

Abstract

Chromatin modifying proteins are attractive drug targets in oncology, given the fundamental reliance of cancer on altered transcriptional activity. Multiple transcription factors can be impacted downstream of primary target inhibition, thus making it challenging to understand the driving mechanism of action of pharmacologic inhibition of chromatin modifying proteins. This in turn makes it difficult to identify biomarkers predictive of response and pharmacodynamic tools to optimize drug dosing. In this report, we show that 89Zr-transferrin, an imaging tool we developed to measure MYC activity in cancer, can be used to identify cancer models that respond to broad spectrum inhibitors of transcription primarily due to MYC inhibition. As a proof of concept, we studied inhibitors of BET bromodomain containing proteins, as they can impart antitumor effects in a MYC dependent or independent fashion. In vitro, we show that transferrin receptor biology is inhibited in multiple MYC positive models of prostate cancer and double hit lymphoma when MYC biology is impacted. Moreover, we show that bromodomain inhibition in one lymphoma model results in transferrin receptor expression changes large enough to be quantified with 89Zr-transferrin and positron emission tomography (PET) in vivo. Collectively, these data further underscore the diagnostic utility of the relationship between MYC and transferrin in oncology, and provide the rationale to incorporate transferrin-based PET into early clinical trials with bromodomain inhibitors for the treatment of solid tumors.
Keywords: PET, lymphoma, prostate cancer, MYC, transferrin receptor, BRD4
Introduction
Several decades of research have shown that the altered transcriptional activity of cancer cells renders them sensitive to small-molecule inhibitors of chromatin regulatory proteins. This work culminated in the approval of the histone deacetylase inhibitor vorinostat for the treatment of diffuse large B-cell lymphoma.1 Since this milestone, several other promising targets have been identified that regulate other dimensions of chromatin biology, including EZH2 (a histone-lysine N-methyltransferase), and more recently BET bromodomain containing proteins, which bind to acetylated chromatin to regulate transcription.2,3
BET bromodomain containing protein, family member 4 (BRD4) was recently discovered from a large scale RNAi screen to promote the viability of hematological cancers.4 Coincident with this finding, (+)-JQ1, the first compound to potently and selectively inhibit BRD4, was disclosed, and its impressive antitumor effects have stimulated great excitement for BRD4 as a drug target.5 Adding to the enthusiasm, early mechanistic studies showed that BRD4 inhibition resulted in tumor cell death via the suppression of MYC expression and activity, providing a new approach to the longstanding challenge of inhibiting this highly important but “undruggable” oncogene.6−10 Since these observations, several other bioactive bromodomain inhibitors have been disclosed in the literature, and BRD4 has been found to regulate the survival of other malignancies, including many common solid tumors. There are now >10 open clinical trials accruing patients in the USA to evaluate bromodomain inhibitors in hematological malignancies or solid tumors.
In this era of precision medicine, the importance of companion diagnostics to maximize therapeutic benefit with effective drug dosing is understood and emphasized. In the case of MYC, we appreciated that better biomarkers to identify patients with hyperactive MYC and dynamic response assessment to therapy are urgently needed. To this end, we previously developed 89Zr-transferrin, a positron emitting radiotracer that measures intracellular MYC activity through the transferrin receptor (TFRC), which is a direct MYC target gene. We showed in preclinical prostate cancer models with genetically engineered MYC alleles that 89Zr-transferrin could measure MYC activity.11,12 The goal of this study was to determine if 89Zr-transferrin could be used to monitor the suppression of MYC with bromodomain inhibitors, a finding that we expect would have immediate implications for the design of ongoing and future clinical trials.
