

Abstract
In a 1914 book entitled The Respiratory Function of the Blood, Joseph Barcroft stated that “the cell takes what it needs and leaves the rest.” He postulated that there must be both a “call for oxygen” and a “mechanism by which the call elicits a response…” In the past century, intensive investigation has provided significant insights into the hemodynamic and biophysical mechanisms involved in supplying oxygen to skeletal muscle. However, the identification of the mechanism by which tissue oxygen needs are sensed and the affector responsible for altering the upstream vasculature to enable the need to be appropriately met has been a challenge. In 1995, Ellsworth et al proposed that the oxygen carrying erythrocyte, by virtue of its capacity to release the vasoactive mediator ATP in response to a decrease in oxygen saturation, could serve both roles. Several in vitro and in situ studies have established that exposure of erythrocytes to reduced oxygen tension induces the release of ATP which does result in a conducted arteriolar vasodilation with a sufficiently rapid time course to make the mechanism physiologically relevant. The components of the signaling pathway for the controlled release of ATP from erythrocytes in response to exposure to low oxygen tension have been determined. In addition, the implications of defective ATP release on human pathological conditions have been explored. This review provides a perspective on oxygen supply and the role that such a mechanism plays in meeting the oxygen needs of skeletal muscle.
Keywords: ATP, erythrocyte, Oxygen Transport
Skeletal muscle requires not only an adequate convective supply of oxygen (O2) to the muscle as a whole, but also a mechanism to appropriately distribute that O2 within the muscle to meet localized tissue needs. August Krogh (1919A,B) recognized, over 90 years ago, that a limitation resulting from the distance O2 can diffuse from an O2 source into O2 consuming tissue requires that O2 be appropriately distributed down to the level of the capillary. Krogh's work focused on diffusion from a single idealized capillary into a surrounding volume of tissue but did not address how sufficient O2 supply would be delivered to these capillaries to support the O2 needs of the tissue. The goal of this review is to provide an overview of microvascular O2 supply to the O2 consuming tissue and the potential role erythrocyte derived ATP may play in its regulation.
Based on many decades of research, we know that there must be an effective interaction between the convective and diffusive O2 transport processes within the microvasculature and the surrounding tissue to ensure a level of tissue oxygenation that meets but, importantly, does not exceed local metabolic needs. The results of experimental studies and computational modeling approaches have resulted in a basic working framework for the process shown in Figure 1. If blood were a simple homogenous fluid, then microvascular blood flow could be easily calculated from knowledge of microvascular architecture (diameter and length of individual vascular segments, and network organization), blood viscosity and the systemic blood pressure gradient across the organ. Knowing the haemoglobin O2 saturation of the blood and systemic haemoglobin concentration one could then calculate convective transport of O2 to the capillary bed and, based on the relationship between O2 saturation and partial pressure of O2 (pO2), the diffusive transport from the blood to supply the O2 needs of the tissue. Ultimately the density of capillaries in the muscle, O2 consumption rate in the tissue and the O2 supply in the capillaries determine the level of tissue oxygenation. However, as discussed below, the unique characteristics of microvascular rheology and the three-dimensional architecture of the microvasculature have a substantial affect on both the convective and diffusive transport of O2 and hence on tissue oxygenation.
Figure 1. Flow diagram of basic aspects of oxygen supply to skeletal muscle.
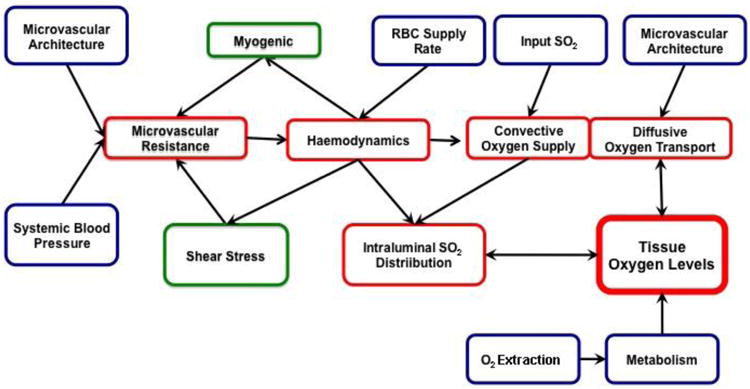
Skeletal muscle oxygen transport is focused on supplying adequate amounts of oxygen to meet tissue needs. As such, tissue oxygen levels can be considered the controlled variable. If tissue oxygen levels are to be maintained at a steady state, then the amount of oxygen delivered by the feeding vasculature must match tissue oxygen utilization (metabolism). The amount of oxygen delivered to the tissue is determined by systemic and local factors that influence both microvascular resistance and input oxygen saturation. Microvascular resistance in turn impacts haemodynamics and convective oxygen supply. The microvascular architecture and the oxygen levels within the feeding vessels each impact the magnitude of diffusive oxygen transport and ultimately tissue oxygen levels. Parameters which we can directly measure are indicated in blue, those which need to be computed based on known relationships and models are presented in red while those which must be assumed based on models which are in various stages of development are indicated in green.
The Complexities of Basic Microvascular Oxygen Transport
Microvasculature architecture is not static, even in the short term, as regulatory control based on pressure (myogenic control) and flow rate (shear stress) modulate arteriolar diameter while neural, humoral and metabolic regulatory systems attempt to adjust systemic and local microvascular pressure or blood flow. An additional complicating factor is that blood itself is not a homogenous fluid. As the microvessel diameter approaches the dimensions of the erythrocytes, the two phase nature of blood (erythrocytes suspended in plasma) becomes evident and the properties of convective transport change dramatically from what one would expect from larger blood vessels. In all vessels, erythrocyte concentration decreases near the vessel wall resulting in the mean erythrocyte velocity being higher than that of plasma. In the vessel, the higher erythrocyte velocity relative to plasma results in a reduction in vessel haematocrit (Fahraeus Effect) based on a simple mass balance. At arteriolar bifurcations, if blood flow is not uniformly distributed within branches, more of the blood volume with the high erythrocyte concentration will enter the high flow branch while the blood volume with the lower concentration will be “skimmed” into the lower flow branch. The effect of this preferential distribution of erythrocytes increases as the arteriolar diameter decreases. As blood flows through a cascade of bifurcations, vessel hematocrit falls from large arterioles to the capillary bed and erythrocytes become heterogeneously distributed. Since viscosity in microvessels is strongly dependent on hematocrit, an overall decrease in hematocrit causes a decrease in flow resistance (Fahraeus-Lindqvist effect). The heterogeneous distribution of erythrocytes impacts convective O2 transport which is further affected by the loss of O2 from arterioles (Duling & Berne 1970). Oxygen freely diffuses from the high pO2 of the arteriolar blood to the lower pO2 in the vessel wall and surrounding tissue. In addition, less well oxygenated erythrocytes flowing through nearby capillaries or paired venules also decrease O2 saturation in erythrocytes near the vessel wall (Ellsworth & Pittman 1990). At bifurcations, the radial gradient in O2 saturation has an effect similar to the gradient in haematocrit resulting in an overall decrease in O2 saturation from larger to smaller arterioles and substantially lower O2 saturations entering the capillary bed (Ellsworth et al 2009).
