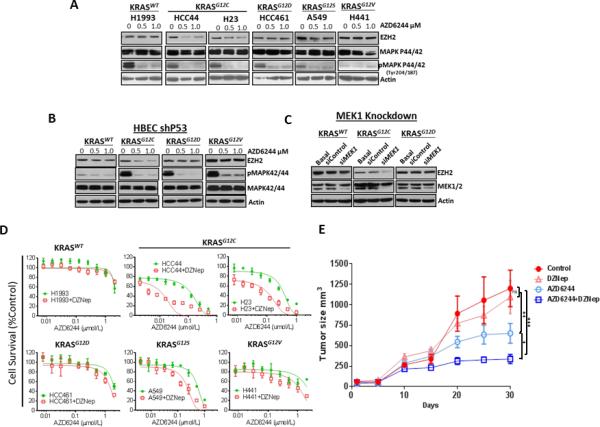
Disruption of the signaling MEK-ERK pathway affects EZH2 expression in NSCLC cell lines with mutant KRASG12C, and the combination of MEK1i with EZH2i results in a significantly increased sensitivity to MEK-ERK targeted therapy in cells expressing mutant KRASG12C and KRASG12S. A and B, EZH2, MAPK P44/42, and phospho-MAPK P44/42 expression in NSCLC and HBEC cell lines. KRASWT and KRAS mutants were treated with different doses of the MEK1i AZD6244 (0, 0.5, and 1.0 μM). C, EZH2 and MEK1/2 expression in NSCLC cell lines upon knockdown of MEK1 expression by treatment with siMEK1. D, EZH2 inhibition with DZNep in combination with MEK1i. (Data are graphed as the mean percent increase ± percent standard deviation). Treatment with DZNep decrease AZD6244 IC50, 7.2-fold (P < 0.03) in HCC44 cells, a 3.6-fold (P < 0.05) in H23 cells, and a 2.6-fold (P < 0.05) in A549 cells. E, Athymic nude mice were inoculated with H23 cell lines expressing KRASG12C mutant and then treated with vehicle, DZNep, AZD6244, or a combination of DZNep plus AZD6244. Tumor volume was determined for each treatment. (*P < 0.05; **P < 0.03, ***P < 0.001).
We previously demonstrated that EZH2 depletion sensitizes lung adenocarcinoma cell lines to both platinum and VEGFR-2 targeted therapies (9). We asked whether treatment with the combination of an EZH2i and MEK-ERK–targeting provides additional therapeutic benefits to lung adenocarcinoma cell lines. We pretreated lung adenocarcinoma cell lines with KRASWT and KRAS mutant using DZNep and then treated them with AZD6244 at increasing concentrations in the presence or absence of DZNep. We found that the in vitro sensitivity to AZD6244 was significantly higher in cell lines with mutant KRASG12C (P < 0.05) and slightly higher in cell line with mutant KRASG12S in the presence of DZNep than in cells treated with AZD6244 in the absence of DZNep (Fig. 2D). This type of response was not seen in cell lines with KRASWT, KRASG12D, or KRASG12V. Similar results were observed when we used highly selective EZH2i, GSK343 and two EZH2i of the last generation EPZ6438 and GSK126 (Supplementary Fig. 2A and 2B). We found that the in vitro sensitivity to AZD6244 was significantly increased in the presence of GSK343 in lung adenocarcinoma cell lines harboring KRASG12C mutation, with a partially increased sensitivity in KRASWT cell lines, and no change in cell lines with mutant KRASG12D (Supplementary Fig. 2A). The response was more marked in the presence of two highly selective EZH2i EPZ6438 and GSK126 (Supplementary Fig. 2B). In vivo studies using lung adenocarcinoma cell line xenografts confirmed our in vitro studies. The in vivo sensitivity to AZD6244 in nude mice inoculated with H23 cell lines with mutant KRASG12C was significantly increased in the presence of DZNep. We observed a greater inhibition of tumor growth when we combined DZNep and AZD6244, compared with the results for mouse control groups treated with DZNep or AZD6244 alone (Fig. 2E). These results suggested that inhibition of the MEK-ERK signaling pathway in combination with EZH2 inhibition greatly increases the sensitivity of lung adenocarcinoma KRASG12C mutant to MEK-ERK–targeted therapy.