Experimental Section
General Methods
Unless otherwise stated, all cell lines were acquired from ATCC. CWR22Pc was provided by Marja Nevalainen of Thomas Jefferson University, and the TDM8 and L428 cells were provided by Dr. Anas Younes at Memorial Sloan Kettering Cancer Center. Lymphoma lines and CWR22Pc were grown in RPMI 1640 supplemented with 10% FBS, 100 U of penicillin/streptomycin, and 2 mM l-glutamine. MDA PCa 2b cells were grown in HPC1 (AthenaES, Halethorpe, MD) with 10% FBS, 100 U of penicillin/streptomycin, and 2 mM l-glutamine. (+)-JQ1 was a generous gift from Dr. James Bradner of Dana Farber Cancer Institute. (+)-JQ1 carrier (10% 2-hydroxypropyl-β-cyclodextrin) was purchased from Sigma-Aldrich. PFI-1 was purchased from Sigma-Aldrich. Prevalidated real time PCR primers were purchased from Qiagen. Antibodies to MYC, TFRC, and actin were purchased from Cell Signaling Technologies and secondary antibodies from Abcam. Human holo-transferrin was purchased from Sigma-Aldrich, and DFO was obtained from Macrocyclics. Zirconium-89 was produced at MSKCC via the 89Y(p,n)89Zr transmutation reaction on a TR19/9 variable-beam energy cyclotron (Ebco Industries Inc.). Iodine-131 was purchased from PerkinElmer.
In Vitro Assays
Cells were counted and plated at fixed cell concentrations between treatment arms. Cells were treated with (+)-JQ1, PFI-1, or vehicle for 48 h, and RNA was harvested with a RNeasy mini kit (Qiagen). Amount of RNA was quantified using a NanoDrop (ThermoScientific), and 1.5 μg of RNA was then converted to cDNA with a high capacity cDNA reverse transcription kit (AppliedBiosystems). Relative changes in MYC and TFRC mRNA levels were assessed with a PikoReal real time PCR cycler (Thermo Fisher Scientific). The data was normalized to the GAPDH control and then to MYC and TFRC levels in vehicle treated cells. For immunoblot and cellular uptake assays, cells were harvested at 72 h. For immunoblot, cell pellets were lysed in RIPA buffer with protease and phosphatase inhibitor cocktails (Calbiochem) and then resolved using 1D SDS PAGE. Antibodies were used at the following concentrations to probe the blots: 1:500 MYC, 1:750 TFRC, 1:20,000 actin. Appropriate secondary antibodies were used at 1:5000 for MYC and TFRC and 1:7500 for actin. Chemiluminescence was used to visualize protein expression. For transferrin uptake assays, intact cells adhered to tissue culture plates were washed with PBS and incubated with ∼0.5 μg of 131I-transferrin in PBS–1% BSA for 60 min at 37 °C. Standard wells with no cells were also incubated for an hour. After washing twice with PBS–1% BSA, the cell associated activity was harvested in 1 M NaOH (aq). For suspension cell lines a similar protocol was utilized. In brief, cells were resuspended in PBS–1% BSA at a concentration of 1 × 106 million cells/mL. ∼0.5 μg of 131I-transferrin was added to 1 mL aliquots of the cell suspension as well as to 1 mL of no-cell standards and incubated for 60 min on a shaker at 37 °C. The cells were then spun down at 3000 rpm for 3 min at 4 °C. Two washes with PBS–1% BSA were performed. The radioactivity was counted on a Wizard2 gamma counter, and the data was expressed as a % of total activity in the cell as compared to standards. For adherent cell lines, data was also normalized to cell count. Experiments were run in quintuplet.
Animal Studies
All animal studies were conducted in compliance with the Institutional Animal Care and Use Committee at MSKCC. Five to seven week old male CB-17 SCID were obtained from Taconic Farms (Deerwood, MD). The animals weighed approximately 20–25 g. These intact male mice were inoculated with 5 × 106 TDM8 human lymphoma cells into one or both flanks in a 1:1 mixture (v/v) of media and Matrigel (Corning). Tumors were palpable within 14 days of injection. A stock of 100 mg/mL JQ1 in DMSO was 20-fold diluted by dropwise addition of the (+)-JQ1 carrier, vortexed, and sonicated, yielding a 5 mg/mL final solution. Mice were then intraperitoneally injected every 12 h with freshly diluted (+)-JQ1 at 50 mg/kg or a similar volume of carrier containing 5% DMSO for a total of 5 doses. Tumors were then processed for protein extraction by mechanical homogenization in RIPA supplemented with protease and phosphatase inhibitors. Protein expression was then analyzed by immunoblot with the same protocol as for in vitro Western blots.