As O2 needs in local tissue regions change, the supply of O2 must similarly change to reestablish a new “steady state”. However, any change in flow distribution, especially at small arteriolar bifurcations, will have an amplified effect on convective O2 transport as both hematocrit and O2 saturation change in the same direction (i.e., O2 supply proportional to product of haematocrit and O2 saturation). Thus, small changes in arteriolar diameter may result in larger changes in O2 supply due to this amplified preferential distribution of O2 supply relative to the change in flow distribution. Taking all of these factors into account (i.e., multiple regulatory systems, complex microvascular rheology), one should expect that microvascular O2 supply will be dynamically adjusting to match O2 need under all metabolic conditions.
Toward an Understanding of Factors Controlling Microvascular O2 Delivery: Role of Erythrocytes
Although much is understood about the biophysics of the microcirculation, the definition of the mechanism(s) by which the change in tissue O2 need is communicated to the vasculature and by which the vasculature alters O2 supply to continuously meet this need remains to be fully understood. Joseph Barcroft stated that “the cell takes what it needs and leaves the rest” (Barcroft 1914). He went on to suggest a scenario by which this could occur with first “the call for oxygen”, a signal within the tissue that indicates a deficit in O2. This initial signal would then have to be followed by “a mechanism by which the call elicits a response, the immediate response consisting of the carriage of oxygen to the tissue by the blood and its transference from the blood to the cell”. An early attempt to define a mechanism by which O2 supply could be adjusted to meet tissue needs was provided by Krogh (1919B). Although Krogh had no insights into the mechanism by which the tissue signaled its need for O2, he did suggest that a need for an increase in O2 supply could be met by augmentation of the surface area for diffusion by the recruitment of previously unperfused capillaries. However, subsequent studies have failed to identify a sufficient number of unperfused capillaries within skeletal muscle to make this mechanism effective (Poole et al 2011). Researchers have explored several neurovascular and humoral mechanisms by which O2 supply could be affected to meet the O2 need (Duling 1974, Hester 1993, Jackson 1987) but none has been able to provide for the acute spatial and temporal sensitivity required. Importantly, although these mechanisms could modulate perfusion, none provides a means by which O2 need is sensed or O2 supply is monitored.
To begin to address this critical question, Ellis et al (2012) developed a technique in which the surface O2 levels of a rat perfused extensor digitorum longus (EDL) muscle can be manipulated and levels of erythrocyte O2 saturation and supply rate in individual capillaries simultaneously determined. In this preparation, a reduction in the surface O2 tension from ∼150 mmHg to ∼10 mmHg, resulted in the establishment of a sink for O2 effectively decreasing tissue O2 levels. Their results established a close inverse relationship between the level of O2 on the muscle surface and erythrocyte supply rate, suggesting that the decrease in O2 tension induced the increase in O2 supply. Importantly, when surface O2 tension was restored, erythrocyte supply rate returned to previous levels implicating the presence of a dynamic regulatory system that continuously adjusts flow to meet tissue O2 needs. Although tissue O2 tension was not measured in these studies, the measured erythrocyte supply rate and haemoglobin O2 saturation levels in capillaries during the low O2 challenge were close to resting levels for the muscle isolated from its environment, suggesting that the increase in erythrocyte supply rate was able to adequately meet tissue O2 need.
Although the previous work demonstrates that the need for O2 can evoke an increase in O2 supply, it does not provide the means by which the fall in tissue O2 tension is sensed. In 1993, Stein and Ellsworth evaluated O2 transport in the hamster cheek pouch retractor muscle during severe hypoxia induced by a reduction in systemic hematocrit and ventilation with 10% oxygen (Stein & Ellsworth 1993). An important result of their study was that O2 content was more important than O2 tension in supplying O2 to resting skeletal muscle. This result suggested that the erythrocyte itself was somehow involved in determining tissue oxygenation over and above its role as a mobile O2 carrier since the erythrocyte is the only part of the O2 transport cascade that is influenced by O2 content. An important link between the erythrocyte and vasodilation was demonstrated using an experimental model that mimics the in vivo situation in which fully oxygenated erythrocytes enter a region of tissue that is undersupplied with O2. In this system, small isolated arterioles were double cannulated and placed in a chamber surrounded by buffer the O2 content of which was altered to provide a normal and reduced O2 tension. When vessels were perfused with buffer and the O2 tension of surrounding fluid was reduced to <20 mmHg (a model of increased O2 need), the vessel did not dilate. However, when the same vessels were subsequently perfused with fully oxygenated erythrocytes the vessel did dilate in response to a fall in the surrounding O2 tension (Dietrich et al 2000, Sprague et al 2010). This demonstrates that the erythrocyte is necessary for the vasodilation of these resistance vessels in response to reduced O2 tension. One confounding problem is that buffer containing erythrocytes is more viscous than buffer alone. Thus, one possibility in these studies is that the erythrocyte evoked a vasodilation as a result of an increase in shear stress on the endothelium. However, it is unclear how such an effect would be O2 dependent. To address this issue, additional studies were performed in which the viscosity of the perfusing buffer was increased by the addition of dextran. Under these conditions, the vessel again failed to dilate in response to reduced extraluminal O2 tension (Dietrich et al 2000). One interpretation is that the erythrocyte itself is the controlling factor in the dilatory response to reduced O2 tension. It is important to note that when these vessels were perfused with fully oxygenated erythrocytes, the time required for the vasodilation in response to reduced O2 tension was on the order of 500 msec (Dietrich et al 2000) implicating a physiological relevance to this response.