Pharmacological Inhibition of the PI3K/AKT Pathway Affects EZH2 Expression in KRASG12D and KRASG12S Mutant Lung Adenocarcinoma Cell Lines
PI3K/AKT is another major signaling pathway frequently activated by oncogenic KRAS. To determine whether KRAS mutation modulates the expression of EZH2 through this PI3K/AKT pathway in lung adenocarcinoma, we blocked it using MK2206. We found that treatment with MK2206 strongly decreased EZH2 expression in cells harboring KRASG12D, partially decreased expression in cells with KRASG12S, and slightly decreased expression in cells with KRASG12V; we did not observe these changes in KRASWT cells or in cell lines with KRASG12C (Fig. 3A). Additionally, we found that MK2206 treatment strongly decreased EZH2 expression in HBEC lines having KRASG12D, moderately decreased expression in lines with KRASG12C, and slightly decreased expression in cells with KRASWT or KRASG12V (Fig. 3B).
Figure 3.
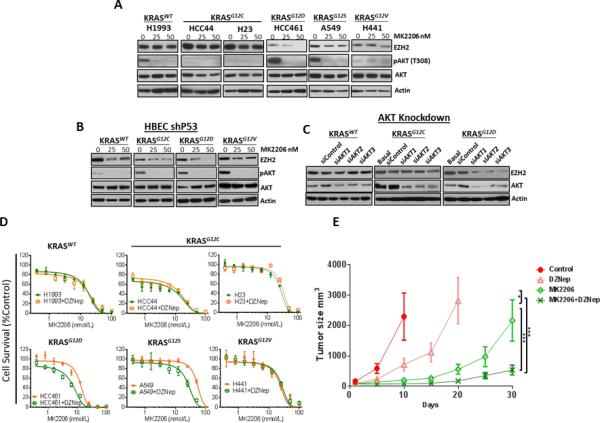
Disruption of the PI3K/AKT pathway affects EZH2 expression in NSCLC cell lines with mutant KRASG12D and KRASG12S, and the combination of AKTi with EZH2i results in a significant increased sensitivity in vitro and in vivo to PI3K/AKT targeted therapy in cells expressing mutant KRASG12D. A and B, EZH2, AKT, and phospho-AKT expression in NSCLC and HBEC cell lines. KRASWT and KRAS mutants were treated with different doses of the AKTi MK2206 (0, 25, and 50 nM). C, EZH2 and AKT expression in NSCLC cell lines upon knockdown of AKT expression by treatment with siAKT. D, Pharmacologic inhibition of EZH2 with DZNep in combination with AKT inhibition. (Data are graphed as the mean percent increase ± percent standard deviation). Treatment with DZNep decrease MK2206 IC50, 2.0-fold (P < 0.05) in cells expressing KRASG12D and a 2.2-fold (P < 0.05) in cells expressing KRASG12S. E, Athymic nude mice were inoculated with HCC461 cell lines expressing KRASG12D and then treated with vehicle, DZNep, MK2206, or a combination of DZNep plus MK2206. Tumor volume was determined for each treatment. (*P < 0.05; **P < 0.03, ***P < 0.001).
To determine if this effect was specifically mediated by AKT signaling inhibition, we knocked down AKT 1, 2, and 3 in lung adenocarcinoma cell lines with KRASWT, KRASG12C, and KRASG12D mutations by treatment with siAKT1, siAKT2, and siAKT3. Importantly, in these experiments we observed results similar to those obtained with MK2206: EZH2 expression decreased only in lung adenocarcinoma cell lines with KRASG12D and not in cell lines with KRASWT or KRASG12C (Fig. 3C). These findings suggest that EZH2 expression in lung cancer cells with mutant KRASG12D EZH2 is regulated by the PI3K/AKT signaling pathway.