Conjugation of Holo-Tf to DFO
A 20 mg/mL solution of holo-Tf was prepared in saline, and 10 mg of the solution was buffer exchanged in a PD10 column (GE Healthcare) that had been prepped with 100 mM potassium bicarbonate at pH 9. A 7.5 mg/mL solution of DFO in DMSO was also prepared. 0.4 mg of this solution was added to the holo-Tf and incubated at 37 °C for 1 h. Another PD10 column was prepped with saline, and the DFO–Tf was then eluted with saline through this column into fraction. Concentrations of fractions were determined by NanoDrop, and concentrated fractions were combined.
Radiolabeling of 89Zr-DFO–Tf
89Zr in oxalic acid was buffered to pH 6.8 using 2 M potassium carbonate. 500 μL of HEPES was added, followed by DFO–Tf to achieve a final concentration of approximately 12 μg/mCi. This mixture was incubated at room temperature for an hour. The reaction was stopped by adding 10 μL of DTPA, and the mixture was added to a PD10 column prepped with saline. Fractions were eluted with saline and counted using a dose calibrator. Appropriate fractions were combined. Purity of the reaction was measured by radio iTLC.
Radiolabeling of 131I-Holo-Tf
A 10 mg/mL solution of holo-Tf in PBS was prepared fresh for each conjugation. 200 μL of this solution and an additional 50 μL of PBS were added to an iodogen-coated tube on ice. 131I was then added, and the mixture was incubated at room temperature for 5 min. The solution was then washed three times in a 30 kDa Amicon Spin Filter (Millipore). Purity was assessed via radio iTLC in 10% TFA. The 131I-holo-Tf was further diluted in 0.2% BSA–PBS before incubation with the cells.
Small-Animal PET Imaging and Biodistribution Studies
PET was conducted using a Focus 120 micro-PET scanner. Mice were injected with approximately 300 μCi of 89Zr-DFO–Tf via the tail veil. While anesthetized with 2% isoflurane the mice were imaged at various time points (4, 24, 48 h post injection). In the treated cohort, scans were timed to approximately 6–8 h after morning drug administration. Acquisitions were collected within an energy window of 350–750 keV and a coincidence-timing window of 6 ns. Mice were imaged for the time necessary to record 20 million coincident events. The data was converted into 2-dimentional histograms, and images were reconstructed by filter back-projection.
To evaluate the uptake of 89Zr-DFO–Tf in human xenografts, biodistribution studies were conducted following imaging. Animals (n = 5) were euthanized by CO2 asphyxiation after scans were completed. Blood, muscle, and tumor were harvested immediately following sacrifice. The tissues were weighed and counted using a gamma counter to assess 89Zr concentration. Calibration with known amounts of 89Zr was performed to determine the amount of activity in each organ. This activity was then decay corrected, and the percentage of the injected dose per gram (% ID/g) of tissue was calculated and reported.
Statistical Analysis
Data were analyzed using the unpaired, two-tailed Student t-test. Differences at the 95% confidence level (P < 0.05) were considered to be statistically significant.