Role of Erythrocyte-released ATP
Although these studies implicate the erythrocytes in the vasodilation that is required to regulate delivery of O2 to skeletal muscle, they do not address the mechanism by which vasodilation is initiated. The first insights into such a mechanism were provided in a 1952 study in which Folkow suggested that arteriolar dilation observed in response to pump perfusion of a denervated cat hindlimb with blood was the result of the release of ATP (Folkow 1952). While it is likely that in these experiments the pump itself was responsible for the release of ATP, one could postulate that there might be physiological stimuli for such release as well. In 1992, Bergfeld and Forrester (1992) reported that healthy human erythrocytes release ATP in a controlled fashion in response to exposure to low O2 in the presence of hypercapnia. Subsequently, Ellsworth et al (1995) confirmed these results using hamster erythrocytes and established that it was reduced O2 tension that was required for ATP release. Low O2-induced ATP release has been shown to occur with erythrocytes from healthy humans (Sprague & Ellsworth 2012), rabbits (Sprague et al 2002) and rats (Jagger et al 2001).
A possible scenario by which erythrocytes could play a role in controlling microvascular perfusion was provided by Ellsworth et al (1995). They suggested that the erythrocyte, by virtue of its capacity to release ATP in response to reduced O2 tension, could serve as both a sensor of O2 need and initiator of a response to direct increased O2 delivery to the local area in need. They proposed that as an erythrocyte enters a region of low tissue O2 tension, ATP is released, binds to purinergic receptors on the endothelium, directing O2 supply to the region in need. It has been previously reported that for a stimulus to be an effective controller of perfusion, it must evoke a response that is conducted to upstream supply arterioles. (Kurjiaka & Segal 1995). Thus, for ATP to be an important factor in blood flow control, it must evoke such a conducted response. A conducted response can result from alterations in membrane potential on the endothelial or smooth muscle cells or a change in the state of contractility of the smooth muscle cells usually the result of alterations in the level of cGMP or intracellular free Ca2+ (Ellsworth et al 2009).
In a study using in situ video microscopy, it was determined that the intraluminal application of ATP, at levels similar to those which could be released by erythrocytes as they perfuse a small arteriole, did evoke a significant increase in upstream diameter (McCullough et al 1997). In a subsequent study, it was shown that similar amounts of ATP applied into the lumen of collecting venules also induced an increase in arteriolar diameter of a similar magnitude (Collins et al 1998). Importantly, these studies suggested that the signal for the arteriolar dilation crossed the intervening capillary bed, most likely via conduction through the endothelial cells. These studies support a role for the erythrocyte in communicating local tissue O2 needs to the vasculature with the result an increase in O2 supply to local tissue regions. This view is strengthened further by the finding that when ATP was applied near a capillary and erythrocyte supply rate in that capillary determined, not only did erythrocyte supply rate increase but O2 tension in the tissue surrounding the capillaries did as well (Ellsworth 2000).
Signaling pathway for low O2-induced ATP release from erythrocytes
If exposure to low O2 tension is an important physiological stimulus for ATP release from erythrocytes, then such exposure must be coupled to a discrete, well regulated signaling pathway. Within the erythrocyte, membrane bound haemoglobin would be the first to off-load its O2. This decrease in O2 saturation induces a conformational change in these haemoglobin molecules that could lead to local membrane deformation and, thereby, serve as an initiator of a signaling pathway (Jagger et al 2001, Wan et al 2008, Sridharan et al 2010, Forsyth et al 2011). Importantly, it has been reported that membrane deformation and stress can lead to the activation of heterotrimeric G proteins of the Gi subtype (Chachisvilis et al 2006, Ngai & Yao 2010, Nobles et al 2005).
Components of a signaling pathway that links exposure of erythrocytes to low O2 tension with ATP release have been identified with the initial step the activation of Gi (Olearczyk et al 2004). It is of interest that in erythrocytes activation of Gi does not inhibit but rather activates adenylyl cyclase leading to increases in intracellular cAMP (Olearczyk et al 2004, Shrideran et al 2010). We propose that in the low O2-signaling pathway, increases in cAMP lead to activation of protein kinase A (PKA) (Sprague et al 2001) and the cystic fibrosis transmembrane conductance regulator (CFTR) (Sprague et al 1998). Although CFTR was suggested to be a conduit for ATP release from cells, recent evidence suggests that this is not the case in this signaling pathway in erythrocytes. Shrideran, et al (2010) demonstrated that the final conduit for low O2-induced ATP release from erythrocytes is pannexin 1. The details of this pathway are depicted in Figure 2 and have been the subject of several recent reviews (Ellsworth et al 2009, Ellsworth & Sprague 2012, Sprague et al 2011, Sprague & Ellsworth 2012).
Figure 2. Proposed mechanism by which human erythrocytes participate in the sensing of tissue oxygen (O2) tension and stimulate increases in blood flow (O2 delivery) to match that need.
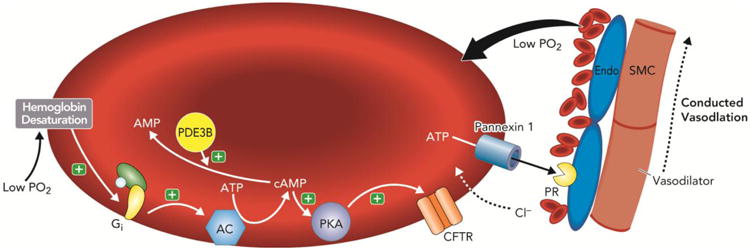
Under this model, when tissue O2 levels fall, O2 is released from haemoglobin contained within circulating erythrocytes. The fall in haemoglobin oxygen saturation results in a conformational change in haemoglobin and subsequent activation of the heterotrimeric G-protein, Gi. In this pathway activation of Gi stimulates adenylyl cyclase (AC) activity resulting in increases in 3′5′-adenosine monophosphate (cAMP), activation of protein kinase A (PKA) and the cystic fibrosis transmembrane conductance regulator (CFTR). The final conduit for ATP release in response to exposure to low O2 is pannexin 1. The cAMP required for ATP release is hydrolyzed by phosphodiesterase 3 (PDE3B). Released ATP binds to purinergic receptors (PR) on the endothelium (Endo) stimulating production of vasodilators that cause both local relaxation of vascular smooth muscle (SMC) and dilation that is conducted to upstream resistance vessels. (+) = activation.