Then, we investigated the efficacy of combined EZH2 and PI3K/AKT inhibition in lung adenocarcinoma cell lines with KRASWT and various KRAS mutations. We pretreated KRASWT and KRAS mutant cell lines with DZNep and then we treated them with MK2206 at increasing concentrations in the presence or absence of DZNep at a fixed concentration. We found that the in vitro sensitivity to MK2206 was significantly increased in the presence of DZNep only in cells harboring mutant KRASG12D and KRASG12S (P < 0.05), compared with cells treated with MK2206 in the absence of DZNep (Fig. 3D). This effect was not observed in lung adenocarcinoma cell lines with KRASWT or other KRAS mutation subtypes. The use of the highly selective EZH2i, GSK343 and two EZH2i of the last generation EPZ6438 and GSK126 provided similar results than those obtained with DZNep treatment. The sensitivity to MK2206 was significantly increased in the presence of GSK343 in lung adenocarcinoma cells with mutant KRASG12D, partially increased in KRASWT cells, and not increased in cells with mutant KRASG12C (Supplementary Fig. 2A). The effect was more marked in the presence of EPZ6438 and GSK126 (Supplementary Fig. 2B). Similar to our in vitro results, the reduction of tumors treated with MK2206 in mice inoculated with the HCC461 KRASG12D mutant cell line was significantly higher in the presence of DZNep compared with the results for the control group or with results for mice treated with DZNep or MK2206 alone (Fig. 3E). These findings suggest that the inhibition of the PI3K/AKT pathway in combination with an EZH2i greatly increases the sensitivity of lung adenocarcinoma cells with KRASG12D and KRASG12S mutations to PI3K/AKT-targeted therapy.
EZH2 Expression is Regulated by MEK-ERK and PI3K/AKT Signaling Pathways in KRAS Mutant Colon and Pancreatic Cancer Cell Lines
To examine whether our finding in lung cancer cell lines could be reproduced in other tumors with high frequencies of KRAS mutation, we evaluated the effect of blocking MEK-ERK and PI3K/AKT signaling pathways on the expression of EZH2, by using the inhibitors AZD6244 and MK2206 on a panel of KRAS mutant colon and pancreatic cancer cell lines. We found that AZD6244 treatment decreased the expression of EZH2 in a dose-dependent manner in colon and pancreatic cancer cell lines harboring KRASG12C and KRASG12V mutations, and we did not observe this effect in the cells with KRASWT or KRASG12D (Fig. 4A, upper panel). MK2206 treatments decreased EZH2 expression in a dose-dependent manner strongly in colon and pancreatic cancer cell lines harboring KRASG12D, moderately in cell lines expressing KRASG12C mutations, and only minimally inhibited EZH2 expression in KRASWT cell lines, and we did not in KRASG12V (Fig. 4A, bottom panel). Interestingly, we found in colon and pancreatic cancer cell lines that treatment with AZD6244 and MK2206 decrease the expression of EZH2 in cell lines expressing KRASG12V and KRASG12C, respectively, expanding the spectrum of response in these tumors. These results suggest that, similar to our results for lung adenocarcinoma, in colon and pancreatic cancer KRAS mutant cell lines the MEK-ERK and PI3K/AKT signaling pathways regulate EZH2 expression. Whereas KRASG12C mutant preferentially regulated EZH2 expression through the MEK-ERK signaling pathway, mutant KRASG12D preferably regulated EZH2 expression through the PI3K/AKT signaling pathway. We found that sensitivity to AZD6244 was significantly increased in the presence of the EZH2i DZNep in cells with mutant KRASG12C; this phenomenon was not observed in colon and pancreatic cancer cell lines with KRASWT or KRASG12D mutations (Fig. 4B, upper panel). Also, in colon and pancreatic cancer cells with KRASG12C and KRASG12D mutations, the sensitivity to MK2206 was significantly increased in the presence of DZNep; no change in sensitivity was detected in KRASWT cell lines (Fig. 4B, bottom panel). These results suggest that, like the results for lung adenocarcinoma, MEK-ERK and PI3K/AKT-targeted therapies have synergistic effects when used in combination with an EZH2i in KRAS mutant colon and pancreatic cancer cell lines.