Results
We first evaluated the impact of bromodomain inhibition in human lymphoma and prostate cancer models, two disease states known to frequently harbor hyperactive MYC and previously shown to be sensitive to BRD4 inhibition in select genetic backgrounds. We utilized four models: TDM8 (diffuse large B-cell lymphoma), Raji (Burkitt’s lymphoma), and the hormone sensitive prostate cancer cell lines CWR22Pc and MDA PCa 2b. TDM8 harbors the t(8;14)(q24;q32) chromosomal translocation characteristic of the highly morbid “double hit” lymphoma that results in hyperactive MYC.13 Raji also harbors this translocation, and was previously annotated to be sensitive to BRD4 inhibition with (+)-JQ1.8,14−16 No data was available for CWR22Pc and MDA PCa 2b, though prostate cancer models were previously shown to be sensitive to bromodomain inhibition.17 We chose to study the models with two bromodomain inhibitors, (+)-JQ1 and PFI-1, to minimize the likelihood that any effects we observed might be due to off target pharmacology associated with either drug.5,18
We first treated the cell lines with doses of drug over three logs to define a minimal bioactive dose. We found that the proliferation of all cell lines was potently inhibited by 1 μM or less (+)-JQ1, and 10 μM PFI-1 (Supplemental Figure 1). Lower doses did not result in measurable changes in cell number over 5 days (data not shown). Using these data, we next asked if MYC and TFRC mRNA were suppressed after 48 h of treatment. In all cases, we observed >50% suppression of MYC mRNA compared to control, and consistent suppression of TFRC mRNA (Figure 1A). Immunoblot analysis at 72 h showed that MYC and TFRC protein levels were reduced and, in most cases, completely ablated (Figure 1B). These data led us to investigate whether (+)-JQ1 and PFI-1 suppress the uptake of 131I-labeled transferrin in vitro. Consistent with the mRNA and protein data, a 72 h treatment with inhibitor resulted in statistically significant changes in transferrin uptake in all cell lines (Figure 1C, P < 0.05).
Figure 1.

BRD4 inhibitors suppress MYC and TFRC expression, and inhibit the uptake of transferrin in vitro. (A) Real time PCR data showing that (+)-JQ1 (500 nM TDM8, Raji, MDA PCa 2b; 1 μM CWR 22Pc) and PFI-1 (10 μM) suppress MYC and TFRC mRNA levels after 48 h of treatment. The data is expressed as percent changes in ΔCT values compared to control. (B) Immunoblot data showing that MYC and TFRC expression are reduced by treatment with BRD4 inhibitors for 72 h. (C) In vitro uptake data showing that BRD4 inhibitors suppress 131I-transferrin uptake 72 h post treatment.
Bromodomain inhibitors do not always impart antiproliferative effects by inhibiting MYC, altering instead the activity of other transcription factors.19−23 This presented to us the opportunity to ask whether TFRC expression and transferrin uptake are impacted by bromodomain inhibition in a genetic background in which MYC is not inhibited. During our survey of cell lines, we found that L428, a human, MYC positive Hodgkins’s lymphoma cell line, was highly sensitive to BRD4 inhibitors, but these antiproliferative effects were not due to MYC suppression. Indeed, neither MYC mRNA nor protein levels were measurably reduced by bioactive doses of (+)-JQ1 or PFI-1 (Figures 2A and 2B). Importantly, TFRC mRNA and protein were also not suppressed by drug, and 131I-transferrin uptake was not inhibited (Figure 2C). We next examined if either drug impacted MYC or TFRC mRNA levels in A549, a lung adenocarcinoma model whose proliferation was previously shown to be inhibited by (+)-JQ1 in a MYC-independent fashion.19MYC and TFRC were unaffected by (+)-JQ1 and PFI-1 (Supplemental Figure 2). Collectively, these data underscore the specificity of the relationship between MYC and TFRC expression and activity, and suggest that TFRC could serve as a biomarker to probe the mechanistic basis for tumor response to BRD4 inhibition.
Figure 2.

TFRC expression and transferrin biology is unaffected in a MYC positive cell line sensitive to BRD4 inhibition in a MYC-independent fashion. (A) Real time PCR data showing that MYC and TFRC mRNA levels are unaffected by bioactive doses of (+)-JQ1 and PFI-1 48 h after treatment. (B) Immunoblot data showing that protein levels of MYC and TFRC are not impacted by BRD4 inhibitors 72 h after treatment. (C) In vitro uptake data showing that 131I-transferrin uptake is not reduced by BRD4 inhibition.
Molecular changes in vitro are not always sufficiently large to be quantified in vivo with nuclear imaging, and on this basis, we next asked if pharmacological inhibition of MYC and TFRC can be measured in vivo with 89Zr-transferrin and PET. We first inoculated male SCID mice with TDM8 subcutaneous xenografts and injected treatment naive mice with 89Zr-transferrin to monitor uptake over time. Analysis of microPET data acquired serially over 48 h showed that tumor resolution was optimal at 48 h, and saturated between 24 and 48 h (Supplemental Figures 3A and 3B). Ex vivo biodistribution data showed that tumor uptake (∼9.5% ID/g) exceeded uptake in the muscle (∼1.5% ID/g) and blood pool activity (∼4.5% ID/g), providing the rationale to assay 89Zr-transferrin biodistribution at this time point postinjection (Supplemental Figure 3C).