Several studies have confirmed that the release of ATP via this erythrocyte pathway is critically dependent upon an increase in intracellular cAMP (Sprague et al 2001, Sprague et al 2011). The local concentration of this second messenger is the result of the balance between its synthesis by adenylyl cyclase and its hydrolysis by phosphodiesterase 3 (PDE3). Any imbalance between the synthetic and degradation rates of cAMP could result in impaired low O2-induced ATP release (Hanson et al 2010). Thus, increased degradation of cAMP via increases in PDE3 activity would limit ATP release while the failure to adequately limit cAMP synthesis and localize this second messenger could result in unwanted activation of other signaling cascades (Adderley et al 2011). Clearly, the precise regulation of cAMP levels and the localization of cAMP to this signaling pathway are of critical importance for low O2-induced ATP release from erythrocytes. The central role of PDE3 in regulating cAMP levels in this pathway suggests an intriguing site for pharmacological interaction with the process.
One of the more challenging problems has been, and remains, elucidation of the mechanism by which desaturation of haemoglobin leads to activation of Gi in the low O2 signaling pathway. As noted above, one possibility is that the initiating event is related to the conformational change in membrane-associated haemoglobin induced by the offloading of O2 (Jagger et al 2001, Wan et al 2008, Sridharan et al 2010, Forsyth et al 2011). Under this hypothesis, desaturation of haemoglobin would result in local membrane deformation leading to biomechanical activation of Gi. Jagger et al (2001) demonstrated that ATP release in response to low O2 levels was prevented if haemoglobin was maintained in its fully saturated conformation by carbon monoxide. In addition, Shrideran demonstrated that reductions in erythrocyte deformability result in impaired low O2-induced ATP release (Sridharan et al 2010)
Although the signaling pathway described above has several components that must be activated for ATP release to occur, if low O2-induced ATP release is physiologically relevant, the response must occur in msec. Recently, Goldman et al (2012) constructed a dynamic, computational model of this pathway using reported time constants for activation of the various components. The model predicts levels of ATP release consistent with measurements obtained over a wide range of haemoglobin O2 saturations (sO2). Importantly the release is predicted to occur within msec. The physiological relevance of this ATP release with respect to the control of microvascular flow was shown in a separate computational model (Goldman et al 2012). Results of the model confirmed that intravascular ATP release from erythrocytes in response to low O2 tension could result in local vasodilation in arterioles and conducted vasodilation from capillaries since the levels of O2 saturation present in the microcirculation were capable of stimulating this response.
Timing of Response to Reduced Tissue O2 Tension
As stated above, if low O2-induced ATP release from erythrocytes is an important regulator of local O2 delivery, then the time course for sensing a local reduction in tissue O2 tension and the vascular response must be rapid (Figure 3). In arterioles in which flow velocities can be as high as 1 cm/sec, the O2 saturation of slow moving erythrocytes near the wall reflects tissue O2 levels (Moschandreou et al 2010) making them ideal sensors for regulating O2 supply. Since the plasma gap between the erythrocyte and the endothelium is less than 1 μm, the erythrocyte would only travel ∼0.5 mm before the ATP which was released from the erythrocyte reaches the purinergic receptor on the endothelium. The time from receptor activation and vasodilation at an upstream location is determined by the characteristics of the vascular control system.
Figure 3. Timing of erythrocyte signaling.

When a well-oxygenated erythrocyte enters a region of lower oxygen tension (pO2), oxygen diffuses from the erythrocyte reducing its oxygen saturation (∼25 msec). The reduction in oxygen saturation initiates the release of ATP from the erythrocyte (∼25-75 msec) which diffuses across the plasma layer to bind with purinergic receptors on the endothelium (< 1.0 msec). At an average erythrocyte velocity in a capillary of 125-μm/sec, the erythrocyte will travel approximately 6 to 12 μm from the point it experiences the decrease in tissue pO2 until it signals the endothelium, that is, from one to two erythrocyte lengths or less than the length of a capillary endothelial cell.
In capillaries, computational modeling suggests that the time from the instant the erythrocyte experiences a significant decrease in tissue O2 tension until it signals the endothelium via the release of ATP is ∼ 50 to 75 msec (or ∼125 to 175 msec for a peak response) based on the assumption that timing for O2 saturation dependent release is similar to that reported for shear dependent release (Wan et al 2008). If one assumes a mean transit time across a capillary bed of 4000-5000 msec, then the effect can be considered nearly instantaneous. At an average erythrocyte velocity of 125 μm/sec the cell will travel ∼ 6 to 12 μm before the signal reaches the purinergic receptor. Since capillary diameters are less than the diameter of the erythrocyte, the cells must deform to pass through the capillary bed, the effect of which is to enhance O2 offloading by exposing a large area of the erythrocyte to the surrounding tissue. From an O2 regulatory perspective this also means that almost all ATP released from the erythrocyte is only a fraction of a micron from the endothelial wall and hence the ability of each erythrocyte to communicate with the endothelium is also enhanced. The continual arrival of new erythrocytes to the capillary bed provides for a dynamic system in which the status of tissue oxygenation can be continuously monitored and rapidly signaled to the capillary endothelium to communicate to the arteriolar tree for flow adjustments to maintain appropriate tissue oxygenation (Ellis et al 2012).
Recently a micro-delivery system to control O2 availability to highly localized regions of the microvascular bed within intact skeletal muscle tissue has been developed (Ghonaim 2013). The goal of these studies was to determine if changing haemoglobin O2 saturation of erythrocytes flowing through capillaries was sufficient to stimulate a microvascular flow response. Using this approach with a small exchange area (100 μm in diameter) it was shown that changing O2 saturation in individual capillaries was not sufficient to cause a flow response. However, a reduction in O2 tension in a larger exchange area (1000 × 200 μm) containing 3-4 capillaries within a single network was sufficient to induce conducted micro-vascular responses. Computational modeling showed that the larger exchange area was required to increase ATP release not only in the immediate region of the exchange area but also downstream resulting in a larger proportion of the endothelium contributing to the conducted signal (Ghonaim et al 2013). Thus, these studies provide convincing evidence supporting an erythrocyte-initiated O2 saturation-dependent signaling mechanism that can be localized to discrete tissue regions.