Figure 4.
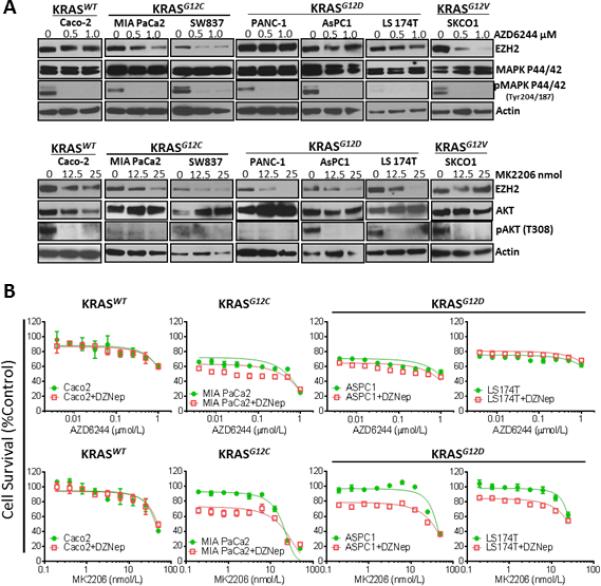
EZH2 expression is regulated by MEK-ERK and PI3K/AKT signaling pathways in colon and pancreatic tumor KRAS mutants. A, top panel, EZH2, MAPK P44/42, and phospho-MAPK P44/42 expression in colon and pancreatic cell lines. KRASWT and KRAS mutants were treated with different doses of the MEK1i AZD6244 (0, 0.5, and 1.0 μM). A, bottom, EZH2, AKT, and phospho-AKT expression in colon and pancreatic cell lines. KRASWT and KRAS mutants were treated with different doses of the AKTi MK2206 (0, 25, and 50 nM). B, top, EZH2 inhibition with DZNep in combination with MEK1i. (Data are graphed as the mean percent increase ± percent standard deviation). Treatment with DZNep caused a 16-fold (P < 0.03) decrease in the AZD6244 IC50 in MIA PaCa2 cells. B, bottom, EZH2 inhibition with DZNep in combination with AKTi. (Data are graphed as the mean percent increase ± percent standard deviation). Treatment with DZNep caused a decrease in MK2206 IC50, 10.6-fold (P < 0.03) in cells expressing KRASG12C, a 6.4-fold (P < 0.05) in cells expressing KRASG12D and a 6.8-fold (P < 0.05) in cells expressing KRASG12D.
Blockage of the AKT-EZH2 Axis Decreases the Activity of STAT3 in Lung Adenocarcinoma Cell Lines with KRASG12D Mutation
An association has been recently described in glioblastoma stem-like cells between EZH2 and STAT3 (29). This association is dependent on phosphorylation of EZH2 at serine 21 by AKT, which stimulates direct methylation of STAT3 by EZH2, enhancing STAT3 activity (29). To determine the ability of oncogenic KRAS to mediate the AKT-dependent phosphorylation of EZH2 and subsequent methylation and activation of STAT3, we blocked the PI3K/AKT signaling pathway using MK2206 and found a significant reduction of pS21-EZH2 in the KRASG12D mutant cell line, with a partial reduction in KRASG12S and KRASG12V cells; no change was detected in the KRASG12C mutant and KRASWT cell lines (Fig. 5A). Additionally, we found that AKT inhibition greatly decreased phospho-STAT3 expression in the KRASG12D cell line, with a partial decrease in KRASG12S cells and no change in KRASG12C mutant and KRASWT cell lines (Fig. 5A). Because it has been shown that EZH2 methylates specific lysine residues of protein not histones (29, 33), we evaluated the levels of methyl lysine (methyl K) in lung adenocarcinoma cell lines undergoing AKT inhibition. We found that methyl K levels were reduced in KRASG12D and KRASG12S cell lines but not in cell lines with KRASG12C or KRASG12V mutations or with KRASWT (Fig. 5A). Similar results were observed in HBEC lines; we found that MK2206 treatment strongly decreased pS21-EZH2, phospho-STAT3 expression, and lysine methylation in HBEC lines having KRASG12D (Fig. 5B). Additionally, pS21-EZH2 and phospho-STAT3 expressions and lysine methylation were significantly reduced at high concentrations of AKTi in HBEC cells with KRASG12C and not in cell lines with KRASG12V mutation or KRASWT. AKT knockdown confirmed these observations in lung adenocarcinoma cell lines, particularly in cell lines with KRASG12D (Fig. 5C).