A separate cohort of mice bearing bilateral subcutaneous TDM8 xenografts were treated with vehicle or (+)-JQ1 at 50 mg/kg, and the response to treatment was assessed via PET. The animals were administered the appropriate treatment and then 89Zr-DFO-transferrin approximately 2 h later. The mice were then treated with fresh (+)-JQ1 every 12 h until they had received 5 total doses. MicroPET images were acquired during this time. These images showed that tumors in the (+)-JQ1 treated arm had visually less 89Zr-DFO-transferrin uptake (Figure 3A). Ex vivo biodistribution confirmed this observation (Figure 3B). Lastly, immunoblot analysis of TDM8 tumors from a separate cohort of mice that received drug treatment but not the radiotracer showed that MYC and TFRC protein expression levels were suppressed by (+)-JQ1 treatment, as expected (Figure 3C).
Figure 3.
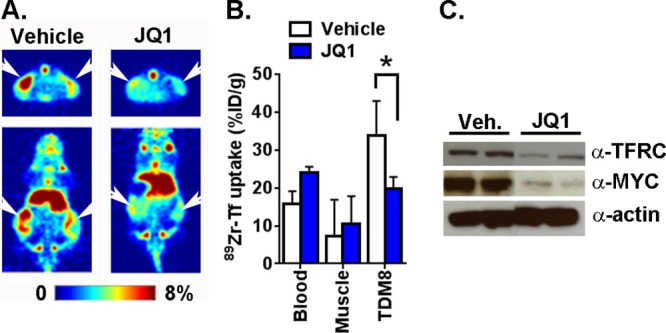
89Zr-DFO-transferrin measures treatment induced changes in MYC and TFRC expression. (A) Representative coronal and transverse slices of microPET images from mice bearing subcutaneous TDM8 xenografts and treated with vehicle or (+)-JQ1 (50 mg/kg, intraperitoneal injection, BID, 5 total doses). The site of the tumor is indicated with white arrowheads. (B) Biodistribution data showing the suppression of 89Zr-transferrin uptake in the TDM8 tumors treated with (+)-JQ1. *P < 0.01. (C) Immunoblot data from two xenografts per treatment arm showing the suppression of MYC and TFRC protein by (+)-JQ1 compared to vehicle.
Discussion
In this report, we show that MYC and TFRC expression are both inhibited in human lymphoma and prostate cancer models sensitive to bromodomain inhibitors. Moreover, we show that TFRC expression is not inhibited in two models known to be sensitive to BRD4 inhibition in a MYC-independent fashion. Finally, our data demonstrates that 89Zr-transferrin can noninvasively measure the suppression of intracellular MYC by the BRD4 inhibitor (+)-JQ1.
While the enthusiasm for clinical trials with bromodomain inhibitors grows, the importance of establishing biomarkers to define effective therapeutic intervention early is becoming more urgent. As clinical trials with targeted therapies have shown, molecular imaging tools are a powerful complement to tissue-based predictive biomarkers (e.g., 18F-FDG to monitor early response to imatinib in GIST or vemurafenib in BRAF V600E melanoma).24,25 Another unique consideration for bromodomain inhibitors is that the mechanism of tumor response can be complex and difficult to predict a priori. In this regard, our data suggesting that tumor uptake of transferrin is a faithful biomarker of MYC inhibition suggests that 89Zr-transferrin could be used as a pharmacodynamic biomarker in MYC driven indications, as well as research tool in humans to define the overall importance of MYC inhibition to tumor response to bromodomain inhibitors in tumors with more complicated genetic backgrounds. Lastly, the transferrin “response” could be used to profile preclinical models for alternate mechanisms by which bromodomain inhibition alters TFRC biology.