The Physiological Importance of Tightly Regulated O2 Delivery to Skeletal Muscle
There are two primary approaches to achieve an increase in tissue O2 levels in skeletal muscle that differ primarily in the proximity of the flow change to the tissue region in need. Specifically, flow could either be increased to all regions of the muscle or it could be specifically increased only within the vessels supplying the tissue region in need of O2. Using a computational model of O2 transport (Goldman and Popel 2000) based on a single arteriole feeding four capillary networks, the efficiency of each of these approaches was evaluated (Sprague et al 2010). The premise used was that the flow in three of the four capillary networks adequately supplied their local tissue region while the 4th tissue region was undersupplied. The model convincingly demonstrated that if blood flow were increased to all 4 capillary networks equally, as would occur with an increase in total flow, tissue O2 levels would increase in all 4 regions. Although this response to a local increase in O2 need in one region did achieve appropriate tissue oxygenation in that region, the 3 previously adequately supplied regions were now oversupplied with O2 which can be detrimental. If instead of increasing blood flow within all 4 capillary networks, flow was increased only in the network feeding the region in need, then all tissue regions could be appropriately supplied with O2. The latter mechanism for selected increases in blood flow is consistent with the appropriate response to Barcroft's “call for oxygen” to specific areas with increased O2 requirements.
Erythrocyte-Released ATP Alters the O2 Supply Model
If one incorporates the concept of the erythrocyte as the sensor of O2 need and, via the release of ATP, an effector of a mechanism to direct blood flow to specific regions based on O2 need within an organ, then Figure 1 above becomes significantly more complex (Figure 4). When tissue O2 need exceeds the capacity of the microvasculature to supply O2 by convection and diffusion, tissue oxygen tension falls, O2 extraction from nearby erythrocytes is increased, haemoglobin O2 saturation decreases, the amount of ATP released increases and a conducted signal is initiated which enhances oxygen supply. The efficiency in this response is influenced not only by the kinetics of ATP release but also by the rate of diffusion of ATP from the releasing erythrocyte to the purinergic receptor on the endothelium. In arterioles, ATP released from erythrocytes near the vessel wall would have a short distance to diffuse to initiate a local vasodilation which would also be conducted upstream. If the O2 flux from the arteriole was sufficient to decrease O2 saturation of erythrocytes further from the vessel wall, the impact on flow would be prolonged since these erythrocytes would be carried downstream before releasing ATP. This released ATP would have a greater diffusion distance but ultimately would reach the endothelium and contribute to the signal for dilation. In capillaries, ATP released from erythrocytes has only a small gap to diffuse across to reach purinergic receptors since the erythrocytes must deform to traverse the capillary bed. Although capillaries do not dilate, the conducted signal initiated on the endothelium can be transmitted to arterioles upstream of these capillaries thereby increasing O2 supply to this region of tissue. Other factors will also influence the impact of erythrocyte ATP release including the rate of ATP degradation, the kinetics of purinergic signaling and mediator release as well as the rate and extent of conduction. The more widespread the deficiency in tissue O2 supply, the more ATP would be released at all levels of the microvascular bed and the greater the increase in convective O2 supply to meet that need.
Figure 4. Flow diagram of dynamic regulation of oxygen supply to skeletal muscle incorporating erythrocyte released ATP.
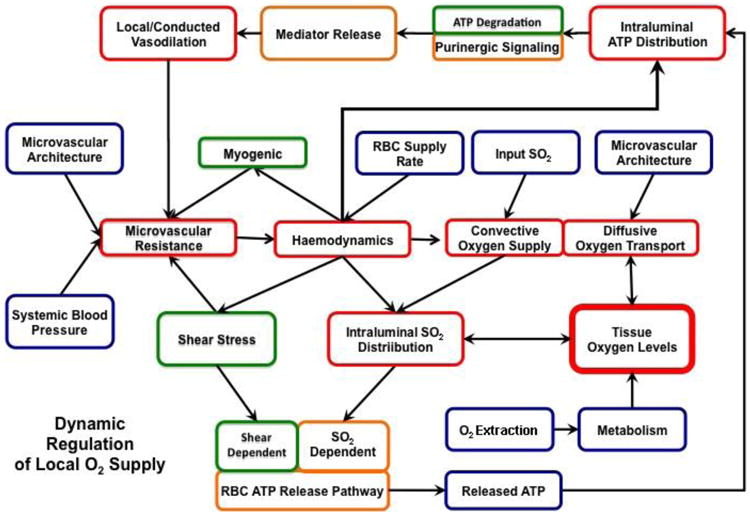
Skeletal muscle oxygen transport is focused on supplying appropriate amounts of oxygen to meet localized tissue needs. Incorporating erythrocyte-derived ATP into the oxygen transport construct presented in Figure 1, provides a mechanism by which oxygen can be directed to specific localized regions of the tissue to meet local oxygen needs. In this scenario, ATP released from the erythrocyte in response to reduced oxygen tension interacts with purinergic receptors on the endothelium and induces a local and conducted vasodilation that decreases microvascular resistance. The resulting local increase in flow enhances convective oxygen supply to the specific region of the tissue that initiated the “call for oxygen”. Parameters which we can directly measure are indicated in blue, those which need to be computed based on known relationships and models are presented in red while those which must be assumed based on models which are in various stages of development are indicated in green.
Defects in low O2-induced ATP release from human erythrocytes may have pathological consequences
If erythrocyte-derived ATP is an important contributor to the matching of O2 delivery with need in skeletal muscle, then it could be anticipated that pathological conditions in which the distribution of O2 supply relative to demand is compromised might have, as a component, a defect in the release of ATP from erythrocytes. One such human condition is type 2 diabetes (DM2). Patients with diabetes have decreased muscle blood flow both at rest and with exercise and a major complication of their disease is peripheral vascular insufficiency and delayed wound healing (McVeigh et al 1992, Mokdad et al 2000). Although direct studies of the skeletal muscle microcirculation are not possible in humans, such studies have been performed in several animal models of diabetes (Behnke et al 2002, Frisbee 2001, Padilla el al, 2006). These studies demonstrate reductions in capillary erythrocyte flux, convective O2 delivery and diffusive O2 transport. Importantly, in contrast to erythrocytes from healthy humans, cells from humans with DM2 do not release ATP in response to low O2 and fail to stimulate dilation of isolated-perfused skeletal muscle arterioles exposed to reduced extraluminal O2 tension, a model of increased tissue O2 need (Sprague et al 2010, Sprague et al 2011). Although the exact defect in signaling that results in the loss of low O2-induced ATP release remains under investigation, it is clear that expression of the heterotrimeric G protein, Gαi2 is selectively reduced in erythrocytes of humans with DM2 (Sprague et al 2006). Such a defect would lead to reduced ATP release when the cells are exposed to reduced O2 tension.