Figure 5.
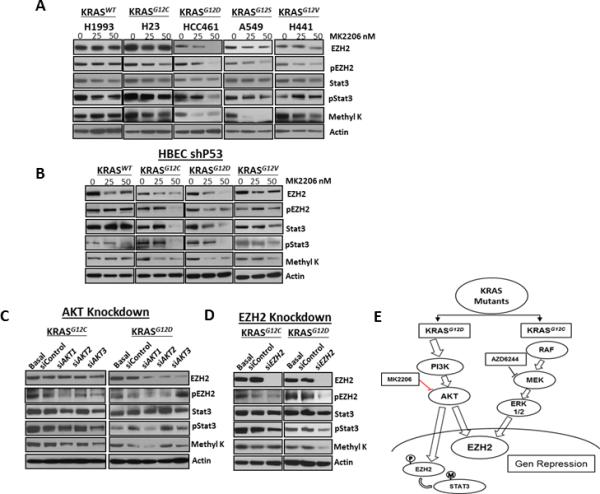
AKT depletion decreased pS21-EZH2, phospho-STAT3, and methyl K levels strongly in cell lines expressing KRASG12D mutants. A and B, EZH2, pS21-EZH2, STAT3, phospho-STAT3, and methyl K level expression in NSCLC cell lines and HBEC cell lines. KRASWT and KRAS mutants were treated with different doses of the AKTi MK2206 (0, 25, and 50 nM). C, EZH2, pS21-EZH2, STAT3, phospho-STAT3, and methyl K level expression in NSCLC cell lines expressing KRASG12C and KRASG12D mutants upon knockdown of AKT1, AKT2, and AKT3 expression by treatment with siAKT. D, EZH2, pS21-EZH2, STAT3, phospho-STAT3, and methyl K level expression in NSCLC cell lines expressing KRASG12C and KRASG12D mutants upon knockdown of EZH2. E, Proposed model showing EZH2 as a downstream effector of KRAS signaling in malignant cell KRAS mutants.
Furthermore, to determine the role of EZH2 in methylated lysine residues in STAT3, we knocked down EZH2 by siRNA and evaluated the levels of methyl K in lung adenocarcinoma cell lines. We found that EZH2 knockdown decreased methyl K and phospho-STAT3 levels in cell lines with KRASG12D but not in cell lines with KRASG12C mutation (Fig. 5D). Altogether, our findings suggest that oncogenic KRAS phosphorylates EZH2 through the PI3K/AKT signaling pathway and regulates STAT3 methylation and activation via EZH2 phosphorylation.
DISCUSSION
Although EZH2 overexpression occurs in a wide variety of cancers, the mechanisms driving EZH2 expression in lung cancer are not fully understood. Gene inactivation by epigenetic mechanisms has been described as a critical step in tumorigenesis, but whether this phenomenon occurs randomly or as consequence of a specific pathway activation is still unclear. In the current study, we demonstrated that oncogenic KRAS regulates EZH2 expression in lung cancer. Specifically, we have demonstrated that different amino acid substitutions in KRAS mutants differentially modulate EZH2 expression: the KRASG12C mutant preferentially regulates EZH2 expression through MEK-ERK signaling pathway, KRASG12D mutant preferentially regulates the expression of EZH2 by the PI3K/AKT signaling pathway, and KRASG12S uses both signaling pathways to regulate EZH2 expression. Our findings show that depletion of MEK1 potently decreased EZH2 expression in lung adenocarcinoma cells expressing the KRASG12C mutant. In turn, depletion of the AKT signaling pathway potently decreased EZH2 expression in lung adenocarcinoma cells expressing the KRASG12D mutant. Additionally, we extended these findings to colon and pancreatic cancers and observed similar results. Interestingly, we found in lung adenocarcinoma tumors that EZH2 expression levels were significantly higher in tumors with KRASG12C compared with other amino acid changes. Overall, this finding suggesting that oncogenic KRASG12C and KRASG12D mutations in different kind of tumors differentially modulate EZH2 expression through MEK-ERK and PI3K/AKT signaling, respectively. Identifying to EZH2 as a downstream effector of KRAS signaling pathway that contribute to tumorigenesis.