Acknowledgments
The authors acknowledge Valerie Longo and Dr. Pat Zanzonico of the Small Animal Imaging Core at MSKCC for technical assistance. M.G.D., D.E.S., and M.J.E. were supported by the Imaging and Radiation Sciences Bridge Program of MSKCC. D.E.S. was supported by the 2014 Rebecca and Nathan Milikowsky Prostate Cancer Foundation Young Investigator Award. M.J.E. was supported by the 2013 David H. Koch Young Investigator Award from the Prostate Cancer Foundation, and by the National Institutes of Health (R00CA172695, 1R01CA17661-01). Technical services provided by the MSKCC Small-Animal Imaging Core Facility and the MSKCC Radiochemistry & Molecular Imaging Probe Core, supported in part by NIH Cancer Center Support Grant P30CA008748-48, are gratefully acknowledged. NIH Shared Instrumentation Grant S10 RR020892-01, which provided funding support for the purchase of the Focus 120 microPET, is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.5b00882.
Antiproliferative data, real time PCR, and imaging data (PDF)
Author Contributions
‡ M.G.D. and K.E.C. contributed equally.
The authors declare the following competing financial interest(s): M.J.E. receives consulting fees from ORIC Pharmaceuticals, Inc.
Supplementary Material
References
- Kavanaugh S. M.; White L. A.; Kolesar J. M. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am. J. Health-Syst. Pharm. 2010, 67 (10), 793–7. 10.2146/ajhp090247. [DOI] [PubMed] [Google Scholar]
- Chang C. J.; Hung M. C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106 (2), 243–7. 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Y.; Chiang C. M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282 (18), 13141–5. 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478 (7370), 524–8. 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468 (7327), 1067–73. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandopadhayay P.; Bergthold G.; Nguyen B.; Schubert S.; Gholamin S.; Tang Y.; Bolin S.; Schumacher S. E.; Zeid R.; Masoud S.; Yu F.; Vue N.; Gibson W. J.; Paolella B. R.; Mitra S. S.; Cheshier S. H.; Qi J.; Liu K. W.; Wechsler-Reya R.; Weiss W. A.; Swartling F. J.; Kieran M. W.; Bradner J. E.; Beroukhim R.; Cho Y. J. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20 (4), 912–25. 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146 (6), 904–17. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler T.; Ghatak P.; Price D. H.; Conaway R.; Conaway J.; Chiang C. M.; Bradner J. E.; Shilatifard A.; Roy A. L. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PLoS One 2014, 9 (1), e87003. 10.1371/journal.pone.0087003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown M. R.; Bradner J. E. Therapeutic Strategies to Inhibit MYC. Cold Spring Harbor Perspect. Med. 2014, 4 (10), a014266. 10.1101/cshperspect.a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott C. J.; Kopp N.; Bird L.; Paranal R. M.; Qi J.; Bowman T.; Rodig S. J.; Kung A. L.; Bradner J. E.; Weinstock D. M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120 (14), 2843–52. 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland J. P.; Evans M. J.; Rice S. L.; Wongvipat J.; Sawyers C. L.; Lewis J. S. Annotating MYC status with 89Zr-transferrin imaging. Nat. Med. 2012, 18 (10), 1586–91. 10.1038/nm.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. J. Measuring oncogenic signaling pathways in cancer with PET: an emerging paradigm from studies in castration-resistant prostate cancer. Cancer Discovery 2012, 2 (11), 985–94. 10.1158/2159-8290.CD-12-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Grubor V.; Love C. L.; Banerjee A.; Richards K. L.; Mieczkowski P. A.; Dunphy C.; Choi W.; Au W. Y.; Srivastava G.; Lugar P. L.; Rizzieri D. A.; Lagoo A. S.; Bernal-Mizrachi L.; Mann K. P.; Flowers C.; Naresh K.; Evens A.; Gordon L. I.; Czader M.; Gill J. I.; Hsi E. D.; Liu Q.; Fan A.; Walsh K.; Jima D.; Smith L. L.; Johnson A. J.; Byrd J. C.; Luftig M. A.; Ni T.; Zhu J.; Chadburn A.; Levy S.; Dunson D.; Dave S. S. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (4), 1398–403. 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant B. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (40), 16669–74. 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emadali A.; Rousseaux S.; Bruder-Costa J.; Rome C.; Duley S.; Hamaidia S.; Betton P.; Debernardi A.; Leroux D.; Bernay B.; Kieffer-Jaquinod S.; Combes F.; Ferri E.; McKenna C. E.; Petosa C.; Bruley C.; Garin J.; Ferro M.; Gressin R.; Callanan M. B.; Khochbin S. Identification of a novel BET bromodomain inhibitor-sensitive, gene regulatory circuit that controls Rituximab response and tumour growth in aggressive lymphoid cancers. EMBO molecular medicine 2013, 5 (8), 1180–95. 10.1002/emmm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K.; Erikson J.; ar-Rushdi A.; Huebner K.; Croce C. M. The translocated c-myc oncogene of Raji Burkitt lymphoma cells is not expressed in human lymphoblastoid cells. Proc. Natl. Acad. Sci. U. S. A. 1985, 82 (9), 2900–4. 10.1073/pnas.82.9.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani I. A.; Dommeti V. L.; Wang X.; Malik R.; Cieslik M.; Yang R.; Escara-Wilke J.; Wilder-Romans K.; Dhanireddy S.; Engelke C.; Iyer M. K.; Jing X.; Wu Y. M.; Cao X.; Qin Z. S.; Wang S.; Feng F. Y.; Chinnaiyan A. M. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510 (7504), 278–82. 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Da Costa D.; Thanasopoulou A.; Filippakopoulos P.; Fish P. V.; Philpott M.; Fedorov O.; Brennan P.; Bunnage M. E.; Owen D. R.; Bradner J. E.; Taniere P.; O’Sullivan B.; Muller S.; Schwaller J.; Stankovic T.; Knapp S. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 73 (11), 3336–46. 10.1158/0008-5472.CAN-12-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood W. W.; Zejnullahu K.; Bradner J. E.; Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (47), 19408–13. 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A.; Frumm S. M.; Alexe G.; Bassil C. F.; Qi J.; Chanthery Y. H.; Nekritz E. A.; Zeid R.; Gustafson W. C.; Greninger P.; Garnett M. J.; McDermott U.; Benes C. H.; Kung A. L.; Weiss W. A.; Bradner J. E.; Stegmaier K. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discovery 2013, 3 (3), 308–23. 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Walker S. R.; Nelson E. A.; Cerulli R.; Xiang M.; Toniolo P. A.; Qi J.; Stone R. M.; Wadleigh M.; Bradner J. E.; Frank D. A. Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Mol. Cancer Ther. 2014, 13 (5), 1194–205. 10.1158/1535-7163.MCT-13-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z.; Huang B.; Wu X.; Zhang H.; Qi J.; Bradner J.; Nair S.; Chen L. F. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33 (18), 2395–404. 10.1038/onc.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Gholamin S.; Schubert S.; Willardson M. I.; Lee A.; Bandopadhayay P.; Bergthold G.; Masoud S.; Nguyen B.; Vue N.; Balansay B.; Yu F.; Oh S.; Woo P.; Chen S.; Ponnuswami A.; Monje M.; Atwood S. X.; Whitson R. J.; Mitra S.; Cheshier S. H.; Qi J.; Beroukhim R.; Tang J. Y.; Wechsler-Reya R.; Oro A. E.; Link B. A.; Bradner J. E.; Cho Y. J. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20 (7), 732–40. 10.1038/nm.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte L. H.; Costa L.; Teixeira J. P.; Lopes F.; Soares O.; Bastos A. L. 18F-FDG PET/CT in the management of GIST. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, S163–S163. [Google Scholar]
- McArthur G. A.; Puzanov I.; Amaravadi R.; Ribas A.; Chapman P.; Kim K. B.; Sosman J. A.; Lee R. J.; Nolop K.; Flaherty K. T.; Callahan J.; Hicks R. J. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J. Clin. Oncol. 2012, 30 (14), 1628–34. 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.