Since a local increase in cAMP is critical for low O2-induced ATP release from erythrocytes, inhibiting the degradation of this second messenger should restore ATP release from DM2 erythrocytes and stimulation of isolated arterioles. In a recent study, it was determined that after pretreatment with the PDE3 inhibitor, cilostazol, DM2 erythrocytes released ATP in response to exposure to low O2 and that the magnitude of that release was not different from that seen with cells from healthy humans (Sprague et al 2011). Of greater physiological importance, cilostazol rescued the dilatory effect of DM2 erythrocytes in isolated skeletal muscle arterioles exposed to reduced O2 tension in a concentration-dependent manner (Sprague et al 2011). Taken together, these results suggest that use of a PDE3 inhibitor in humans with DM2 could be of benefit in the prevention and/or treatment of the associated peripheral vascular disease.
It is important to recognize that there are other physiologically relevant stimuli for ATP release from erythrocytes in addition to exposure to low O2. One such stimulus is activation of the erythrocyte prostacyclin (PGI2) receptors (Sprague et al 2008). PGI2 is released from the endothelium in response to exposure to shear stress and in response to application of ATP in some vascular beds (Ragazzi & Chinellato 1992). The pathway for PGI2-mediated ATP release has been well characterized (Figure 5).
Figure 5. Proposed signaling pathway for ATP release from erythrocytes in response to activation of the prostacyclin (PGI2) receptor (IPR).
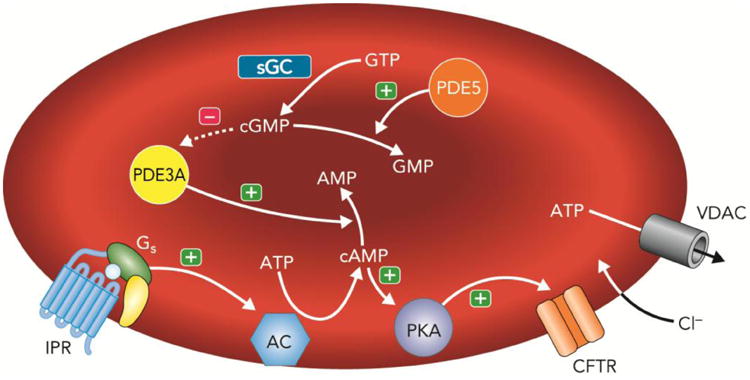
Exposure of human erythrocytes to PGI2 or its analogs results in activation of the heterotrimeric G protein, Gs. In this pathway activation of Gi stimulates adenylyl cyclase (AC) activity resulting in increases in 3′5′-adenosine monophosphate (cAMP) leading to activation of protein kinase A (PKA) and the cystic fibrosis transmembrane conductance regulator (CFTR). The final conduit for ATP release in response to IPR activation is the voltage dependent anion transporter (VDAC). The cAMP required for ATP release is hydrolyzed by phosphodiesterase 3 (PDE3A). Increases in intracellular cGMP, which is hydrolyzed by PDE5, inhibit the activity of PDE3A and enhance IPR-mediated ATP release. (+) = activation; (−) = inhibition.
Although several components are shared by both signaling pathways, ATP release from the PGI2 pathway differs in that it involves direct receptor-mediated activation of Gs and a distinct final conduit for ATP release, namely the voltage-dependent anion channel (VDAC) (Sridharan et al 2012). It is interesting to note that in erythrocytes of humans with DM2 in which low O2-induced ATP release is compromised, the response to PGI2 is significantly potentiated (Knebel et al 2013). While this “compensation” could help increase flow to skeletal muscle, unlike the response to low O2, the increase in flow would not be directed selectively to areas of increased O2 need (Sprague et al 2010). Thus, the response to PGI2 would result in increases in bulk blood flow and the over-perfusion of some areas of the tissue. This illustrates the potentially critical role of low O2-induced ATP release from erythrocytes in the maintenance of normal tissue oxygenation.
Summary and Conclusions
We have established that erythrocytes from healthy humans, rats, hamsters and rabbits release ATP in response to reduced O2 tension in vitro and have determined the components of a signaling pathway within the erythrocyte which is likely responsible for this controlled release. In addition, we have determined that ATP, at concentrations similar to those which would be released in vivo, induces a conducted vasodilation when applied intraluminally in both arterioles and venules in situ. Using an in vitro system to mimic the in vivo situation in which oxygenated blood enters a region of low oxygen tension, we established that only in the presence of erythrocytes that release ATP does low O2 exposure result in a significant vasodilation. We have evidence to support the physiological relevance of this mechanism in terms of the time course for the response. Although evidence exists regarding the measured time for ATP release from erythrocytes in response to shear stress, the time required for the O2 saturation dependent release has not been determined with sufficient accuracy to verify the dynamic model of the signaling pathway (Goldman et al 2012). Sove et al (2013) recently proposed a method for measuring the dynamics of ATP release using a microfluidic device along with a computational model demonstrating the feasibility of detecting release times on the order of 10s of milliseconds. Importantly, knowledge of the alterations in the response time associated with diabetes could provide valuable insights into the dynamics of the signaling pathway and may help identify therapeutic targets to help ameliorate the perfusion defects associated with this pathology.
The establishment of erythrocyte-derived ATP as an integral component in the appropriate supply of oxygen to skeletal muscle via alteration in the distribution of perfusion will require that the mechanism be confirmed in vivo.