The high frequency of KRAS mutants observed in human cancer and the association with resistance to existing therapies in different tumors make it an attractive therapeutic target (11). Because KRAS is not considered directly druggable, most efforts have focused on targeting downstream effectors of the KRAS pathway, including MEK-ERK and PI3K/AKT either singly or a combination. Nevertheless, the therapeutic efficacy and toxicity of these strategies are yet to be clarified, increasing the need to identify new downstream effectors of KRAS or combination approaches to target KRAS mutant tumors. In addition to the complexity of KRAS signaling, there is evidence that the different amino acid substitutions in KRAS mutants could determine response to therapy and the behavior of lung adenocarcinoma (21, 22). Recently a study reported the effect of mutant KRAS stable knockdown on cell proliferation and showed that KRAS knockdown inhibited cell proliferation more markedly in cell lines mutant KRASG12C, indicating that those mutants seem to be more dependent on oncogenic KRAS signaling (34). Consistent with this, in the current study we found that EZH2 inhibition increased the sensitivity to MEK-ERKi and PI3K/AKTi in lung adenocarcinoma cell lines with specific subtypes of KRAS mutations. EZH2 depletion enhanced the sensitivity to MEK1i in KRASG12C mutant tumor cells compared with the other mutations. In contrast, EZH2 inhibition enhanced the sensitivity to MK2206 in tumor cells with KRASG12D mutation compared with other types of KRAS mutations. These findings suggest that EZH2 depletion sensitizes cancer cells to MEK-ERK and PI3K/AKT inhibition in specific KRAS mutation. These findings reinforce the concept of the clinical importance of identifying the specific mutation present in KRAS to identify and guide therapy in cancer patients.
We and other have previously shown that EZH2 overexpression may play a role in drug resistance (9, 35). Accumulating evidence indicates that EZH2 overexpression silences important tumor-suppressor genes (36-39). EZH2 depletion reverses the expression of genes silenced by EZH2, favoring response to therapy. Overall, we found that EZH2 inhibition in combination with MEK-ERK or PI3K/AKT targeting positively affects the response to these therapies. Several preclinical models suggest that epigenetic targeted therapy can reactivate gene expression and reverse tumor growth (43, 44). The identification of epigenetic components that act downstream of KRAS pathways can provide potential new targets for therapy and can be used in combination with current available therapies to increase efficiency and decrease toxicity.