Acknowledgments
The authors which to thank the numerous students, research assistants and colleagues who have contributed to the ideas presented here. Work from the author's laboratories was supported by the National Institutes of Health, the American Diabetes Association, the American Heart Association, the Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council and the Heart and Stroke Foundation of Ontario. We thank J.L. Sprague for inspiration
Footnotes
Conflict of Interest: There are no conflicts of interest
References
- Adderley SP, Sridharan M, Bowles EA, Stephenson AH, Sprague RS, Ellsworth ML. Inhibition of ATP release from erythrocytes: a role for EPACs and PKC. Microcirc. 2011;18:128–135. doi: 10.1111/j.1549-8719.2010.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcroft J. The Respiratory Function of Blood. Cambridge University Press; Cambridge: 1914. [Google Scholar]
- Behnke BJ, Kinding CA, McDonough P, Poole DC, Sexton WL. Dynamics of microvascular oxygen pressure during rest-contraction transition in skeletal muscle of diabetic rats. Am J Physiol. 2002;283:H926–932. doi: 10.1152/ajpheart.00059.2002. [DOI] [PubMed] [Google Scholar]
- Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovas Res. 1992;26:40–47. doi: 10.1093/cvr/26.1.40. [DOI] [PubMed] [Google Scholar]
- Chachisvilis M, Zang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci. 2006;103:15463–68. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DM, McCullough WT, Ellsworth ML. Conducted vascular responses: Communication across the capillary bed. Microvasc Res. 1998;56:43–53. doi: 10.1006/mvre.1998.2076. [DOI] [PubMed] [Google Scholar]
- Dietrich HH, Ellsworth ML, Sprague RS, Dacey RG., Jr Red blood cell regulation of microvascular tone through adenosine triphosphate. Am J Physiol. 2000;278:H1294–H1298. doi: 10.1152/ajpheart.2000.278.4.H1294. [DOI] [PubMed] [Google Scholar]
- Duling B. Oxygen sensitivity of vascular smooth muscle. II. In vivo studies. Am J Physiol. 1974;227(1):42–49. doi: 10.1152/ajplegacy.1974.227.1.42. [DOI] [PubMed] [Google Scholar]
- Duling BR, Berne RM. Longitudinal gradients in periarteriolar oxygen tension. A possible mechanism for the participation of oxygen in local regulation of blood flow. Circ Res. 1970;27:669–678. doi: 10.1161/01.res.27.5.669. [DOI] [PubMed] [Google Scholar]
- Ellis CG, Milkovich S, Goldman D. What is the efficiency of ATP signaling from erythrocytes to regulate distribution of o(2) supply within the microvasculature? Microcirc. 2012;19(5):440–50. doi: 10.1111/j.1549-8719.2012.00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML. The red blood cell as an oxygen sensor: what is the evidence? Acta Physiol Scand. 2000;168:551–9. doi: 10.1046/j.1365-201x.2000.00708.x. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology. 2009;24:107–16. doi: 10.1152/physiol.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol. 1995;269:H2155–61. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Pittman RN. Arterioles supply oxygen to capillaries by diffusion as well as by convection. Am J Physiol. 1990;258:H1240–43. doi: 10.1152/ajpheart.1990.258.4.H1240. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Sprague RS. Regulation of blood flow distribution in skeletal muscle: role of erythrocyte-released ATP. J Physiol. 2012;590(20):4985–91. doi: 10.1113/jphysiol.2012.233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkow B. A critical study of some methods used in investigations on the blood circulation. Acta Physiol Scandinav. 1952;27:118–20. doi: 10.1111/j.1748-1716.1953.tb00929.x. [DOI] [PubMed] [Google Scholar]
- Forsyth AM, Wan J, Owrutsky PD, Abkarian M, Stone HA. Multiscale approach to link red blood cell dynamics, shear viscosity, and ATP release. Proc Natl Acad Sci. 2011;108(27):10986–91. doi: 10.1073/pnas.1101315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisbee JC. Impaired dilation of skeletal muscle microvessels to reduce oxygen tension in diabetic obese Zucker rats. Am J Physiol. 2001;281:H1568–74. doi: 10.1152/ajpheart.2001.281.4.H1568. [DOI] [PubMed] [Google Scholar]
- Ghonaim NW. University of Western Ontario - Electronic Thesis and Dissertation Repository. 2013. Investigating Conducted Microvascular Response To Localized Oxygen Delivery In Vivo Using A Novel Micro-Delivery Approach. Paper 1643. [Google Scholar]
- Ghonaim NW, Fraser GM, Ellis CG, Yang J, Goldman D. Modeling steady state SO2-dependent changes in capillary ATP concentration using novel O2 micro-delivery methods. Front Physiol. 2013;4:260. doi: 10.3389/fphys.2013.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D, Fraser GM, Ellis CG, Sprague RS, Ellsworth ML, Stephenson AH. Toward a multiscale description of microvascular flow regulation: o(2)-dependent release of ATP from human erythrocytes and the distribution of ATP in capillary networks. Front Physiol. 2012;3:246. doi: 10.3389/fphys.2012.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D, Popel AS. A computational study of the effect of capillary network anastomoses and tortuosity on oxygen transport. J Theor Biol. 2000;206(2):181–94. doi: 10.1006/jtbi.2000.2113. [DOI] [PubMed] [Google Scholar]
- Hanson MS, Stephenson AH, Bowles EA, Sprague RS. Insulin inhibits human erythrocyte cAMP accumulation and ATP release: role of phosphodiesterase 3 and phosphoinositide 3-kinase. Exp Biol & Med. 2010;235:256–62. doi: 10.1258/ebm.2009.009206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester RL. Uptake of metabolites by postcapillary venules: mechanism for the control of arteriolar diameter. Microvasc Res. 1993;46:254–61. doi: 10.1006/mvre.1993.1050. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Arteriolar oxygen reactivity: where is the sensor. Am J Physiol. 1987;253(22):H1120–26. doi: 10.1152/ajpheart.1987.253.5.H1120. [DOI] [PubMed] [Google Scholar]
- Jagger JE, Bateman RM, Ellsworth ML, Ellis CG. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am J Physiol. 2001;280:H2833–39. doi: 10.1152/ajpheart.2001.280.6.H2833. [DOI] [PubMed] [Google Scholar]
- Knebel SM, Elrick MM, Bowles EA, Zdanovec AK, Stephenson AH, Ellsworth ML, Sprague RS. Synergistic effects of prostacyclin analogs and phosphodiesterase inhibitors on cyclic adenosine 3′,5′ monophosphate accumulation and adenosine 3′5′ triphosphate release from human erythrocytes. Exp Biol Med. 2013;238(9):1069–74. doi: 10.1177/1535370213498981. [DOI] [PubMed] [Google Scholar]
- Krogh A. The number and distribution of capillaries in muscles with calculations of the oxygen pressure head necessary for supplying the tissue. J Physiol (Lond) 1919A;52:409–15. doi: 10.1113/jphysiol.1919.sp001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A. The supply of oxygen to the tissues and the regulation of the capillary circulation. J Physiol (Lond) 1919B;52:457–74. doi: 10.1113/jphysiol.1919.sp001844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurjiaka DT, Segal SS. Conducted vasodilation elevates flow in arteriole networks of hamster striated muscle. Am J Physiol. 1995;269:H1723–28. doi: 10.1152/ajpheart.1995.269.5.H1723. [DOI] [PubMed] [Google Scholar]
- McCullough WT, Collins DM, Ellsworth ML. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol. 1997;272:H1886–91. doi: 10.1152/ajpheart.1997.272.4.H1886. [DOI] [PubMed] [Google Scholar]
- McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, Andrews JW, Hayes JR. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetol. 1992;35:771–6. doi: 10.1007/BF00429099. [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Ford ES, Bowman BA, Nelson DE, Engelgau MM, Vinicor F, Marks JS. Diabetes trends in the U.S.:1990-1998. Diab Care. 2000;23:1278–83. doi: 10.2337/diacare.23.9.1278. [DOI] [PubMed] [Google Scholar]
- Moschandreou TE, Ellis CG, Goldman D. Mass transfer in a rigid tube with pulsatile flow and constant wall concentration. J Fluids Eng. 2010;132(8):81202. doi: 10.1115/1.4002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngai CY, Yao X. Vascular responses to shear stress: the involvement of mechanosensors in endothelial cells. Open Circ & Vasc J. 2010;3:85–94. [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci. 2005;102:18706–11. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Heterotrimeric G protein Gi is involved in a signal transduction pathway for ATP release from erythrocytes. Am J Physiol. 2004;286:H940–45. doi: 10.1152/ajpheart.00677.2003. [DOI] [PubMed] [Google Scholar]
- Padilla DJ, McDonough P, Behnke BJ, Kano Y, Hageman KS, Musch TI, Poole DC. Effects of Type II diabetes on capillary hemodynamics in skeletal muscle. Am J Physiol. 2006;291(5):H2439–44. doi: 10.1152/ajpheart.00290.2006. [DOI] [PubMed] [Google Scholar]
- Poole DC, Copp SW, Hirai DM, Musch TI. Dynamics of muscle microcirculatory and blood-myocyte O2 flux during contractions. Acta Physiol (Oxf) 2011;202:293–310. doi: 10.1111/j.1748-1716.2010.02246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragazzi E, Chinellato A. Purinergic receptors, prostacyclin and atherosclerosis. Pharmacol Res. 1992;26:123–34. doi: 10.1016/s1043-6618(05)80125-2. [DOI] [PubMed] [Google Scholar]
- Sove RJ, Ghonaim N, Goldman D, Ellis CG. A computational model of a microfluidic device to measure the dynamics of oxygen-dependent ATP release from erythrocytes. PLoS One. 2013;8(11):e81537. doi: 10.1371/journal.pone.0081537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Achilleus D, Stephenson AH, Ellis CG, Ellsworth ML. A selective phosphodiesterase 3 inhibitor rescues low PO2-induced ATP release from erythrocytes of humans with type 2 diabetes: implication for vascular control. Am J Physiol. 2011;301(6):H2466–72. doi: 10.1152/ajpheart.00729.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Hanson MS, DuFaux EA, Sridharan M, Adderley S, Ellsworth ML, Stephenson AH. Prostacyclin analogs stimulate receptor-mediated cAMP synthesis and ATP release from rabbit and human erythrocytes. Microcirc. 2008;15(5):461–71. doi: 10.1080/10739680701833804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Olearczyk JJ, Stephenson AH, Lonigro AJ. The role of G protein β subunits in the release of ATP from human erythrocytes. J Physiol Pharmacol. 2002;53:667–74. [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML. Erythrocyte-derived ATP and perfusion distribution: Role of intracellular and intercellular communication. Microcirc. 2012;19:430–9. doi: 10.1111/j.1549-8719.2011.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Kleinhenz ME, Lonigro AJ. Deformation-induced ATP release from red blood cells requires cystic fibrosis transmembrane conductance regulator activity. Am J Physiol. 1998;275:H1726–32. doi: 10.1152/ajpheart.1998.275.5.H1726. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP release. Am J Physiol. 2001;281:C1158–1164. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Goldman D, Bowles EA, Achilleus D, Stephenson AH, Ellis CG, Ellsworth ML. Divergent effects of low O2 tension and iloprost on ATP release from erythrocytes of humans with type 2 diabetes: Implications for O2 supply to skeletal muscle. Am J Physiol. 2010;299:H566–73. doi: 10.1152/ajpheart.00430.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Stephenson AH, Bowles EA, Stumpf MS, Lonigro AJ. Reduced expression of Gi in erythrocytes of humans with type 2 diabetes is associated with impairment of both cAMP generation and ATP release. Diabetes. 2006;55:33588–93. doi: 10.2337/db06-0555. [DOI] [PubMed] [Google Scholar]
- Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol. 2010;299:H1146–52. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan M, Bowles EA, Richards JP, Krantic M, Davis KL, Dietrich KA, Stephenson AH, Ellsworth ML, Sprague RS. Prostacyclin Receptor-Mediated ATP Release from Erythrocytes Requires the Voltage-Dependent Anion Channel (VDAC) Am J Physiol. 2012;302:H553–9. doi: 10.1152/ajpheart.00998.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan M, Sprague RS, Adderley SP, Bowles EA, Ellsworth ML, Stephenson AH. Diamide decreases deformability of rabbit erythrocytes and attenuates low oxygen tension-induced ATP release. Exp Biol Med. 2010;235:1142–8. doi: 10.1258/ebm.2010.010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JC, Ellsworth ML. Capillary oxygen transport during severe hypoxia: role of hemoglobin oxygen affinity. J Appl Physiol. 1993;75:1601–7. doi: 10.1152/jappl.1993.75.4.1601. [DOI] [PubMed] [Google Scholar]
- Wan J, Ristenpart WD, Stone HA. Dynamics of shear-induced ATP release from red blood cells. Proc Natl Acad Sci. 2008;105(43):16432–7. doi: 10.1073/pnas.0805779105. [DOI] [PMC free article] [PubMed] [Google Scholar]