Recently it has been shown that EZH2 is phosphorylated at S21 by AKT; this modification allows EZH2 to bind and methylate STAT3, leading to increased STAT3 activity (29). Blockade of AKT signaling decreases the EZH2-STAT3 interaction and STAT3 methylation (29). Interestingly, this phenomenon occurs preferentially in glioblastoma stem-like cells and not in non-stem tumor cells. These findings identify STAT3 as a downstream effector of the AKT-EZH2 axis to control stem cell self-renewal and tumorigenesis (29). In our study, we demonstrated that oncogenic KRAS mediated EZH2 phosphorylation and enhanced STAT3 activity differentially, depending on the amino acid substituted on the KRAS mutant. Importantly, we demonstrated that depletion of AKT decreased pS21-EZH2, phospho-STAT3, and methyl K levels strongly in cell lines expressing KRASG12D mutants compared with cell lines expressing other KRAS mutants. This suggests that AKT signaling in cell lines expressing KRASG12D mutants is necessary for EZH2 phosphorylation and STAT3 activation. Additionally, we found that EZH2 knockdown decreased strongly the phospho-STAT3 and methyl K levels in cell lines expressing KRASG12D, suggesting that methylation of STAT3 by EZH2 is key to improving the activation of STAT3. Although the mechanisms by which STAT3 methylation increase its activation are not fully understood, it has been suggested that EZH2-mediated K180 methylation of STAT3 prevents its dephosphorylation, allowing STAT3 to remain phosphorylated and active (33, 45). Accumulation of pSTAT3 has been reported many tumors including NSCLC, suggesting that STAT3 behaves as a bona fide oncogene and is therefore an attractive therapeutic target (46). However, specific inactivation of STAT3 has been difficult to achieve (47). Increased EZH2 phosphorylation and enhanced STAT3 activity in cancer cells with KRASG12D mutants would provide advantage in survival, proliferation and other processes compared to other KRAS mutants. In this study, we showed that inhibition of AKT signaling and EZH2 depletion strongly decreased the activation of STAT3 in cell lines expressing the KRASG12D mutant. Additionally, we showed that the combined inhibition of AKT and EZH2 increased sensitivity in vitro and in vivo to AKT targeting in the KRASG12D mutant, suggesting that part of this response is due to inhibition of STAT3 activation. These findings suggest that targeting of AKT/EZH2 pathways may be an attractive approach to improving therapeutic response and survival of patients harboring the KRASG12D mutant, affecting simultaneously AKT signaling, EZH2 function, and STAT3 activity.
In summary, we found evidence that oncogenic KRAS regulates EZH2 expression via the MEK-ERK and PI3K/AKT signaling pathways (Fig. 5E). Interestingly, the signaling pathways used by KRAS depend on the amino acid substituted at KRAS. The KRASG12C mutant preferentially regulates EZH2 expression through the MEK-ERK signaling pathway, the KRASG12D mutant uses the PI3K/AKT signaling pathway to regulate EZH2 expression, while KRASG12S can use both signaling pathways to regulate EZH2 expression. Disruption of MEK1 and AKT signaling decreases EZH2 expression in a mutant-specific way. EZH2 depletion sensitizes of the cancer cells KRAS mutant to MEK-ERKi and PI3K/AKTi KRAS-dependent mutation. In addition, our findings indicate that AKT inhibition or EZH2 depletion decreases the activation of STAT3 in the malignant cell mutant KRASG12D (Fig. 5E). These data presented here identify EZH2 as a downstream effector of KRAS signaling in malignant cell KRAS mutants. Currently, multiple EZH2i are being tested in different tumors. Understanding the context in which EZH2i will be useful is vital to select patients who may benefit from these therapies. These findings identify EZH2 as a potential target in patients with KRAS mutants that can be used in combination with MEK-ERK and PI3K/AKT targeting therapies to increase efficiency and overcome resistance to these therapies.
Supplementary Material
Acknowledgments
Financial Support: This study was supported part by a Department of Defense PROSPECT grant (W81XWH-07-1-0306; to I. I. Wistuba and J. D. Minna); the UT Lung Specialized Programs of Research Excellence grant P50CA70907 from the NIH National Cancer Institute (to I.I. Wistuba and J. D. Minna); R01 CA155196 (to I. I. Wistuba and V. Papadimitrakopoulou) from the NIH National Cancer Institute; Cancer Prevention Research Institute of Texas (CPRIT) grant RP110708 (to I. I. Wistuba); and, MD Anderson's Institutional Tissue Bank 2P30CA016672 from the NIH National Cancer Institute.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Yu J, Cao Q, Mehra R, Laxman B, Tomlins SA, Creighton CJ, et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell. 2007;12:419–31. doi: 10.1016/j.ccr.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 2.Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27:7274–84. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 4.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 5.Fujii S, Ochiai A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008;99:738–46. doi: 10.1111/j.1349-7006.2008.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huqun, Ishikawa R, Zhang J, Miyazawa H, Goto Y, Shimizu Y, et al. Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer. 2012;118:1599–606. doi: 10.1002/cncr.26441. [DOI] [PubMed] [Google Scholar]
- 8.Behrens C, Solis LM, Lin H, Yuan P, Tang X, Kadara H, et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin Cancer Res. 2013;19:6556–65. doi: 10.1158/1078-0432.CCR-12-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riquelme E, Suraokar M, Behrens C, Lin HY, Girard L, Nilsson MB, et al. VEGF/VEGFR-2 upregulates EZH2 expression in lung adenocarcinoma cells and EZH2 depletion enhances the response to platinum-based and VEGFR-2-targeted therapy. Clin Cancer Res. 2014;20:3849–61. doi: 10.1158/1078-0432.CCR-13-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 11.Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756:81–2. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–3 e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–84. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 14.Siegfried JM, Gillespie AT, Mera R, Casey TJ, Keohavong P, Testa JR, et al. Prognostic value of specific KRAS mutations in lung adenocarcinomas. Cancer Epidemiol Biomarkers Prev. 1997;6:841–7. [PubMed] [Google Scholar]
- 15.Raponi M, Winkler H, Dracopoli NC. KRAS mutations predict response to EGFR inhibitors. Curr Opin Pharmacol. 2008;8:413–8. doi: 10.1016/j.coph.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 17.Miller MS, Miller LD. RAS Mutations and Oncogenesis: Not all RAS Mutations are Created Equally. Front Genet. 2011;2:100. doi: 10.3389/fgene.2011.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt JD, Strimas A, Martin JE, Eyer M, Haddican M, Luckett BG, et al. Differences in KRAS mutation spectrum in lung cancer cases between African Americans and Caucasians after occupational or environmental exposure to known carcinogens. Cancer Epidemiol Biomarkers Prev. 2002;11:1405–12. [PubMed] [Google Scholar]
- 19.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–91. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cespedes MV, Sancho FJ, Guerrero S, Parreno M, Casanova I, Pavon MA, et al. K-ras Asp12 mutant neither interacts with Raf, nor signals through Erk and is less tumorigenic than K-ras Val12. Carcinogenesis. 2006;27:2190–200. doi: 10.1093/carcin/bgl063. [DOI] [PubMed] [Google Scholar]
- 21.Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235–7. doi: 10.1093/annonc/mdq680. [DOI] [PubMed] [Google Scholar]
- 22.Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–39. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 24.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 25.Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011;2:261–74. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–10. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 27.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–32. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 28.Stark GR, Wang Y, Lu T. Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 2011;21:375–80. doi: 10.1038/cr.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–52. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010;70:9937–48. doi: 10.1158/0008-5472.CAN-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, et al. Multiple oncogenic changes (KRAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–28. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 32.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010;107:21499–504. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10:336–46. doi: 10.1158/1535-7163.MCT-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther. 2011;10:325–35. doi: 10.1158/1535-7163.MCT-10-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paul TA, Bies J, Small D, Wolff L. Signatures of polycomb repression and reduced H3K4 trimethylation are associated with p15INK4b DNA methylation in AML. Blood. 2010;115:3098–108. doi: 10.1182/blood-2009-07-233858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–30. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, et al. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One. 2009;4:e5011. doi: 10.1371/journal.pone.0005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang R, Wang R, Chang H, Wu F, Liu C, Deng D, et al. Downregulation of Ezh2 expression by RNA interference induces cell cycle arrest in the G0/G1 phase and apoptosis in U87 human glioma cells. Oncol Rep. 2012;28:2278–84. doi: 10.3892/or.2012.2033. [DOI] [PubMed] [Google Scholar]
- 41.Wu ZL, Zheng SS, Li ZM, Qiao YY, Aau MY, Yu Q. Polycomb protein EZH2 regulates E2F1-dependent apoptosis through epigenetically modulating Bim expression. Cell Death Differ. 2010;17:801–10. doi: 10.1038/cdd.2009.162. [DOI] [PubMed] [Google Scholar]
- 42.Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–7. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wrangle J, Wang W, Koch A, Easwaran H, Mohammad HP, Vendetti F, et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget. 2013;4:2067–79. doi: 10.18632/oncotarget.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 47.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.